Introduction
Atherosclerosis is a chronic metabolic disorder,
which is characterized by the formation and accumulation of lipid
plaques in the arteries and by inflammatory responses (1–4). The
main lipid-laden cells are known as foam cells and are a key
attribute of atherosclerotic lesions (2–4). In
atherosclerotic lesions, macrophages that express scavenger
receptors on their plasma membranes uptake low-density lipoprotein
(LDL) deposited into blood vessel walls, and are converted into
foam cells. Foam cells can secrete various inflammatory cytokines
and promote the development of atherosclerosis (5). Therefore, preventing foam cell
formation by decreasing cholesterol influx and increasing
cholesterol efflux may be a potential treatment strategy for
atherogenesis.
Macrophage-specific reverse cholesterol transport
(RCT) is one of the most important high density lipoprotein
(HDL)-mediated cardioprotective mechanisms (6). RCT is a process during which
cholesterol in peripheral cells is effluxed into circulating HDL
and transported back into the liver for excretion in bile and feces
(7). The cholesterol efflux from
cells to HDL is a rate-limiting step of RCT (8). The family of ATP-binding cassette (ABC)
transporters, including ABCA1 and ABCG1, has been demonstrated to
serve a critical role in the RCT pathway by promoting the
translocation of cholesterol across cellular bilayer membranes
(9,10). ABCA1 enhances the cholesterol efflux
to lipid-poor apolipoproteins, such as apolipoprotein A-I (apoAI),
while ABCG1 increases the cholesterol efflux to HDL (11). The expression levels of ABCA1 and
ABCG1 are regulated, to a certain extent, by the peroxisome
proliferator-activated receptor γ (PPARγ) and liver X receptor α
(LXRα) (12).
Interleukin-32 (IL-32) was originally identified as
natural killer (NK) 5 transcript 4, induced by IL-18 in NK cells
(13). Due to its cytokine-like
characteristics and its critical role in inflammation, it was
renamed as IL-32 (13). IL-32 is
promoted by interferon-γ, IL-18 and IL-12 in various cell types,
such as epithelial cells. In addition, it induces the production of
various pro-inflammatory cytokines, including tumor necrosis
factor-alpha (TNF-α), IL-8, IL-1β and IL-6, as well as of numerous
chemokines in monocytes and macrophages (13). There are six isoforms of IL-32 by
alternative splicing, namely IL-32α, IL-32β, IL32γ, IL-32δ, IL-32ε,
and IL-32ζ (13), with IL-32α being
the most abundant transcript (14).
As IL-32 is a pro-inflammatory cytokine, its effects on various
inflammatory diseases, such as rheumatoid arthritis and
inflammatory bowel disease, have been explored (15). However, the role of IL-32 on
cholesterol efflux remains unknown. In the present study, IL-32α
was used to treat oxidized LDL (ox-LDL)-stimulated THP-1
macrophages in order to investigate the effects of IL-32α on
cholesterol efflux.
Materials and methods
Cell culture
The human monocyte THP-1 cell line was purchased
from the American Type Culture Collection (Manassas, VA, USA).
THP-1 cells were cultured in RPMI 1640 medium supplemented with 10%
(v/v) fetal calf serum, 0.1% nonessential amino acids, 100 U/ml
penicillin and 100 mg/ml streptomycin. Cells were incubated at 37°C
in a humidified atmosphere of 5% CO2. THP-1 cells were
treated with 160 nmol/l phorbol 12-myristate 13-acetate (PMA) for
72 h and fully differentiated into macrophages. The differentiated
THP-1 macrophages were then washed extensively with
phosphate-buffered saline (PBS), and cells were incubated in
serum-free medium containing 0.1% bovine serum albumin (BSA) and 50
µg/ml human ox-LDL (Anhui Yiyuan Biotechnology Co., Ltd., Anhui,
China) for a further 48 h. All reagents for cell culture were
obtained from Invitrogen (Thermo Fisher Scientific, Inc., Waltham,
MA, USA). PMA was purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). Recombinant human IL-32α was obtained from
R&D Systems (Minneapolis, MN, USA).
Morphological examination
THP-1 macrophages were incubated in chamber slides
in serum-free medium (containing 0.1% BSA) with or without IL-32α
(20, 50, and 100 ng/ml) for 6 h, and then exposed to 50 µg/ml
ox-LDL for 24 h. Cells were washed with PBS for three times, fixed
with 5% formalin solution in PBS for 30 min, and stained with Oil
Red O for a further 30 min. Subsequently, cells were counterstained
with hematoxylin for 5 min. Finally, images of the stained cells
were captured using a Leica optical microscope (Leica Microsystems
GmbH, Wetzlar, Germany). The number of stained cells was counted in
five random fields (magnification, ×100) for each group.
Plasmid construction
The pcDNA3.1-PPAR-γ plasmid was constructed by
inserting the human PPAR-γ cDNA (Genbank no., NM_138711.3) into a
pcDNA3.1 mammalian expression vector. The full-length cDNA sequence
was amplified using Takara Ex Taq DNA polymerase (Takara Bio, Inc.,
Otsu, Japan), according to the manufacturer's protocol. The primers
used were as follows: PPAR-γ forward, 5′-GGGGGTACCATGACCATGGTTG-3′
and reverse, 5′-GGGCTCGAGCTAGTACAAGTCC-3′. Thermocycling conditions
were as follows: Initial denaturation at 95°C for 5 min, followed
by 35 cycles of 95°C for 30 sec, 59°C for 30 sec, and 72°C for 45
sec. All constructs were sequenced to ensure accuracy.
Cell transfection
THP-1 macrophages (2×106 cells/well) were seeded in
6-well plates and incubated overnight prior to transfection.
pcDNA3.1-PPAR-γ or pcDNA3.1 vector was added to each well at a
final concentration of 50 nM and was incubated with Lipofectamine
2000 (Invitrogen; Thermo Fisher Scientific, Inc.) at room
temperature for 30 min. The pcDNA3.1-transfected group served as a
negative control.
Cholesterol efflux
THP-1 macrophages were assigned to the following
groups: Untreated control, ox-LDL, ox-LDL+IL-32α, ox-LDL+15d-PGJ2,
ox-LDL+DMSO, ox-LDL+IL-32α+15d-PGJ2, and ox-LDL+IL-32α+DMSO. THP-1
macrophages were cultured until they reached 60% confluence, and
were then labeled with 0.2 µCi/ml [3H] cholesterol
(PerkinElmer, Inc., Waltham, MA, USA) for 24 h. Fresh serum-free
medium with 0.1% BSA containing the PPARγ agonist 15d-PGJ2 (5 µM;
Merck & Co., Inc., Whitehouse Station, NJ, USA) or 0.1%
dimethyl sulfoxide (DMSO; Sigma-Aldrich; Merck KGaA) was added to
THP-1 macrophages for 1 h, followed by exposure to IL-32α (50
ng/ml) for a further 6 h. Ox-LDL was then added to cells and
incubated for another 24 h. Equilibrated [3H]
cholesterol-labeled cells were washed with PBS and incubated in
efflux medium [RPMI 1640 medium and 0.1% BSA with 25 µg/ml human
apoA-I (Sigma-Aldrich; Merck KGaA)] for 4 h. Cells were centrifuged
at 14,000 × g for 15 min, and the supernatants were then collected.
Single layer cells were dissolved in isopropanol. Medium and
cell-associated [3H] cholesterol was examined by liquid
scintillation counting. The percentage of apoA-I-mediated
cholesterol efflux was calculated as follows: Cholesterol efflux
(%)=(total media counts)/(total cellular counts + total media
counts) ×100%.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), and 2 µg total RNA
was used as the template for reverse transcription using a
PrimeScript RT reagent kit (Takara Bio, Inc., Otsu, Japan) cDNA was
used as template for qPCR on the Light Cycler system (Roche
Diagnostics, Shanghai China) using SYBR-Green I Master mixture
(Roche Diagnostics). Cycling conditions were as follows: Initial
denaturation at 95°C for 5 min, then 35 cycles of denaturation at
95°C for 20 sec, annealing at 58°C for 30 sec and extension at 72°C
for 50 sec. The primers used for qPCR in the present study were
synthesized by Sangon Biotech (Shanghai, China) and are listed in
Table I. β-actin was used as the
housekeeping gene control for qPCR. The threshold cycle (Cq) values
were used to calculate the fold change in transcript levels using
the 2−ΔΔCq method (16),
as follows: Fold change=2-[(Cq target-Cq β-actin) of
siRNA]-[(Cq target-Cq β-actin) of control].
 | Table I.Primers for quantitative polymerase
chain reaction analysis. |
Table I.
Primers for quantitative polymerase
chain reaction analysis.
| Gene | Sequence |
|---|
| ABCA1 | Forward:
5′-AACAGTTTGTGGCCCTTTTG-3′ |
|
| Reverse:
5′-AGTTCCAGGCTGGGGTACTT-3′ |
| ABCG1 | Forward:
5′-GGTTCTTCGTCAGCTTCGAC-3′ |
|
| Reverse:
5′-GTTTCCTGGCATTCAGGTGT-3′ |
| PPARγ | Forward:
5′-GGCAATTGAATGTCGTGTCTGTGGAGATAA-3′ |
|
| Reverse:
5′-AGCTCCAGGGCTTGTAGCAGGTTGTCTTGA-3′ |
| LXRα | Forward:
5′-GCGAGGGCTGCAAGGGATTCT-3′ |
|
| Reverse:
5′-ATGGGCCAAGGCGTGACTCG-3′ |
| β-actin | Forward:
5′-TCATGAAGTGTGACGTTGACATCCGT-3′ |
|
| Reverse:
5′-CTTAGAAGCATTTGCGGTGCACGATG-3′ |
Western blot assay
Ox-LDL-stimulated THP-1 macrophages were washed with
PBS for three times and lysed in lysis buffer (20 mM HEPES, 300 mM
NaCl, 0.2 mM EDTA, 1.5 mM MgCl2 and 1% Triton X-100)
containing 2% phenylmethylsulfonyl fluoride. Total protein
concentrations were measured with a Pierce BCA Protein Assay kit
(Thermo Fisher Scientific, Inc.). Subsequently, 20 µg cell lysates
were subjected to 10% SDS-polyacrylamide gel electrophoresis, and
proteins were transferred to nitrocellulose membranes (EMD
Millipore, Billerica, MA, USA). Membranes were blocked with 5%
non-fat milk for 1 h at room temperature and probed with antibodies
against ABCA1 (sc-81779; 1:500 dilution), ABCG1 (sc-20795; 1:500
dilution), PPARγ (sc-6285; 1:500 dilution), LXRα (sc-1202; 1:500
dilution) and anti-human β-actin (sc-47778; 1:1,000 dilution; all
from Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) in
antibody dilution buffer [5% non-fat milk in 1X Tris-buffered
saline/0.1% Tween-20 (TBST)] at 4°C overnight. Next, the membranes
were washed with TBST for three times and then incubated with
horseradish peroxidase-conjugated goat anti-rabbit IgG (sc-2004;
1:2,000 dilution) or goat anti-mouse IgG (sc-2005; 1:1,000
dilution; Santa Cruz Biotechnology, Inc.) at room temperature for 1
h. After washing with TBST, the membranes were subjected to an
enhanced chemiluminescence advanced system (GE Healthcare Life
Sciences, Chicago, IL, USA). β-actin was used as a loading control.
The protein bands were analyzed using Quantity One software
(version 4.6.2; Bio-Rad Laboratories Inc., Hercules, CA, USA).
Statistical analysis
SPSS software version 13.0 (SPSS, Inc., Chicago, IL,
USA) was used to perform statistical analysis. Data are presented
as the means ± standard deviation of three experiments, or are
representative of experiments repeated at least three times.
Comparisons were conducted with a one-way analysis of variance and
post-hoc Tukey's tests. P<0.05 was considered to indicate a
statistically significant difference.
Results
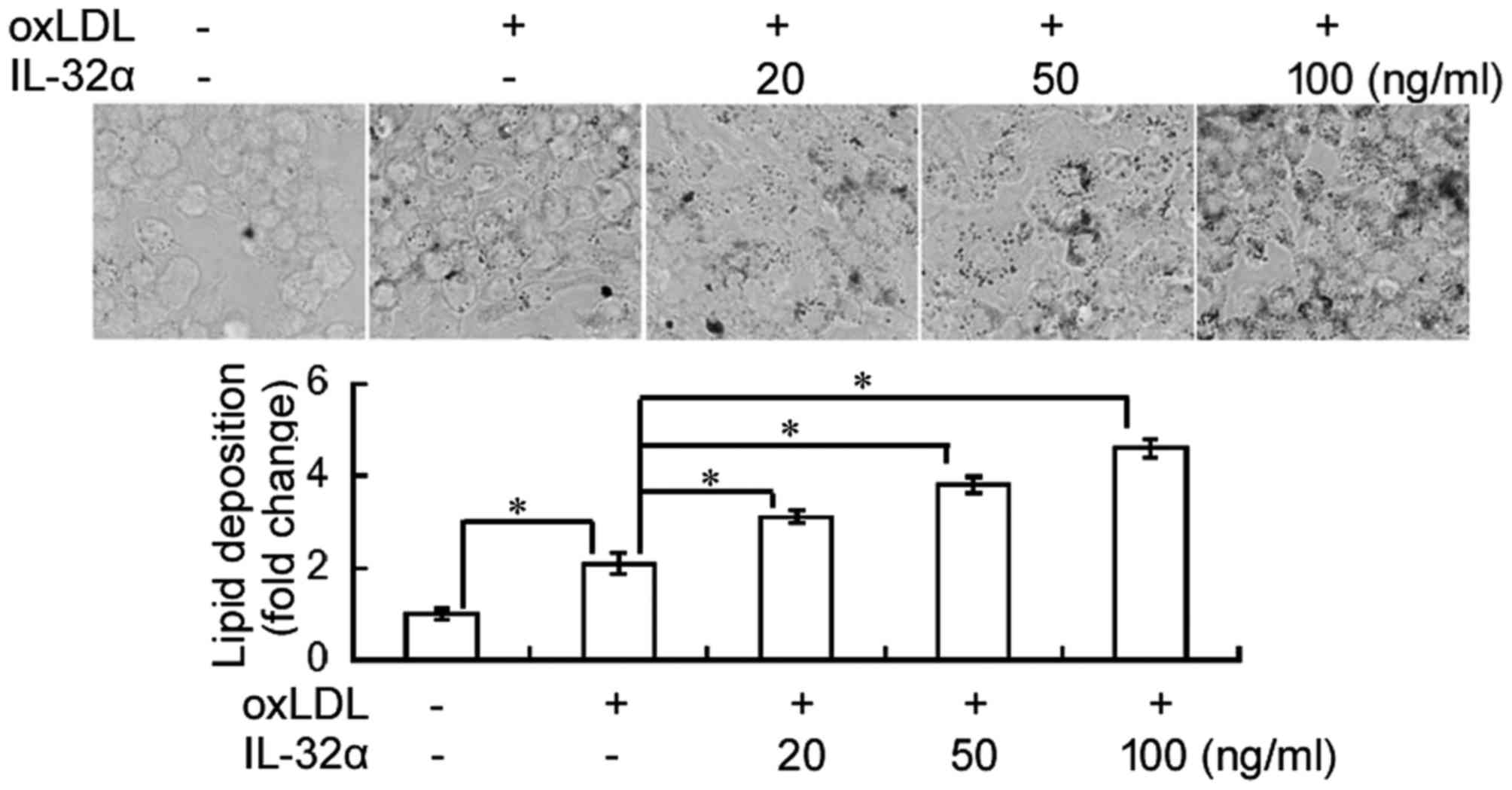
IL-32α induces lipid deposition in
THP-1 macrophages
The effect of IL-32α on lipid deposition in THP-1
macrophages was evaluated using Oil Red O staining. As shown in
Fig. 1, ox-LDL exposure
significantly induced lipid accumulation in THP-1 macrophages,
compared with the untreated control (P<0.05). Notably, addition
of recombinant IL-32α protein significantly induced lipid
deposition in ox-LDL-induced THP-1 macrophages in a
concentration-dependent fashion.
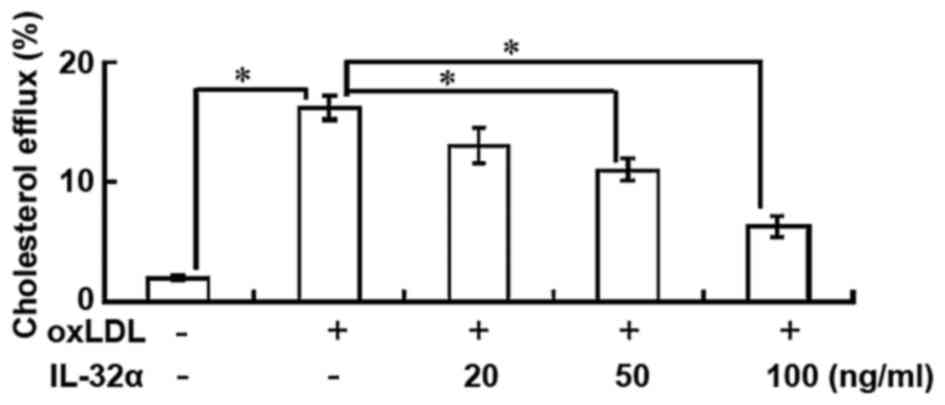
IL-32α reduces cholesterol efflux
The results indicated that the percentage of
cholesterol efflux was significantly increased following ox-LDL
exposure in THP-1 macrophages (P<0.05; Fig. 2). However, IL-32α treatment
attenuated the apoA-I-mediated cholesterol efflux in a
dose-dependent manner in ox-LDL-stimulated THP-1 macrophages, with
a significant effect observed for the 50 and 100 ng/ml IL-32α doses
(P<0.05; Fig. 2). This suggests
that IL-32α increased intracellular lipid accumulation by
decreasing the apoA-I-mediated cholesterol efflux.
IL-32α reduces the expression levels
of ABCA1, LXRα and PPARγ
The importance of ABCA1, ABCG1, LXRα and PPARγ in
apoA-I-mediated cholesterol efflux has previously been established
(13). Therefore, the present study
next analyzed the effects of IL-32α on the mRNA levels of these
molecules by RT-qPCR in ox-LDL-stimulated THP-1 macrophages. The
results demonstrated that ox-LDL treatment significantly raised the
mRNA levels of ABCA1, LXRα and PPARγ, compared with untreated
controls (P<0.05; Fig. 3A). The
addition of 100 ng/ml IL-32α significantly decreased the mRNA
expression levels of ABCA1, LXRα and PPARγ by 59, 52.9 and 61.1%,
respectively, compared with the ox-LDL alone group (P<0.05;
Fig. 3A). However, the level of
ABCG1 was not affected. Subsequently, the study explored whether
the IL-32α-mediated inhibition of ABCA1, LXRα and PPARγ mRNA
expression led to decreased expression of ABCA1, LXRα and PPARγ
protein using western blot analysis. As shown in Fig. 3B, IL-32α also suppressed the
expression of ABCA1, LXRα and PPARγ at the protein level.
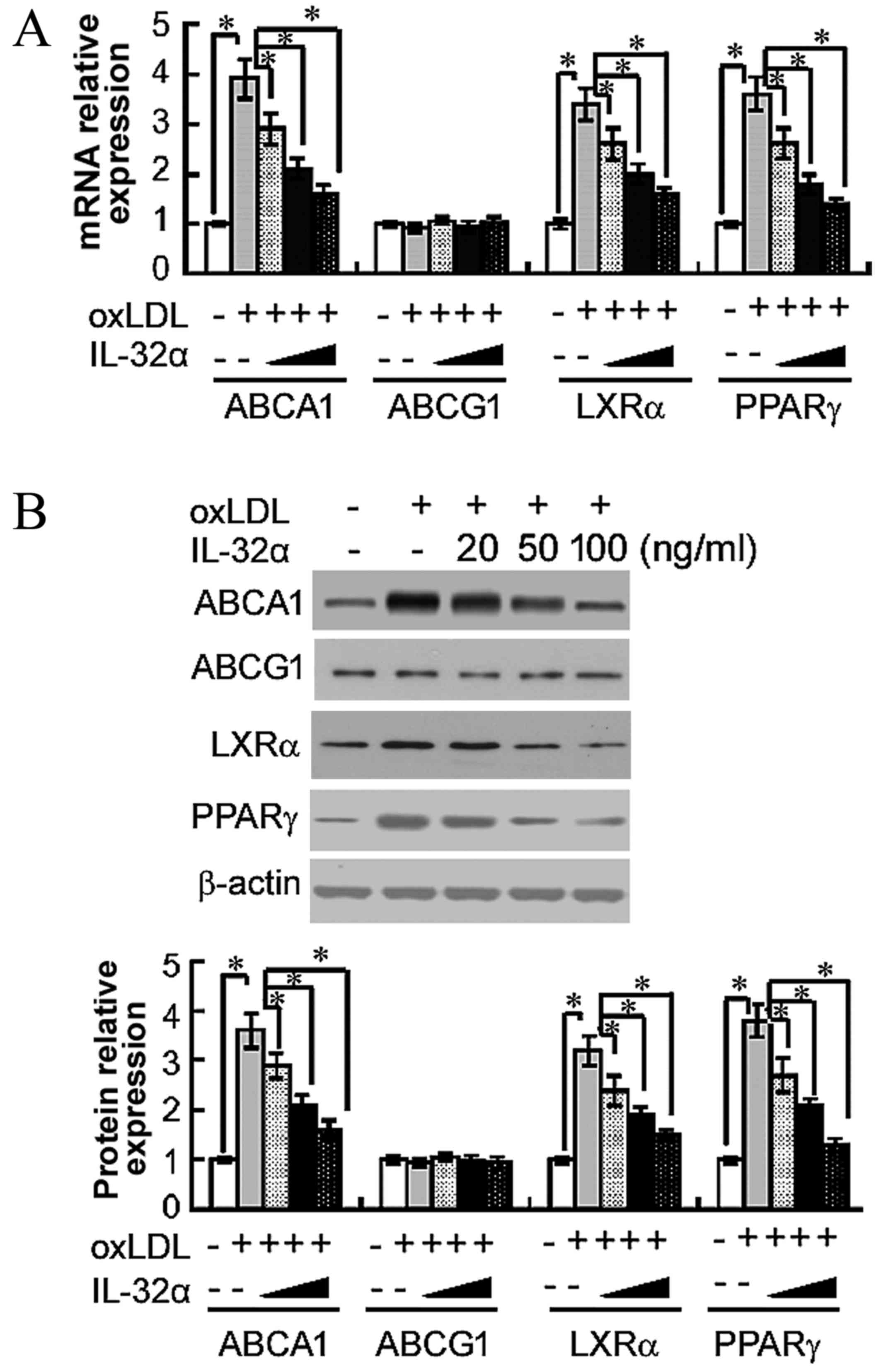 | Figure 3.Effects of IL-32α on the (A) mRNA and
(B) protein expression levels of ABCA1, ABCG1, LXRα and PPARγ in
ox-LDL-stimulated THP-1 macrophages, as determined by quantitative
polymerase chain reaction and western blot analysis, respectively.
THP-1 macrophages were pretreated with different concentrations of
(20, 50 and 100 ng/ml) of IL-32α for 6 h, followed by incubation
with ox-LDL for 72 h. Data are expressed as the mean ± standard
deviation (n=3). *P<0.05. IL-32α, interleukin-32α; ox-LDL,
oxidized low-density lipoprotein; ABC, ATP-binding cassette
transporter; LXRα, liver X receptor α; PPARγ, peroxisome
proliferator-activated receptor γ. |
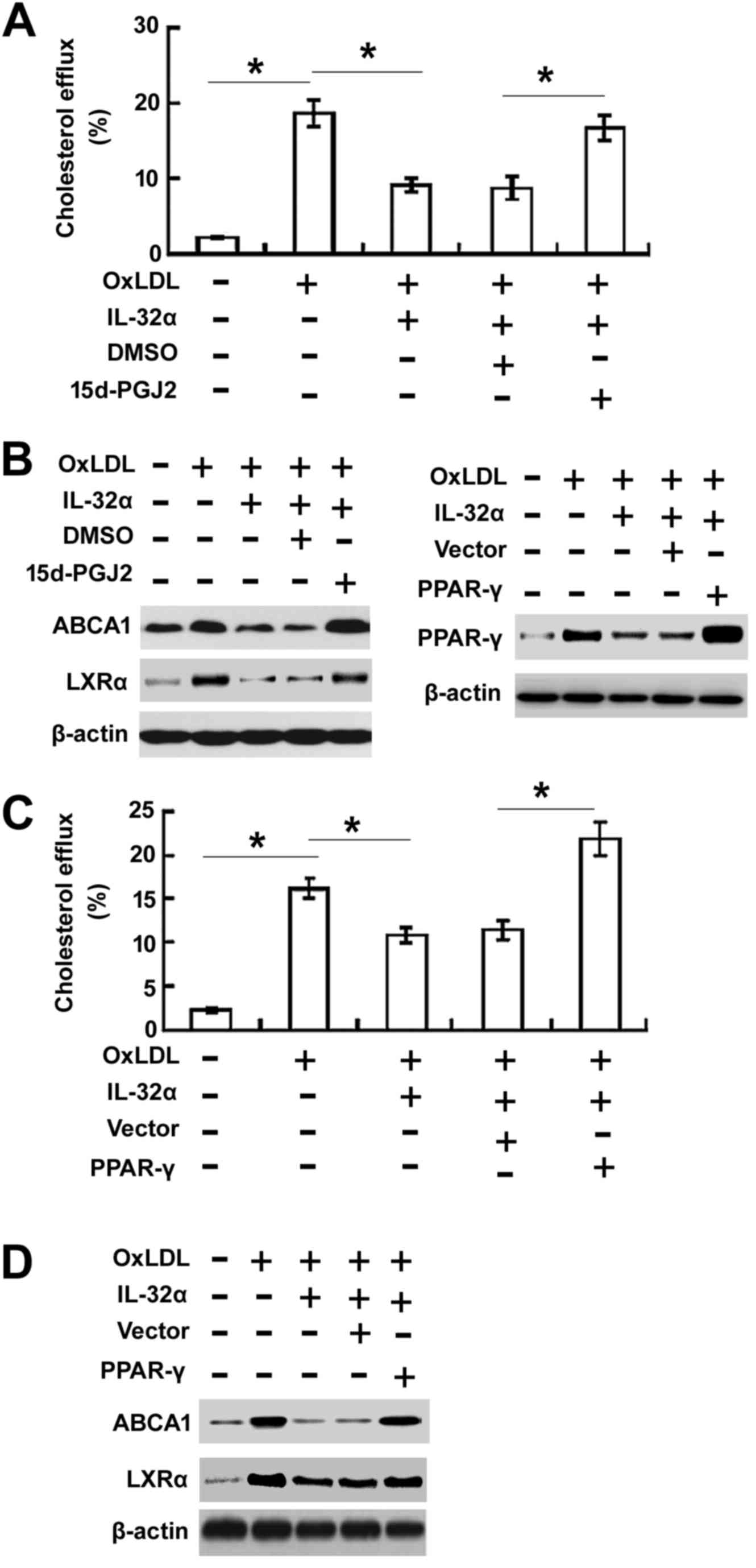
Overexpression of PPARγ rescued
IL-32α-mediated inhibition of cholesterol efflux
To evaluate the effects of PPARγ on IL-32α-mediated
inhibition of cholesterol efflux, the PPARγ agonist 15d-PGJ2 was
used to treat cells (Fig. 4A and B)
or pcDNA3.1-PPARγ (Fig. 4C and D)
was transfected to THP-1 macrophages. The 15d-PGJ2 treatment or
overexpression of PPARγ significantly enhanced cholesterol efflux
(Fig. 4A and C) and increased the
expression levels of ABCA1 and LXRα (Fig. 4B and D) in ox-LDL-stimulated THP-1
cells.
Discussion
In the present study, the effects of treatment with
IL-32α, the most abundant transcript of the pro-inflammatory
cytokine IL-32, were investigated in ox-LDL-stimulated THP-1 cells.
The results revealed that IL-32α increased lipid deposition and
dose-dependently decreased the cholesterol efflux from
ox-LDL-stimulated THP-1 macrophages. In addition, IL-32α attenuated
the expression levels of ABCA1, LXRα and PPARγ. To determine the
mechanism of IL-32α-mediated inhibition of cholesterol efflux,
PPARγ overexpression was performed in THP-1 macrophages by
transfection. PPARγ overexpression abrogated the IL-32α-mediated
inhibition of cholesterol efflux and reversed the IL-32α-mediated
downregulation of ABCA1 and LXRα. Thus, it was demonstrated that
IL-32α inhibits the cholesterol efflux from ox-LDL-exposed THP-1
macrophages by regulating the PPARγ-LXRα-ABCA1 pathway.
IL-32 was identified as NK cell transcript 4 (NK4),
since it was selectively expressed in activated T cells or NK cells
(17). The biological role of IL-32
remained unclear until 2005, when Kim et al (13) demonstrated that recombinant NK4 was
able to induce several pro-inflammatory cytokines, including TNF-α
and IL-8. Therefore, NK4 was renamed to IL-32. The IL-32 gene has
six splice variants, including IL-32α, IL-32β, IL32γ, IL-32δ,
IL-32ε, and IL-32ζ (18), with
IL-32α being the most abundant transcript (14). IL-32 can induce a range of
pro-inflammatory mediators and contribute to autoimmune diseases,
such as rheumatoid arthritis and inflammatory bowel disease
(19). Inflammation serves an
important role in the initiation and progression of atherosclerosis
and immune disease (20). A range of
immune cells, such as macrophages and T-lymphocytes, orchestrate
the inflammatory response in atherosclerosis through the action of
various cytokines (21). However,
the effects of IL-32α as an inflammatory cytokine in
atherosclerosis remain unknown. The present study investigated the
effects of IL-32α on lipid deposition and cholesterol efflux. As
expected, IL-32α enhanced lipid deposition and inhibited
cholesterol efflux in ox-LDL-stimulated THP-1 macrophages, which
simulated a foam cell formation model.
ABCA1 and ABCG1 are integral participants in
cholesterol efflux and reverse cholesterol transport (21). To investigate the mechanism of the
IL-32α-mediated negative effects on cholesterol efflux, western
blot analysis was conducted in the present study to explore the
protein expression of ABCA1 and ABCG1. The results showed that
IL-32α inhibited the expression of ABCA1, but did not affect the
expression of ABCG1, indicating that IL-32α inhibits cholesterol
efflux through the ABCA1 signaling pathway. PPARγ and LXRα are
nuclear receptors that serve a pivotal role in macrophage
cholesterol homeostasis and inflammation (22). Western blot analysis in the current
study demonstrated that IL-32α attenuated the expression levels of
PPARγ and LXRα. These results indicate that IL-32α may inhibit
cholesterol efflux through the PPARγ-LXRα-ABCA1 signaling
pathway.
Inflammatory cytokines are involved in all stages of
atherosclerosis and have a profound influence on the pathogenesis
of atherosclerosis, including cholesterol efflux. IL-6 and TNF-α
inhibit cholesterol efflux by suppressing ABCA1 expression
(23). IL-1β has been demonstrated
to promote lipid accumulation and inhibit cholesterol efflux in
human mesangial cells by dysregulating the expression of
lipoprotein receptors, thus inhibiting cholesterol efflux through
the PPAR-LXRα-ABCA1 pathway (24).
Furthermore, PPARγ agonists protect mesangial cells from
IL-1β-induced intracellular lipid accumulation by activating the
ABCA1 cholesterol efflux pathway (24). To further investigate the role of
PPARγ on IL-32α-meidated inhibition of cholesterol efflux, the
present study demonstrated that the PPARγ agonist 15d-PGJ2 and
PPARγ overexpression through transfection enhanced cholesterol
efflux in ox-LDL-stimulated THP-1 macrophages. These findings
indicate that inhibition of PPARγ serves an important role in
IL-32α-mediated suppression of cholesterol efflux.
In conclusion, the present study demonstrated that
IL-32α promotes lipid deposition and inhibits cholesterol efflux
through the suppression of PPARγ-LXRα-ABCA1 signaling pathway in
ox-LDL-stimulated THP-1 macrophages. However, the role of other
isoforms exclusive of IL-32α in cholesterol efflux remains unknown.
Therefore, more studies are required to determine the function of
each IL-32 isoform in lipid accumulation and cholesterol
efflux.
References
|
1
|
Detmers PA, Hernandez M, Mudgett J,
Hassing H, Burton C, Mundt S, Chun S, Fletcher D, Card DJ, Lisnock
J, et al: Deficiency in inducible nitric oxide synthase results in
reduced atherosclerosis in apolipoprotein E-deficient mice. J
Immunol. 165:3430–3435. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schmitz G, Kaminski WE, Porsch-Ozcürümez
M, Klucken J, Orsó E, Bodzioch M, Büchler C and Drobnik W:
ATP-binding cassette transporter A1 (ABCA1) in macrophages: A dual
function in inflammation and lipid metabolism? Pathobiology.
67:236–240. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bodzioch M, Orsó E, Klucken J, Langmann T,
Böttcher A, Diederich W, Drobnik W, Barlage S, Büchler C,
Porsch-Ozcürümez M, et al: The gene encoding ATP-binding cassette
transporter 1 is mutated in Tangier disease. Nat Genet. 22:347–351.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Glass CK and Witztum JL: Atherosclerosis.
The road ahead. Cell. 104:503–516. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yu XH, Fu YC, Zhang DW, Yin K and Tang CK:
Foam cells in atherosclerosis. Clin Chim Acta. 424:245–252. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Favari E, Zimetti F, Bortnick AE, Adorni
MP, Zanotti I, Canavesi M and Bernini F: Impaired ATP-binding
cassette transporter A1-mediated sterol efflux from oxidized
LDL-loaded macrophages. FEBS Lett. 579:6537–6542. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Berrougui H, Ikhlef S and Khalil A: Extra
virgin olive oil polyphenols promote cholesterol efflux and improve
HDL functionality. Evid Based Complement Alternat Med.
2015:2080622015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Escolà-Gil JC, Llaverias G, Julve J,
Jauhiainen M, Méndez-González J and Blanco-Vaca F: The cholesterol
content of Western diets plays a major role in the paradoxical
increase in high-density lipoprotein cholesterol and upregulates
the macrophage reverse cholesterol transport pathway. Arterioscler
Thromb Vasc Biol. 31:2493–2499. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao GJ, Yin K, Fu YC and Tang CK: The
interaction of ApoA-I and ABCA1 triggers signal transduction
pathways to mediate efflux of cellular lipids. Mol Med. 18:149–158.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee JY, Karwatsky J, Ma L and Zha X: ABCA1
increases extracellular ATP to mediate cholesterol efflux to
ApoA-I. Am J Physiol Cell Physiol. 301:C886–C894. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ji A, Wroblewski JM, Cai L, de Beer MC,
Webb NR and van der Westhuyzen DR: Nascent HDL formation in
hepatocytes and role of ABCA1, ABCG1, and SR-BI. J Lipid Res.
53:446–455. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chawla A, Boisvert WA, Lee CH, Laffitte
BA, Barak Y, Joseph SB, Liao D, Nagy L, Edwards PA, Curtiss LK, et
al: A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in
cholesterol efflux and atherogenesis. Mol Cell. 7:161–171. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim SH, Han SY, Azam T, Yoon DY and
Dinarello CA: Interleukin-32: A cytokine and inducer of TNFalpha.
Immunity. 22:131–142. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Choi JD, Bae SY, Hong JW, Azam T,
Dinarello CA, Her E, Choi WS, Kim BK, Lee CK, Yoon DY, et al:
Identification of the most active interleukin-32 isoform.
Immunology. 126:535–542. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cheon S, Lee JH, Park S, Bang SI, Lee WJ,
Yoon DY, Yoon SS, Kim T, Min H, Cho BJ, et al: Overexpression of
IL-32alpha increases natural killer cell-mediated killing through
up-regulation of Fas and UL16-binding protein 2 (ULBP2) expression
in human chronic myeloid leukemia cells. J Biol Chem.
286:12049–12055. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Goda C, Kanaji T, Kanaji S, Tanaka G,
Arima K, Ohno S and Izuhara K: Involvement of IL-32 in
activation-induced cell death in T cells. Int Immunol. 18:233–240.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim S: interleukin-32 in inflammatory
autoimmune diseases. Immune Netw. 14:123–127. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Conti P and Shaik-Dasthagirisaeb Y:
Atherosclerosis: A chronic inflammatory disease mediated by mast
cells. Cent Eur J Immunol. 40:380–386. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Buckley ML and Ramji DP: The influence of
dysfunctional signaling and lipid homeostasis in mediating the
inflammatory responses during atherosclerosis. Biochim Biophys
Acta. 1852:1498–1510. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Majdalawieh A and Ro HS: PPARgamma1 and
LXRalpha face a new regulator of macrophage cholesterol homeostasis
and inflammatory responsiveness, AEBP1. Nucl Recept Signal.
8:e0042010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ramji DP and Davies TS: Cytokines in
atherosclerosis: Key players in all stages of disease and promising
therapeutic targets. Cytokine Growth Factor Rev. 26:673–685. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang N, Lei J, Lei H, Ruan X, Liu Q, Chen
Y and Huang W: MicroRNA-101 overexpression by IL-6 and TNF-α
inhibits cholesterol efflux by suppressing ATP-binding cassette
transporter A1 expression. Exp Cell Res. 336:33–42. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ruan XZ, Moorhead JF, Fernando R, Wheeler
DC, Powis SH and Varghese Z: PPAR agonists protect mesangial cells
from interleukin 1beta-induced intracellular lipid accumulation by
activating the ABCA1 cholesterol efflux pathway. J Am Soc Nephrol.
14:593–600. 2003. View Article : Google Scholar : PubMed/NCBI
|