Introduction
Stroke is the leading cause of mortality and
disability in adults in China (1).
To date, few therapeutic options are available for the treatment of
ischemic stroke, and recanalization following ischemia is the most
effective method for ischemic stroke (2). However, it benefits only a small
proportion of patients due to the limited thrombolysis window and
the adverse reactions of thrombolytics (2,3). In
addition, recanalization may cause severe cerebral
ischemia-reperfusion (I/R) injury in the local region (4,5).
Accumulating evidence has suggested that inflammatory and oxidative
damage serves an important role in cerebral I/R injury (6–8).
Excessive inflammatory and oxidative responses lead to the
disruption of the blood-brain barrier (BBB), which further
aggravates the progression of ischemic injury subsequent to stroke
(9–12). Therefore, it is essential to identify
alternative therapies for protection against I/R damage, such as
the use of agents with anti-inflammatory and anti-oxidant effects.
Furthermore, it is urgently necessary to establish new
interventions outside of the thrombolysis time window in order to
save the hypoperfused, nonfunctional, but still viable brain tissue
surrounding the core area of the irreversible cerebral
infarction.
TLR4 is the first reported mammalian toll-like
receptor, which is located on the cell surface and is functionally
similar to Drosophila Toll protein (13). Within the brain, TLR4 is mainly
located on glial cells, including microglia, astrocytes and
oligodendrocytes (14). During the
course of brain damage caused by I/R, it is believed that heat
shock proteins (HSPs), such as HSP60, HSP70 and gp90, are
upregulated and leak into the extracellular compartment to induce
immune response and inflammatory response. Binding of HSPs with
TLR4 contributes to myeloid differentiation protein-88 (MyD88)
recruitment (15). The TLR4-MyD88
association further activates IL receptor-associated kinase (IRAK),
which promotes the transcription and expression of tumor necrosis
factor-α (TNF-α), interleukin (IL)-1 and IL-6 (16). Several studies have revealed that
targeting TLR4 may alleviate brain damage caused by cerebral I/R
(17–19). For instance, TLR4 knockout mice have
significantly smaller infarct volume at reperfusion 24 h after 2 h
of ischemia (17). Therefore,
inhibiting TLR4 and reducing its downstream signaling pathways is
regarded as a promising therapeutic strategy.
Nuclear factor erythroid 2-related factor 2 (Nrf2),
a regulator of the antioxidant cell defense system, is inactive in
the cytoplasm by Kelch-like ECH-associated protein 1 via the
formation of a covalent complex (20). Once activated, Nrf2 translocates into
the nucleus, interacts with a small Maf protein, and forms a
heterodimer that binds to antioxidant response element, which
contributes to cytoprotection in oxidative stress-induced injury
with cerebral ischemia (21).
Furthermore, previous evidence suggests that mice lacking of Nrf2
are more vulnerable to the cytotoxic effects of oxidative
stress-induced brain injury in comparison with wild-type mice
(22). Thus, Nrf2 is fundamental to
the defense against oxidative stress and may be a promising
therapeutic target in stroke.
The kallikrein-kinin system (KKS), composed of
kallikrein, kininogen, kinin and kinin receptors, is implicated in
multiple pathological states and represents an attractive
therapeutic target in ischemic stroke. As an important component of
the KKS, tissue kallikrein (TK) cleaves low-molecular-weight
kininogen to produce the potent vasoactive kinins (23). Intact kinins bind to the kinin
B2 receptor and lead to a series of biological effects
(24). It has been well documented
that TK exerts protective effects against stroke (24,25). The
local or systemic delivery of human TK protects against ischemic
brain injury by inhibiting inflammation and oxidative stress, and
by promoting angiogenesis and neurogenesis (24,25).
Based on these studies, it is hypothesized that TK may protect
against ischemic via the TLR4/nuclear factor-κB (NF-κB) and Nrf2
pathways. Therefore, the aim of the present study was to
investigate the potential effects of TK in ischemic stroke and
explore whether the therapeutic benefit of TK was associated with
the activities of TLR4, NF-κB and Nrf2.
Materials and methods
Test animals
All experiments involving animals and tissue samples
were performed according to the guidelines of the National
Institutes of Health Guide for the Care and Use of Laboratory
Animals, and all procedures were approved by the Institutional
Animal Care and Use Committee (IACUC) of Nanjing Medical University
(Nanjing, China). A total of 60 male Sprague-Dawley rats (aged 8–10
weeks; weight, 250–300 g) were provided by the Experimental Animal
Center in Nanjing Medical University. Prior to the experiments,
rats were housed for at least 1 week in their home cages at a
constant temperature (18–22°C), with controlled illumination (12 h
light/dark cycles) and humidity (30–50%), and ad libitum
access to food and water.
Middle cerebral artery occlusion
(MCAO) model in rats
Rats were anesthetized intraperitoneally with 10%
chloral hydrate (Sinopharm Chemical Reagent Co., Ltd., Shanghai,
China; 300 mg/kg) and subjected to MCAO as previously described
(26). In brief, the right common
carotid artery, the external carotid artery (ECA) and the internal
carotid artery (ICA) were exposed and carefully isolated. A 3–0
monofilament nylon suture with a heat-rounded tip was inserted from
the lumen of the ECA to the right ICA to occlude the origin of the
right middle cerebral artery. Rats were subjected to 2 h of
occlusion and then the filament was withdrawn to restore blood
flow. Sham-operated rats were manipulated in the same surgical
procedures, but without insertion of the filament into the ICA.
Body temperature was maintained at 37±5°C with a heating pad.
Drug treatments
Rats (n=15 per group) were treated with TK (Techpool
Bio-Pharma Co., Ltd, Canton, China) with 0.9% saline solution
through the tail-vein at a dose of 8.75×10−3 PNAU/kg
within 3 min. The TK immediate treatment group received TK
immediately after the reperfusion, while the TK delayed treatment
group received TK at 12 h after reperfusion. In the cases of sham
and MCAO model groups, equal volume 0.9% saline were administered
in the same manner.
Behavioral tests
All behavioral tests in rats were conducted in a
quiet, low-lit room and were evaluated by an investigator blinded
to the experimental groups at 24 h after reperfusion (n=10 from
each group). Neurologic deficit scores were assigned to each rat
according to a previously described scoring system (26), and were as follows: 0, no
neurological deficit (normal behavior); 1, mild neurological
deficit (failure to fully lift forepaw); 2, moderate neurological
deficit (circling to the left); 3, severe neurological deficit
(falling to the left); and 4, very severe neurological deficit
(failure to walk spontaneously, reduced level of consciousness).
The higher the neurological deficit score, the more severe
impairment the motor motion injury was. Rats with neurologic
deficit scores of 0 and 4 following MCAO were excluded, as this
indicated modeling failure.
Determination of infarct volume
At 24 h after MCAO, rats (n=5 from each group) were
sacrificed by removing the brains following anesthesia with 10%
chloral hydrate (400 mg/kg, i.p.). Brains were dissected and cut
into 2-mm thick slices. The slices were incubated in 2%
2,3,5-triphenyltetrazolium chloride (TTC) for 30 min at 37°C,
followed by immersion-fixation with 4% paraformaldehyde. A deep red
color indicated normal tissue and a pale gray color indicated the
infarct area. The stained sections were photographed, and then the
digital images were quantified using an Image analysis software,
Image Pro-Plus 5.1 (Media Cybernetics, Inc., Rockville, MD, USA).
The lesion volume was calculated by multiplying the area by the
thickness of slices. Infarct volume in all slices was expressed as
a percentage of the contralateral hemisphere after correction for
edema, using the following formula (27): Hemisphere lesion volume (%) = [total
infarct volume-(volume of intact ipsilateral hemisphere-volume of
intact contralateral hemisphere)]/contralateral hemisphere volume ×
100%.
Western blot analysis
For western blotting assay, total proteins from the
brain cortex samples were obtained using cold RIPA Lysis Buffer
(Beyotime Institute of Biotechnology, Haimen, China) containing 1
mM phenylmethylsulfonyl fluoride (PMSF). The homogenates were
centrifuged at 12,000 × g for 10 min at 4°C, and the protein
concentration in the supernatants was measured using a BCA protein
assay kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA;
catalogue number 23227). Protein samples (40 µg per lane) were
separated by 12% SDS-PAGE and transferred onto polyvinylidene
fluoride membranes (EMD Millipore, Billerica, MA, USA). The
membranes were blocked with 5% nonfat dried milk for 2 h at room
temperature and then incubated at 4°C overnight with the following
primary antibodies: Monoclonal mouse anti-TLR4 (1:400; ab30667;
Abcam, Cambridge, MA, USA), rabbit anti-Nrf2 (1:1,000; ab31163;
Abcam), monoclonal rabbit anti-heme oxygenase 1 (HO-1) (1:10,000;
ab68477; Abcam), monoclonal rabbit anti-matrix metallopeptidase 9
(MMP-9; 1:5,000; ab76003; Abcam), rabbit monoclonal NF-κB p65
(1:1,000; cat. no. 8242; Cell Signaling Technology, Inc., Danvers,
MA, USA), and β-actin (1:1,000; CMCTAG, Inc., San Diego, CA, USA;
catalogue number AT0001). The membranes were subsequently washed
three times with Tris-buffered saline and 0.1% Tween-20. Next,
membranes were incubated with horseradish peroxidase-conjugated
anti-mouse IgG (1:5,000; CWBiotech, Beijing, China; catalogue
number CW0102S) or anti-rabbit IgG (1:5,000; CWBiotech; catalogue
number CW01035) secondary antibodies for 2 h at room temperature.
Immunoreactivity was detected using an enhanced chemiluminescence
reaction (EMD Millipore; catalogue number WBKLS0500). Densitometric
analysis employed Image J Pro-Plus 6.0 analysis system (NIH,
Bethesda, MD, USA). Densitometry values were normalized with
respect to the β-actin immunoreactivity in order to correct for any
loading and transfer differences among samples. A total of 5
replicates were performed.
Enzyme-linked immunosorbent assay
(ELISA)
In order to detect the IL-1β and TNF-α protein
expression, tissues from the brain cortex were harvested at 24 h
after MCAO and homogenized in 0.1 mM phosphate-buffered saline
lysis buffer containing a cocktail of protease inhibitors
(Sigma-Aldrich, St. Louis, MO, USA) and 10 nM PMSF. The homogenate
was centrifuged at 17,000 × g for 15 min, and the supernatant was
collected. The tissue samples were frozen at −80°C immediately
until analysis of cytokine protein concentrations. Next, the tissue
samples were detected by commercial ELISA kits for IL-1β (Shanghai
ExCell Biology Inc., Shanghai, China; catalogue number ER008-48)
and TNF-α (Shanghai ExCell Biology, Inc.; catalogue number
ER006-96) according to the manufacturer's instructions. Inter- and
intra-assay coefficients of variation were <10%.
Statistical analysis
All statistical results are expressed as the mean ±
standard error of the mean. Neurological deficit assessment data
were analyzed with one-way analysis of variance (ANOVA)-Tukey's
multiple comparison test. Other data were analyzed with ANOVA
followed by the Student-Newman-Keuls test. Statistically
significant differences were considered to be those with
P<0.05.
Results
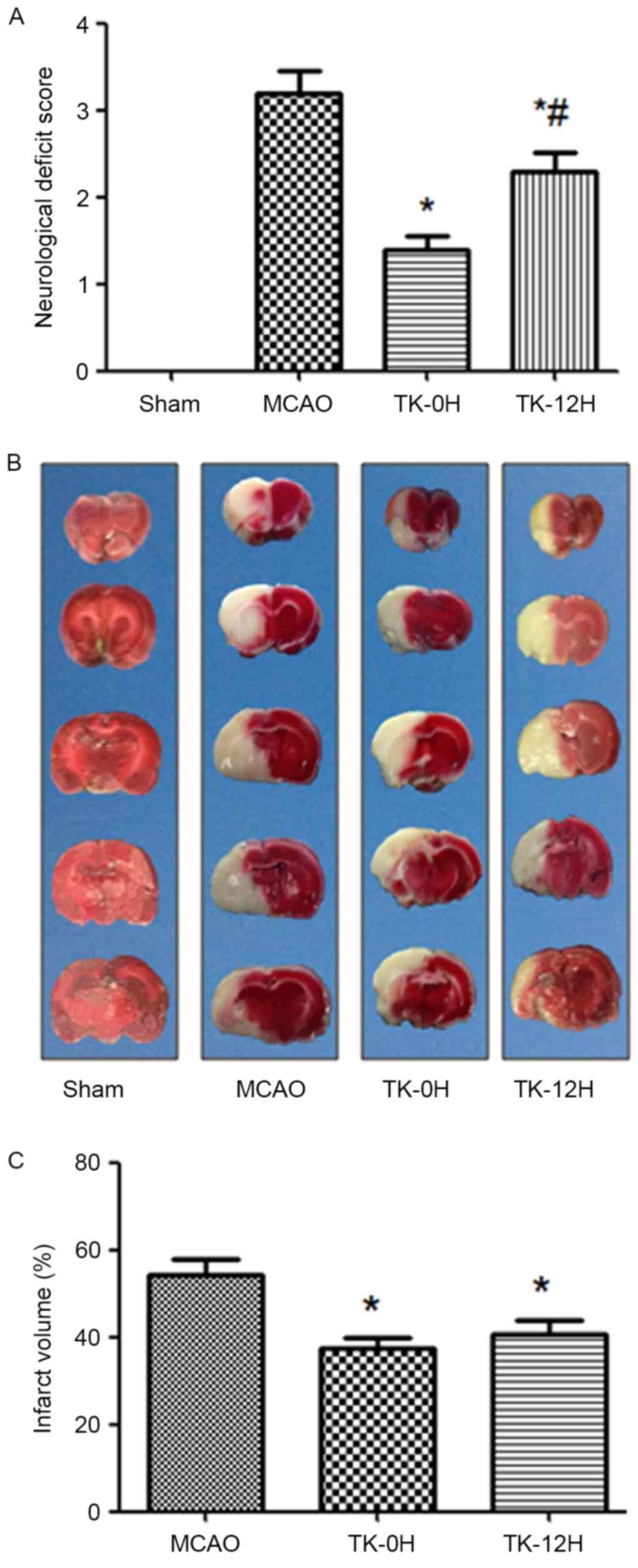
TK improves neurological deficits in
rats following cerebral ischemia
To examine whether TK exerted a neuroprotective
effect on cerebral ischemia, neurologic deficit scoring was
performed at 24 h after cerebral I/R in rats. Compared with the
MCAO model group, the immediate and delayed TK treatment groups
presented a significant improvement in neurological deficit scores
(Fig. 1A; P<0.05), suggesting a
neuroprotective effect of TK treatment in acute stroke. Notably,
compared with the TK delayed treatment group, the TK immediate
treatment group had a more evident reduction of neurological
deficit scores at 24 h after cerebral I/R, with a significant
difference observed between the two TK groups.
TK reduces the infarct volume in rats
following cerebral ischemia
The cerebral infarction following I/R was detected
by TTC staining, as shown in Fig. 1B and
C. No evident infarction was observed in the sham group, while
an extensive lesion was observed in both the striatum and the
cortex in the MCAO model group (Fig.
1B). Compared with the MCAO group, the infarct volume in the TK
immediate and delayed treatment groups was significantly decreased
(Fig. 1C; P<0.05). However, a
tendency towards higher reduction of the infarct volume was
observed in the TK immediate treatment group when compared with TK
delayed treatment group, but this difference was not statistically
significant.
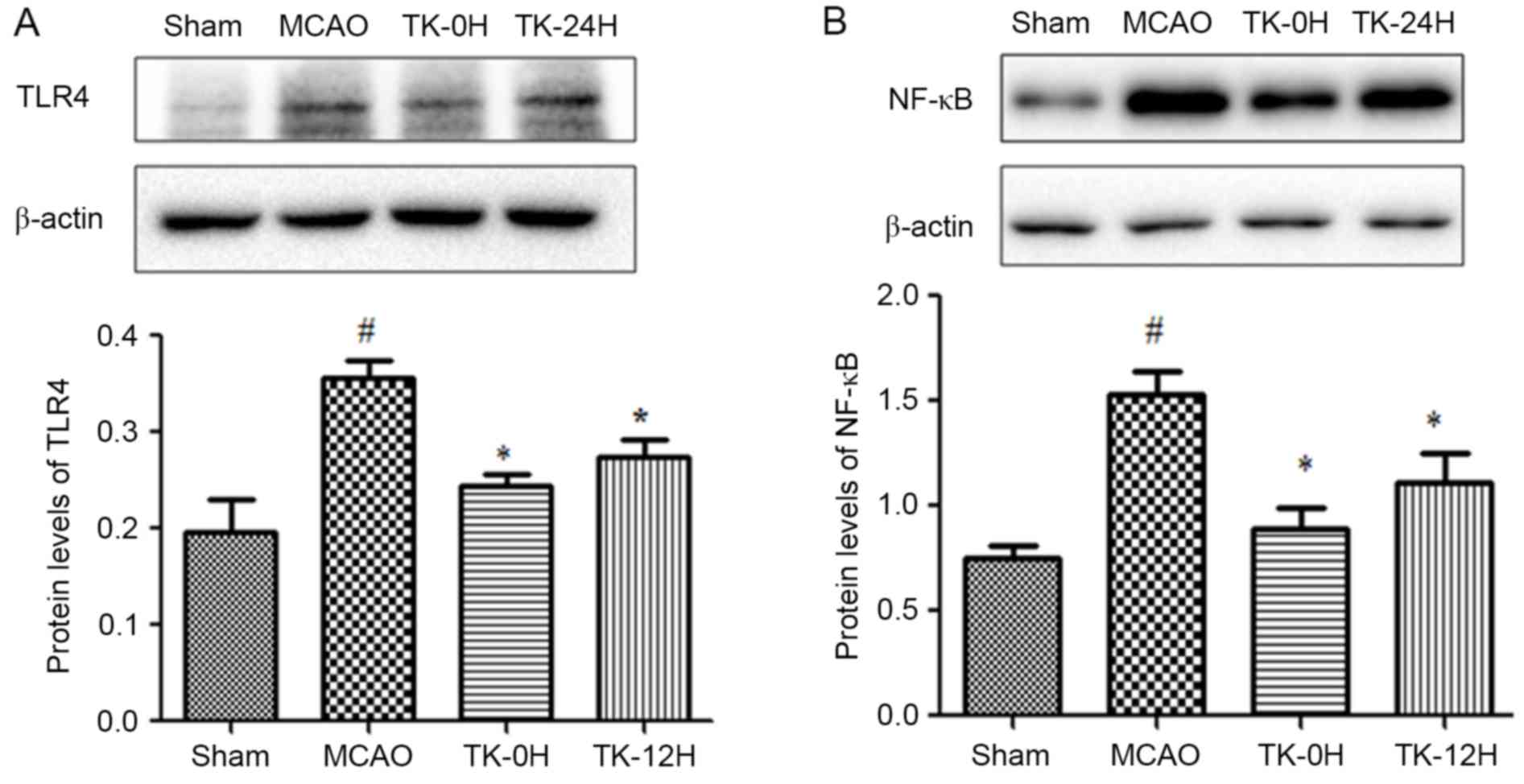
TK decreases inflammatory responses in
ischemic brain
Since inflammation is closely associated with
cerebral infarction, the present study next sought to examine
whether TK exerts an anti-inflammatory effect during the process of
cerebral I/R. As depicted in Fig. 2,
24 h after cerebral I/R, the expression levels of TLR4 and NF-κB in
ischemic brains were detected by western blot analysis. The results
revealed that the protein levels of TLR4 and NF-κB (Fig. 2A and B) were significantly increased
following MCAO. However, this effect was markedly ameliorated by TK
immediate treatment (Fig. 2A and B;
P<0.05). Meanwhile, TK delayed treatment was also able to
significantly inhibit the expression levels of TLR4 and NF-κB.
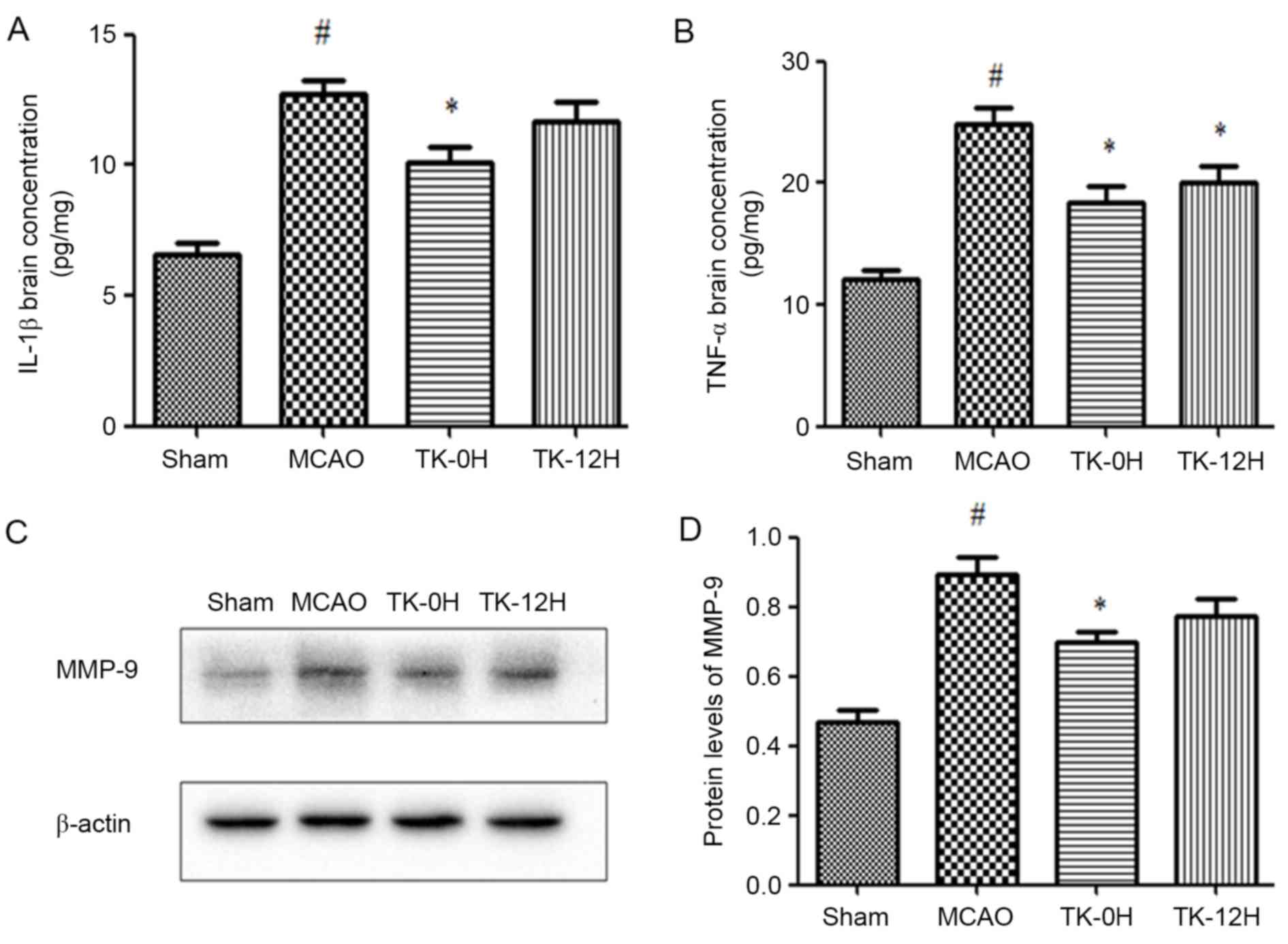
Next, the protein levels of IL-1β and TNF-α in brain
tissue were measured by ELISA. The presented results reveal that
IL-1β and TNF-α levels were significantly increased in the MCAO
model group, in comparison with the sham group (Fig. 3A and B). In consistency with the
aforementioned modulation of TLR4 and NF-κB, the levels of IL-1β
and TNF-α were significantly decreased in the TK immediate
treatment group. Similarly, TK delayed treatment significantly
inhibited the level of TNF-α, while exerting no significant effect
on the level of IL-1β.
Involvement of MMP-9 in the protective
effects of TK against ischemic stroke
To investigate the potential effect of TK on BBB,
the expression of MMP-9 in the ischemic brain was detected by
western blot analysis at 24 h after MCAO. As shown in Fig. 3C and D, MMP-9 expression was markedly
increased in the MCAO group compared with the sham group
(P<0.05). Following the immediate TK treatment, the level of
MMP-9 was significantly reduced (P<0.05). In addition, delayed
TK treatment displayed some effect on inhibiting the expression of
MMP-9 in the ischemic brain, but this effect was not significant
when compared with the MCAO group.
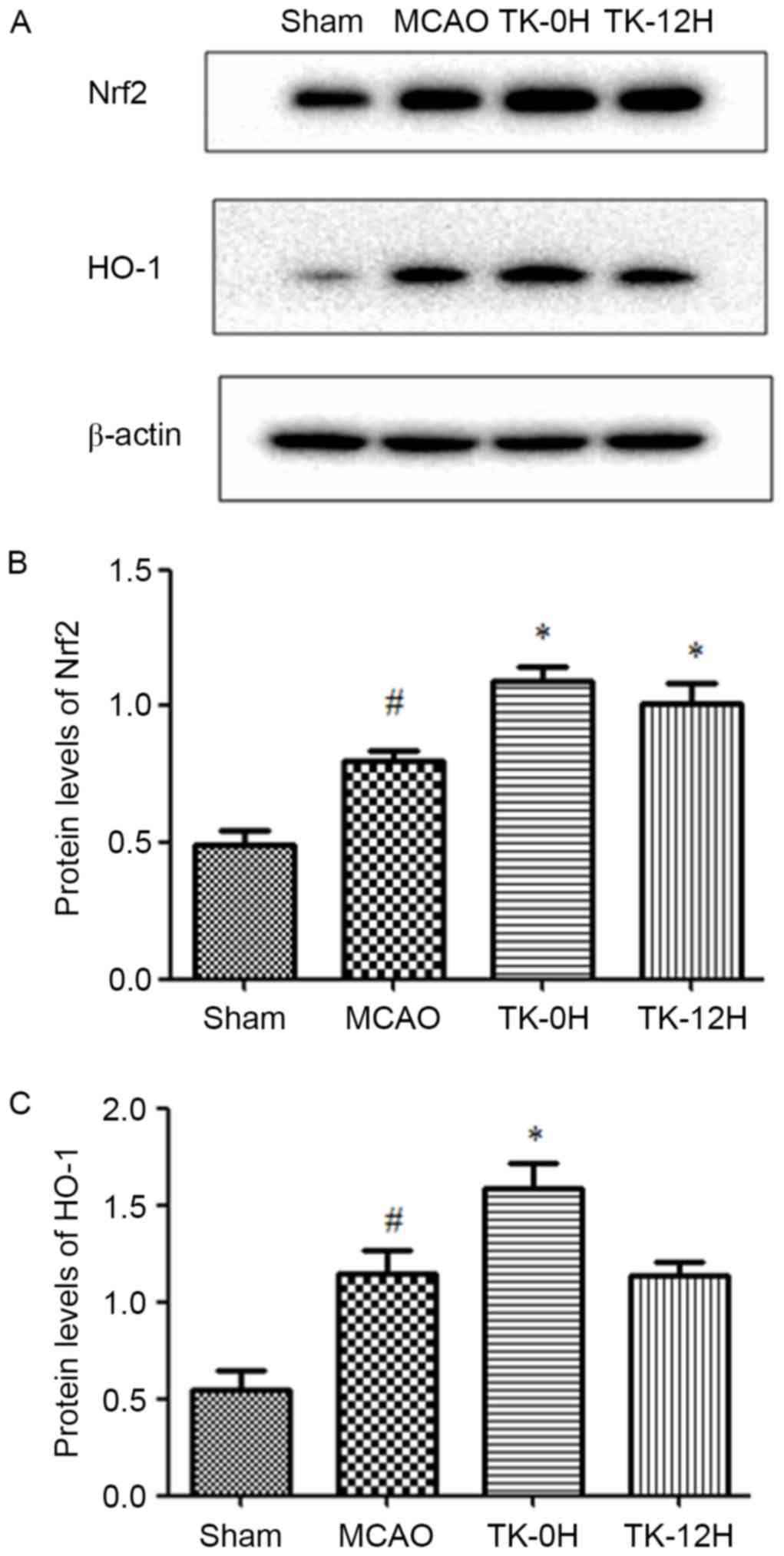
TK increases the expression levels of
Nrf2 and HO-1 proteins in ischemic brain
To further elucidate the mechanisms underlying the
neuroprotective effect of TK, the influence of TK treatment on the
activity of Nrf2 pathway was examined at 24 h after MCAO using
western blot analysis. Compared with the sham group, the levels of
Nrf2 and HO-1 proteins were significantly elevated in the ischemic
brain of the MCAO model group (Fig.
4), suggesting the activation of endogenous antioxidative
system in brain ischemia. Furthermore, TK immediate treatment
significantly increased the protein levels of Nrf2 and HO-1. In the
TK delayed treatment group, Nrf2 protein expression was
significantly increased, however, the HO-1 expression was not
significantly affected when compared with the MCAO group.
Discussion
In the present study, direct evidence that TK
protected rats from I/R-induced brain damage was provided. The
reported data were obtained with an MCAO model, a
well-characterized and classical model used to study the mechanisms
of cerebral ischemic damage in rats (6,26). Using
this model, the present study demonstrated that TK exerted a
neuroprotective effect on cerebral ischemia, which may be partially
due to its anti-inflammatory signaling pathway via downregulation
of TLR4 and NF-κB, accompanied by the activation of antioxidant
protein regulator Nrf2 and HO-1, and decrease of MMP-9
expression.
Cerebral ischemic/reperfusion injury is accompanied
with inflammatory responses and oxidative stress, which serve a
crucial role in the dysfunction and death of neurons in the brain
regions (6–8,28). Brain
ischemia triggers inflammatory responses, which has been associated
with the activation of TLR signaling. Several studies suggest that
TLR4 may serve a more important role in comparison with other
Toll-like receptors during the course of brain damage caused by
ischemia/reperfusion. A previous study demonstrated that the
expression of TLR4 was increased in the ischemic brain (17). In addition, TLR4 knockout mice had
significantly smaller infarct area and volume, and exhibited
improved neurological outcome at 24 h after cerebral I/R compared
with wild-type mice (17). In
previous in vitro studies, TLR4 expression level was also
increased in cultured neurons subjected to glucose deprivation, and
neurons deficient in TLR4 were resistant to death induced by energy
deprivation (29,30). Furthermore, it has been demonstrated
that a constitutively active mutant of human TLR4 transfection into
human cell lines could induce the activation of NF-κB and the
expression of NF-κB-mediated inflammatory cytokines, including
IL-1, IL-6, IL-8 and TNF-α (13).
The transcription factor NF-κB, a key regulator of a variety of
genes involved in cell survival and inflammation, is activated
following cerebral ischemia in neurons, endothelial cells,
astrocytes, microglia and infiltrating inflammatory cells (31). Studies have suggested that NF-κB
activation is primarily mediated by the TLR signaling pathway,
while the activation of NF-κB is required for the induction of
numerous inflammatory cytokines (32). The mammalian NF-κB family comprises
of five members: p65 (RelA), RelB, c-Rel, p50/p105 (NF-κB1), and
p52/p100 (NF-κB2), among which the p65/RelA and p50 are known to be
responsible for a detrimental effect in cerebral ischemia (31). For instance, NF-κB subunit p50
knockout mice displayed a significant reduction in infarct size in
focal cerebral ischemia (33). In
agreement with these studies, the results of the present study
demonstrated that cerebral I/R induced the upregulation of TLR4 and
NF-κB protein expression levels, while immediate and delayed TK
treatment deactivated the activity of TLR4 and NF-κB, emphasizing
the neuroprotective effect of TK on cerebral I/R injury.
It is well documented that oxidative stress is
generated and serves a detrimental role in cerebral I/R injury
(8,34). Nrf2 and its targets genes, known as
phase-II enzymes, function in synergy to remove reactive oxygen
species/reactive nitrogen species through sequential enzymatic
reactions (35). Among phase-II
enzymes, HO-1, a redox-sensitive inducible stress protein, has been
proposed to serve a crucial role in the endogenous anti-oxidative
defense system (36). Studies have
revealed that increasing the activity of Nrf2 pathways and gene
targets exerts highly neuroprotective effect against oxidative and
excitotoxic insults relevant to ischemic stroke in cell culture and
animal models (37,38). Nrf2 knockout mice presented more
severe neurologic dysfunction and larger infarct size caused by
MCAO in comparison with wild-type mice (39). The present study indicated that
systemic administration of TK significantly increased the
expression levels of Nrf2 and HO-1 in the ischemic cortex at 24 h
after MCAO, suggesting the involvement of Nrf2 and HO-1 in the
protective effect of TK in ischemic stroke.
MMPs degrade the extracellular matrix around the
blood vessels, as well as the matrix around neurons, which have
been shown to be associated with increased BBB paracellular
permeability (40). In the early
phase subsequent to cerebral ischemia, MMPs disrupt the BBB,
leading to BBB leakage, leukocyte infiltration, brain edema and
hemorrhage (41). Within the MMP
family, MMP-2 and MMP-9 have been regarded as the key enzymes
associated with secondary damage following ischemic cerebral stroke
(42). Clinical data support that
the activated MMP-9 can be considered as an effective marker for
negative outcome within the early phase of ischemic stroke
development (41). Therefore, MMP-9
is a potential therapeutic target for BBB disruption following
thrombolysis (43). In the present
study, it was observed that MMP-9 expression was increased at 24 h
after ischemic stroke, and TK significantly attenuated the
expression of MMP-9. It is suggested that the MMP-9 suppression was
involved in the neuroprotective effect of TK against stroke, which
may be, at least partly, due to the improvement of the BBB
integrity.
However, certain studies have demonstrated that the
KKS serves a detrimental role in the course of brain injury in
stroke, as well as other brain disease animal models (23). The early activation of the KKS
following cerebral ischemia increased brain vessel permeability,
edema and spread of the ischemic lesion, as kinins and their
receptors have been documented to induce proinflammatory responses
(44). In addition, early
administration of a kinin B2 receptor antagonist may
improve the neurological recovery after focal cerebral I/R
(45). Therefore, the present
results appear to contradict previous findings regarding the role
of kinin in inflammation. However, numerous other studies support
the results of the present study, indicating that TK exerts a
neuroprotective effect in ischemic injury (24,25,46,47).
This type of protective effect may be associated with the following
factors: i) The actions of TK can be mediated by kinin
B2 receptor activation without kinin formation (48); ii) kallikrein-induced
anti-inflammation is dependent on kinin B2 receptor
activation and nitric oxide formation (24); iii) upregulation of endothelial
nitric oxides resulted in dilation of cerebral arterial vessels,
which is critical in maintaining the cerebral blood flow and
conducive to the removal of inflammatory mediators (25); and iv) in the early period following
cerebral I/R, kallikrein induced angiogenesis and improved the
regional cerebral blood flow in the peri-infarction region, and
further reduced the infarction volume and improved the neurological
deficits (46).
In the present study, the protective effect in the
TK delayed treatment group was not as effective as the protection
observed in the TK immediate treatment group, which was not
consistent with the results of Xia et al (24). A possible reason for the discrepancy
between these results was the differential time point selected. In
the present study, 24 h after reperfusion was selected as the only
observation point, not extending to 1–2 weeks after reperfusion.
Furthermore, the pharmaceutical dosage may be another reason for
these different findings. Xia et al (24) used an adenovirus carrying human TK
cDNA, while the present study used the injection of TK directly.
Therefore, additional studies are required to precisely define how
long after ischemic stroke the start of the therapeutic window can
be delayed, and for how long a treatment should be given to achieve
a beneficial prognosis.
In conclusion, the current study demonstrated that
immediate systemic administration of TK decreased the infarct size,
oxidative and inflammatory responses in cerebral ischemia, and
further enriched its anti-inflammatory mechanism against ischemic
stroke. These data also provide important new evidence that delayed
systemic TK treatment following cerebral vascular insult may
achieve neuroprotective effects against ischemia-induced brain
damage. Finally, these results indicated that the upregulation of
the Nrf2 pathway and downregulation of the TLR4/NF-κB pathway
subsequent to ischemia by administration of TK was a potential
mechanism of the neuroprotective effect of TK.
Acknowledgements
This study was supported by a major project from the
Jiangsu Province (grant no. BS2006007), the 333 Program for
high-level personnel training foundation of Jiangsu province of
China (grant no. BRA2012050), funding for the development of
medical clinical science and technology from Jiangsu University
(grant no. JLY2010053), and funding from Changzhou Municipal Bureau
of Health (grant no. WZ201043).
References
|
1
|
Liu L, Wang D, Wong KS and Wang Y: Stroke
and stroke care in China: Huge burden, significant workload, and a
national priority. Stroke. 42:3651–3654. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Song D and Cho AH: Previous and recent
evidence of endovascular therapy in acute ischemic stroke.
Neurointervention. 10:51–59. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Amar AP, Griffin JH and Zlokovic BV:
Combined neurothrombectomy or thrombolysis with adjunctive delivery
of 3K3A-activated protein C in acute ischemic stroke. Front Cell
Neurosci. 9:3442015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Reuter B, Grudzenski S, Chatzikonstantinou
E, Meairs S, Ebert A, Heiler P, Schad LR, Staufenbiel M, Hennerici
MG and Fatar M: Thrombolysis in experimental cerebral amyloid
angiopathy and the risk of secondary intracerebral hemorrhage.
Stroke. 45:2411–2416. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chechneva OV, Mayrhofer F, Daugherty DJ,
Krishnamurty RG, Bannerman P, Pleasure DE and Deng W: A Smoothened
receptor agonist is neuroprotective and promotes regeneration after
ischemic brain injury. Cell Death Dis. 5:e14812014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen X, Zhang X, Wang Y, Lei H, Su H, Zeng
J, Pei Z and Huang R: Inhibition of immunoproteasome reduces
infarction volume and attenuates inflammatory reaction in a rat
model of ischemic stroke. Cell Death Dis. 6:e16262015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ahmad M and Graham SH: Inflammation after
stroke: Mechanisms and therapeutic approaches. Transl Stroke Res.
1:74–84. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nakajima H, Kubo T, Ihara H, Hikida T,
Danjo T, Nakatsuji M, Shahani N, Itakura M, Ono Y, Azuma YT, et al:
Nuclear-translocated Glyceraldehyde-3-phosphate dehydrogenase
promotes poly (ADP-ribose) polymerase-1 activation during
oxidative/nitrosative stress in stroke. J Biol Chem.
290:14493–14503. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yainoy S, Houbloyfa P, Eiamphungporn W,
Isarankura-Na-Ayudhya C and Prachayasittikul V: Engineering of
chimeric catalase-Angiopep-2 for intracellular protection of brain
endothelial cells against oxidative stress. Int J Biol Macromol.
68:60–66. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang L, Li Z, Zhang X, Wang S, Zhu C, Miao
J, Chen L, Cui L and Qiao H: Protective effect of shikonin in
experimental ischemic stroke: Attenuated TLR4, p-p38MAPK, NF-κB,
TNF-α and MMP-9 expression, up-regulated claudin-5 expression,
ameliorated BBB permeability. Neurochem Res. 39:97–106. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Candelario-Jalil E, Yang Y and Rosenberg
GA: Diverse roles of matrix metalloproteinases and tissue
inhibitors of metalloproteinases in neuroinflammation and cerebral
ischemia. Neuroscience. 158:983–994. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vahedi K, Hofmeijer J, Juettler E, Vicaut
E, George B, Algra A, Amelink GJ, Schmiedeck P, Schwab S, Rothwell
PM, et al: Early decompressive surgery in malignant infarction of
the middle cerebral artery: A pooled analysis of three randomised
controlled trials. Lancet Neurol. 6:215–222. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Medzhitov R and Preston-Hurlburt P and
Janeway CA Jr: A human homologue of the Drosophila Toll protein
signals activation of adaptive immunity. Nature. 388:394–397. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jack CS, Arbour N, Manusow J, Montgrain V,
Blain M, McCrea E, Shapiro A and Antel JP: TLR signaling tailors
innate immune responses in human microglia and astrocytes. J
Immunol. 175:4320–4330. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang Y, Ge P and Zhu Y: TLR2 and TLR4 in
the brain injury caused by cerebral ischemia and reperfusion.
Mediators Inflamm. 2013:1246142013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zou N, Ao L, Cleveland JC Jr, Yang X, Su
X, Cai GY, Banerjee A, Fullerton DA and Meng X: Critical role of
extracellular heat shock cognate protein 70 in the myocardial
inflammatory response and cardiac dysfunction after global
ischemia-reperfusion. Am J Physiol Heart Circ Physiol.
294:H2805–H2813. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hyakkoku K, Hamanaka J, Tsuruma K,
Shimazawa M, Tanaka H, Uematsu S, Akira S, Inagaki N, Nagai H and
Hara H: Toll-like receptor 4 (TLR4), but not TLR3 or TLR9,
knock-out mice have neuroprotective effects against focal cerebral
ischemia. Neuroscience. 171:258–267. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Caso JR, Pradillo JM, Hurtado O, Lorenzo
P, Moro MA and Lizasoain I: Toll-like receptor 4 is involved in
brain damage and inflammation after experimental stroke.
Circulation. 115:1599–1608. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cao CX, Yang QW, Lv FL, Cui J, Fu HB and
Wang JZ: Reduced cerebral ischemia-reperfusion injury in Toll-like
receptor 4 deficient mice. Biochem Biophys Res Commun. 353:509–514.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rao J, Qian X, Li G, Pan X, Zhang C, Zhang
F, Zhai Y, Wang X and Lu L: ATF3-mediated NRF2/HO-1 signaling
regulates TLR4 innate immune responses in mouse liver
ischemia/reperfusion injury. Am J Transplant. 15:76–87. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Surh YJ and Na HK: NF-kappaB and Nrf2 as
prime molecular targets for chemoprevention and cytoprotection with
anti-inflammatory and antioxidant phytochemicals. Genes Nutr.
2:313–317. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shah ZA, Li RC, Ahmad AS, Kensler TW,
Yamamoto M, Biswal S and Doré S: The flavanol (−)-epicatechin
prevents stroke damage through the Nrf2/HO1 pathway. J Cereb Blood
Flow Metab. 30:1951–1961. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Albert-Weißenberger C, Sirén AL and
Kleinschnitz C: Ischemic stroke and traumatic brain injury: The
role of the kallikrein-kinin system. Prog Neurobiol 101–102. 1–82.
2013.
|
|
24
|
Xia CF, Yin H, Yao YY, Borlongan CV, Chao
L and Chao J: Kallikrein protects against ischemic stroke by
inhibiting apoptosis and inflammation and promoting angiogenesis
and neurogenesis. Hum Gene Ther. 17:206–219. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xia CF, Yin H, Borlongan CV, Chao L and
Chao J: Kallikrein gene transfer protects against ischemic stroke
by promoting glial cell migration and inhibiting apoptosis.
Hypertension. 43:452–459. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tatlisumak T, Carano RA, Takano K,
Opgenorth TJ, Sotak CH and Fisher M: A novel endothelin antagonist,
A-127722, attenuates ischemic lesion size in rats with temporary
middle cerebral artery occlusion: A diffusion and perfusion MRI
study. Stroke. 29:850–858. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Arumugam TV, Tang SC, Lathia JD, Cheng A,
Mughal MR, Chigurupati S, Magnus T, Chan SL, Jo DG, Ouyang X, et
al: Intravenous immunoglobulin (IVIG) protects the brain against
experimental stroke by preventing complement-mediated neuronal cell
death. Proc Natl Acad Sci USA. 104:pp. 14104–14109. 2007;
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hua F, Ma J, Ha T, Xia Y, Kelley J,
Williams DL, Kao RL, Browder IW, Schweitzer JB, Kalbfleisch JH and
Li C: Activation of Toll-like receptor 4 signaling contributes to
hippocampal neuronal death following global cerebral
ischemia/reperfusion. J Neuroimmunol. 190:101–111. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tang SC, Arumugam TV, Xu X, Cheng A,
Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, et
al: Pivotal role for neuronal Toll-like receptors in ischemic brain
injury and functional deficits. Proc Natl Acad Sci USA. 104:pp.
13798–13803. 2007; View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ridder DA and Schwaninger M: NF-kappaB
signaling in cerebral ischemia. Neuroscience. 158:995–1006. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hayden MS, West AP and Ghosh S: NF-kappaB
and the immune response. Oncogene. 25:6758–6780. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schneider A, Martin-Villalba A, Weih F,
Vogel J, Wirth T and Schwaninger M: NF-kappaB is activated and
promotes cell death in focal cerebral ischemia. Nat Med. 5:554–559.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Morita-Fujimura Y, Fujimura M, Yoshimoto T
and Chan PH: Superoxide during reperfusion contributes to caspase-8
expression and apoptosis after transient focal stroke. Stroke.
32:2356–2361. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shih AY, Li P and Murphy TH: A
small-molecule-inducible Nrf2-mediated antioxidant response
provides effective prophylaxis against cerebral ischemia in vivo. J
Neurosci. 25:10321–10335. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Poss KD and Tonegawa S: Reduced stress
defense in heme oxygenase 1-deficient cells. Proc Natl Acad Sci
USA. 94:pp. 10925–10930. 1997; View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Satoh T, Izumi M, Inukai Y, Tsutsumi Y,
Nakayama N, Kosaka K, Shimojo Y, Kitajima C, Itoh K, Yokoi T and
Shirasawa T: Carnosic acid protects neuronal HT22 cells through
activation of the antioxidant-responsive element in free carboxylic
acid- and catechol hydroxyl moieties-dependent manners. Neurosci
Lett. 434:260–265. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shi H, Jing X, Wei X, Perez RG, Ren M,
Zhang X and Lou H: S-allyl cysteine activates the Nrf2-dependent
antioxidant response and protects neurons against ischemic injury
in vitro and in vivo. J Neurochem. 133:298–308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li L, Zhang X, Cui L, Wang L, Liu H, Ji H
and Du Y: Ursolic acid promotes the neuroprotection by activating
Nrf2 pathway after cerebral ischemia in mice. Brain Res.
1497:32–39. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kunze R, Urrutia A, Hoffmann A, Liu H,
Helluy X, Pham M, Reischl S, Korff T and Marti HH: Dimethyl
fumarate attenuates cerebral edema formation by protecting the
blood-brain barrier integrity. Exp Neurol. 266:99–111. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sapojnikova N, Kartvelishvili T, Asatiani
N, Zinkevich V, Kalandadze I, Gugutsidze D, Shakarishvili R and
Tsiskaridze A: Correlation between MMP-9 and extracellular cytokine
HMGB1 in prediction of human ischemic stroke outcome. Biochim
Biophys Acta. 1842:1379–1384. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Clark AW, Krekoski CA, Bou SS, Chapman KR
and Edwards DR: Increased gelatinase A (MMP-2) and gelatinase B
(MMP-9) activities in human brain after focal ischemia. Neurosci
Lett. 238:53–56. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cai Y, Liu X, Chen W, Wang Z, Xu G, Zeng Y
and Ma Y: TGF-β1 prevents blood-brain barrier damage and
hemorrhagic transformation after thrombolysis in rats. Exp Neurol.
266:120–126. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kamiya T, Katayama Y, Kashiwagi F and
Terashi A: The role of bradykinin in mediating ischemic brain edema
in rats. Stroke. 24:571–576. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zausinger S, Lumenta DB, Pruneau D,
Schmid-Elsaesser R, Plesnila N and Baethmann A: Effects of LF
16–0687 Ms, a bradykinin B(2) receptor antagonist, on brain edema
formation and tissue damage in a rat model of temporary focal
cerebral ischemia. Brain Res. 950:268–278. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lu RY, Luo DF, Xiao SH, Yang LH, Zhao J,
Ji EN, Tao EX, Xing YG, Zhu FY, Luan P and Liu J: Kallikrein gene
transfer induces angiogenesis and further improves regional
cerebral blood flow in the early period after cerebral
ischemia/reperfusion in rats. CNS Neurosci Ther. 18:395–399. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chen ZB, Huang DQ, Niu FN, Zhang X, Li EG
and Xu Y: Human urinary kallidinogenase suppresses cerebral
inflammation in experimental stroke and downregulates nuclear
factor-kappaB. J Cereb Blood Flow Metab. 30:1356–1365. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chao J, Shen B, Gao L, Xia CF, Bledsoe G
and Chao L: Tissue kallikrein in cardiovascular, cerebrovascular
and renal diseases and skin wound healing. Biol Chem. 391:345–355.
2010. View Article : Google Scholar : PubMed/NCBI
|