Introduction
Chronic congestive heart failure (CHF) is the end
outcome of organic heart diseases and one of the major diseases
threatening human health. Heart failure occurs in 0.9% of Chinese
adults (1), and has become a
significant social and public health issue. Cardiac remodeling is
an important pathophysiological process in the event of CHF, and
involves co-action of myocardial apoptosis, cardiac hypertrophy and
myocardial fibrosis (2). Apoptosis
is a gene-controlled autonomous program cell death process
involving activation, expression and regulation of a series of
genes (3). Myocardial apoptosis
occurs in a variety of cardiovascular diseases and is closely
associated with arrhythmia and cardiac dysfunctions (4). Although the detailed mechanisms of the
apoptotic process are not fully understood, members of the caspase
family, including caspases 2, 3, 6, 7, 8, 9 and 10, have been
confirmed to serve an essential role in apoptosis (5). Tumor suppressor gene p53 is another
known regulatory factor closely associated with myocardial
apoptosis (6,7). In addition, overactivation of the
sympathetic nervous system (SNS) and renin-angiotensin-aldosterone
system (RAAS) may lead to myocardial apoptosis (8). Currently, the suppression of excessive
activation of the SNS and RAAS in order to further inhibit
myocardial apoptosis is an important focus for the treatment of
heart failure.
SNS activation is closely associated with myocardial
apoptosis; however, this activation increases the serum
nore-pinephrine concentration, leading to overactivation of the
norepinephrine-tumor necrosis factor-α (TNF-α)-caspase
pathway-mediated myocardial apoptosis (9). In addition, SNS activation increases
the release of angiotensin II (Ang II), leading to nuclear factor
(NF)-κB-mediated myocardial hypertrophy, fibrosis and apoptosis
(10). Zelarayan et al
(11) demonstrated that p53 serves
an important role in Ang II-induced myocardial apoptosis and may
also regulate NF-кB expression. SNS activation is also closely
correlated with the prognosis of patients with heart failure. It is
often accompanied with increased releases of norepinephrine and
neuropeptide Y, peripheral vasoconstriction, reduced renal blood
flow, and decreased sodium and water excretion, all of which will
facilitate the process of heart failure (9).
SNS activation mainly includes cardiac sympathetic
nerve activation (CSNA), renal sympathetic nerve activation (RSNA),
central nervous system activation, as well as skeletal muscle
sympathetic activation (12). The
amount of norepinephrine released in the heart and kidney after SNS
activation accounted for 62% of the total norepinephrine in the
body, suggesting that CSNA and RSNA play essential roles in the
onset and progression of heart failure (13). Following renal sympathetic
activation, the renal afferent nerve may activate multi-organ
sympathetic nerves, including the cardiac sympathetic nerve,
central sympathetic nerve and skeletal muscle sympathetic nerve
(14), which further leads to
increased norepinephrine release and RAS activation, thus
contributing to myocardial remodeling. Renal nerves enter the renal
parenchyma and renal artery, forming a neural network in renal
tubules and glomerulus. Renal nerves have a relatively simple
composition. Anatomical studies have confirmed that 95% of renal
sympathetic nerves are located on the surface of renal artery
(15). To date, parasympathetic
nerves have not been found in renal nerves. Therefore, renal
denervation (RD) is equal to renal sympathetic denervation (RSD). A
previous study has demonstrated that RSD is the anatomical basis
for catheter renal sympathetic nerve ablation within the renal
artery and easy to implement (16).
Transcatheter renal sympathetic ablation has been used for the
treatment of hypertension since 2009, and has been proven to be
safe and effective in reducing the blood pressure in patients with
refractory hypertension (17). Based
on the physiological and pathological effects of RSNA and the fact
that renal sympathetic activation is a common pathophysiological
channel of heart failure and hypertension, it can be speculated
that RSD may also be able to improve myocardial remodeling of
patients with heart failure. The recent REACH-Pilot trial included
patients with CHF and normal blood pressure, and demonstrated that
RSD significantly increased the patients' six-minute walking
distance without significantly affecting their blood pressure, thus
suggesting that renal sympathetic ablation significantly improved
the heart function of patients with heart failure (18). However, renal sympathetic ablation
had little adverse impacts on the hemodynamics and renal function,
suggesting that its impact on the prognosis of patients with heart
failure is not mediated by lowering the blood pressure.
The current RSD therapy for heart failure is
relatively novel, with only a small number of inpatients
participating and a lack of long-term follow-up observations.
Certain researchers have explored the role of RSD in myocardial
remodeling. Wang et al (19)
demonstrated that RSD significantly reduced the serum Ang II and
aldosterone concentrations in a canine model of heart failure
induced by rapid pacing, and inhibited atria reconstruction.
Furthermore, Clayton et al (20) confirmed that RSD inhibited the
expression of myocardial Ang II receptor in a rabbit model of heart
failure, increased urinary sodium excretion, lowered the plasma
brain natriuretic peptide (BNP) level and significantly improved
heart function. Hu et al (21) confirmed that RSD significantly
improved myocardial remodeling in a rat model of myocardial
infarction, increased the urine output and improved cardiac
function of the treated model animals.
Considering the importance of norepinephrine and Ang
II in myocardial apoptosis, it can be speculated that RSD is likely
to inhibit myocardial apoptosis by reducing norepinephrine and Ang
II release. However, to the best of our best knowledge, the effects
of RSD on myocardial fibrosis and its underlying mechanism have not
been previously studied. Therefore, the present study aimed to use
an isoproterenol-induced rat model with pressure overload heart
failure in order to: i) Investigate the impacts of RSD on
myocardial apoptosis to provide a theoretical basis for clinical
treatment of heart failure with RSD; ii) explore the impacts of RSD
on the expression of factors associated with myocardial apoptosis,
including p53, TNF-α, NF-κB, caspase-2 and −3; and iii) analyze the
molecular mechanisms underlying the inhibitory effects of RSD on
myocardial apoptosis, attempting to provide a theoretical basis for
the treatment of heart failure using renal sympathetic ablation.
The present study confirmed that renal sympathetic activation was
accompanied with an increased release of norepinephrine and Ang II.
In addition, norepinephrine was able to induce myocardial apoptosis
through free peroxy radicals-TNF-α-caspase signaling pathway, and
Ang II induced myocardial apoptosis by regulating the expression
levels of p53 and NF-кB.
Materials and methods
Experimental animals
A total of 70 4-week-old male cleaning grade
Sprague-Dawley rats weighing 250–350 g were purchased from Beijing
HFK Bioscience Co., Ltd. (Beijing, China). The rats were housed at
an environment with temperature of 23±2°C, humidity of 61±5% and
light/dark cycle of 12 h, and were fed with a standard diet. All
rats were adapted to the environment for 1 week prior to conducting
any experiments in compliance with the procedures of the Animal
Ethics Committee of Wuhan University (Wuhan, China). The study was
approved by appropriate local institutional review boards at the
Renmin Hospital of Wuhan University (Wuhan, China) and conformed to
the guidelines set forth by the Declaration of Helsinki.
Animal grouping
The rats were randomly divided into four groups,
namely the CHF+sham, CHF+RSD, control (in which the rats were
untreated) and sham groups, with 15 rats each in the control and
sham groups, and 20 rats each in the CHF+sham and CHF+RSD groups.
Rats in the sham group were subjected to pseudo-RSD (in which a
sham procedure was performed on the rats), while rats in the
CHF+sham were subjected to pseudo-RSD followed by induction of
heart failure 1 week later. Rats in the CHF+RSD were subjected to
RSD followed by induction of heart failure 1 week later.
Conduction of RSD and induction of
heart failure
To perform RSD, rats were anesthetized by
intraperitoneal injection of 40 mg/kg sodium pentobarbital
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Abdominal skin was
prepared, disinfected and covered with towels. An incision was made
on the skin along the abdominal linea alba, and subcutaneous tissue
and muscles were separated layer by layer to expose the kidney,
ureter as well as intrathecal arteries, veins and nerves. The
artery and vein sheaths were stripped under a dissecting microscope
(magnification, ×25) to disarticulate all visible nerve fibers,
which along with their surroundings were further wiped with a
cotton ball dipped in 10% phenol solution to achieve sufficient
denervation effect. To ensure thorough denervation, the degree of
renal denervation was evaluated using an electrical stimulation
method (5). Prior to RSD, a
significant sympathetic response (indicated by systolic blood
pressure increase by 5–10 mmHg, heart rate increase by 8–15
beats/min and pale kidney due to renal artery contraction) was
induced by stimulating the proximal renal nerve for 10–30 sec with
square wave provided by ML 1001 (15 V, 0.2 msec, 10 Hz) using a 5Fr
radiofrequency catheter (Ablaze; Japan Lifeline Co., Ltd., Tokyo,
Japan). Subsequent to RSD, the aforementioned responses such as SBP
and heart rate completely disappeared. The procedure strictly
followed the requirements for aseptic surgery. At 3 days after
surgery, 80,000 U penicillin was intraperitoneally injected per day
for 10 days to prevent infection. To induce heart failure, 4 mg/kg
isoproterenol (cat. no. EY0883; Amquar Biological Technology Co.,
Ltd., Shanghai, China) was subcutaneously injected once daily for
10 days at 3 days after surgery with caution to avoid withdraw and
damaging blood vessel.
Determination of plasma
norepinephrine, Ang II and aldosterone levels
At the end of the experimental observation,
peripheral blood was withdrawn from each rat into a tube containing
10% EDTA and aprotinin. Subsequent to centrifugation at 1,350 × g
for 20 min at 4°C, plasma was collected and stored at −70°C until
further use. The plasma contents of norepinephrine, Ang II and
aldosterone were measured using ELISA kits according to the
instructions provided by the manufacturer (cat. no. PH003RAT;
Phygene Life Sciences, Fuzhou, China for Ang II), (ab136933; Abcam,
Cambridge, UK for aldosterone), (PH031UNI; Phygene Life Sciences
for norepinephrine). The results were converted to the actual
content by comparing to the GAPDH as standard.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis of differential mRNA
expression in myocardial tissues
RT-qPCR analysis was used to detect the differential
mRNA expression levels of p53, TNF-α, NF-κB, caspase-2 and −3
between the RSD and normal myocardial tissues. Rats were sacrificed
and myocardial tissues were extracted 6 weeks after establishing
heart failure from rats in the control, sham, CHF+sham and CHF+RSD
groups. The tissues were ground and dissolved in an appropriate
amount of RNAiso Plus solution (Takara, Dalian, China). Total RNA
was then extracted using TRIzol reagent (Takara) and reverse
transcribed into cDNA using the first-strand cDNA kit (cat. no.
FSQ-101; Toyobo Co., Ltd., Tokyo, Japan) and
oligo(dT)15. RT-qPCR analysis was performed using
FastStart Universal SYBR Green kit (Roche Applied Science,
Mannhein, Germany) in a 10 µl reaction system containing 2 µl cDNA
template, 5 µl SYBR Green Mix with ROX, 200 nM forward reaction
primer and 200 nM reverse reaction primer. Table I lists the primer sequences for p53,
TNF-α, NF-κB, caspase 2 and caspase 3. Differential expression
levels of RCR products were analyzed using melting curves and the
2−ΔΔCq method (22). Rat
GAPDH was used as an internal control, and all experiments were
performed for at least three times.
 | Table I.Primers used in RT-qPCR. |
Table I.
Primers used in RT-qPCR.
| Gene (rat) | Forward primer | Reverse primer |
|---|
| p53 |
5′-TCCCCAGCAAAAGAAAAAAC-3′ |
5′-GCACGGGCATCCTTTAATT-3′ |
| TNF-α |
5′-GGCCACCACGCTCTTCTGT-3′ |
5′-CGGGCTTGTCACTCGAGTTT-3′ |
| NF-κB |
5′-ATTTCGATTCCGCTACGTGTG-3′ |
5′-GGGCGTGCAGGTGAATATTT-3′ |
| Caspase-2 |
5′-TGACCAGACTGCACAGGAAAT-3′ |
5′-ACCCCGTAGATGCCACCTT-3′ |
| Caspase-3 |
5′-CCCTGAAATGGGCTTGTGTAT-3′ |
5′-GGGCCATGAATGTCTCTCTGA-3′ |
| GAPDH |
5′-GAGCGAGATCCCGCTAACATC-3′ |
5′-GCGGAGATGATGACCCTTTTG-3′ |
Western blot analysis for the
detection of differences in protein expression in the rat
myocardium
Western blot analysis was used to detect the protein
expression levels of p53, TNF-α, NF-κB, caspase-2 and −3 in the rat
myocardium. First, myocardial tissues were extracted from rats in
the CHF+sham, CHF+RSD, control and sham groups, respectively. The
tissues were dissolved in ice-cold TNEN lysis buffer (50 mM
Tris/HCl, pH 7.5, 150 mM NaCl, 2.0 mM EDTA and 1.0% Nonidet P-40)
supplemented with trace of EDTA, protease inhibitors (Roche Applied
Science) and 1 mmol/l phenylmethylsulfonyl fluoride. Following
incubation at 4°C for 30 min, samples were centrifuged at 12,000 ×
g for 15 min at 4°C to remove pellets. Next, 100 µl supernatant was
mixed with 20 µl of 6X Laemmli buffer (0.3 mol/l Tris-HCl, 6% SDS,
60% glycerol, 120 mmol/l dithiothreitol and tracer dye bromophenol
blue) and incubated at 37°C for 10 min. Subsequently, 20-µl
aliquots of the samples were subjected to 10% SDS-PAGE and
transferred onto a 0.45 µm polyvinylidene fluoride membrane (EMD
Millipore, Billerica, MA, USA). The membrane was then incubated
with monoclonal rabbit antibodies against p53 (cat. no. ab26,
1:1,000, mouse monoclonal antibody), TNF-α (cat. no. ab6671,
1:1,000, rabbit polyclonal antibody), NF-κB (cat. no. ab32360,
1:5,000, rabbit monoclonal antibody), caspase-2 (cat. no. ab179520,
1:1,000, rabbit monoclonal antibody) and caspase-3 (cat. no.
ab13847, 1:500, rabbit polyclonal antibody), as well as with the
internal control GAPDH (1:3,000, mouse monoclonal antibody) (all
from Abcam), followed by incubation with secondary horseradish
peroxidase-conjugated goat anti-rabbit antibody (1:10,000, BL003A)
and goat anti-mouse antibody (1:10,000, BL001A) (both from
Biosharp, Beijing, China). The labeled proteins were then
visualized with SuperSignal West Pico chemiluminescence imaging
system (Pierce; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
The protein levels were quantified by calculating the grayscale
values of the three independent repeats scanned by Quantity One
software (Bio-Rad Laboratories, Inc., Hercules, CA, USA). All
experiments were performed for at least three independent
repeats.
Quantitative measurement of myocardial
apoptosis using flow cytometry
Rat myocardial apoptosis was examined using
FITC-labeled Annexin V/flow cytometry. Annexin V (cat. no. KGA106;
KeyGen Biotech Co., Ltd., Nanjing, China) is a calcium-dependent
phospholipid binding protein with high affinity for binding to
phosphatidylserine on the outer membrane of apoptotic cells.
FITC-labeled Annexin V is commonly used as a sensitive reagent to
quantitatively detect early apoptotic cells by counting the
percentage of positive apoptotic cells. In this experiment,
myocardial tissues were collected from rats in the CHF+sham,
CHF+RSD, control and sham groups and suspended in chilled 1X
phosphate-buffered saline (PBS) solution. The tissues were then
digested with fibrinogen and collagenase to obtain single cells. A
total of 106 cells were mixed with 100 µl binding buffer
and incubated with 10 µl FITC-labeled Annexin V (20 µg/ml) in the
dark at room temperature for 30 min, followed by incubation with 5
µl PI (50 µg/ml) in the dark for 5 min. Cells were then diluted
with 400 µl binding buffer and immediately subjected to FACScan
flow cytometry to quantitatively measure the percentage of
apoptotic cells using cells without FITC-labeled Annexin V and PI
as a negative control.
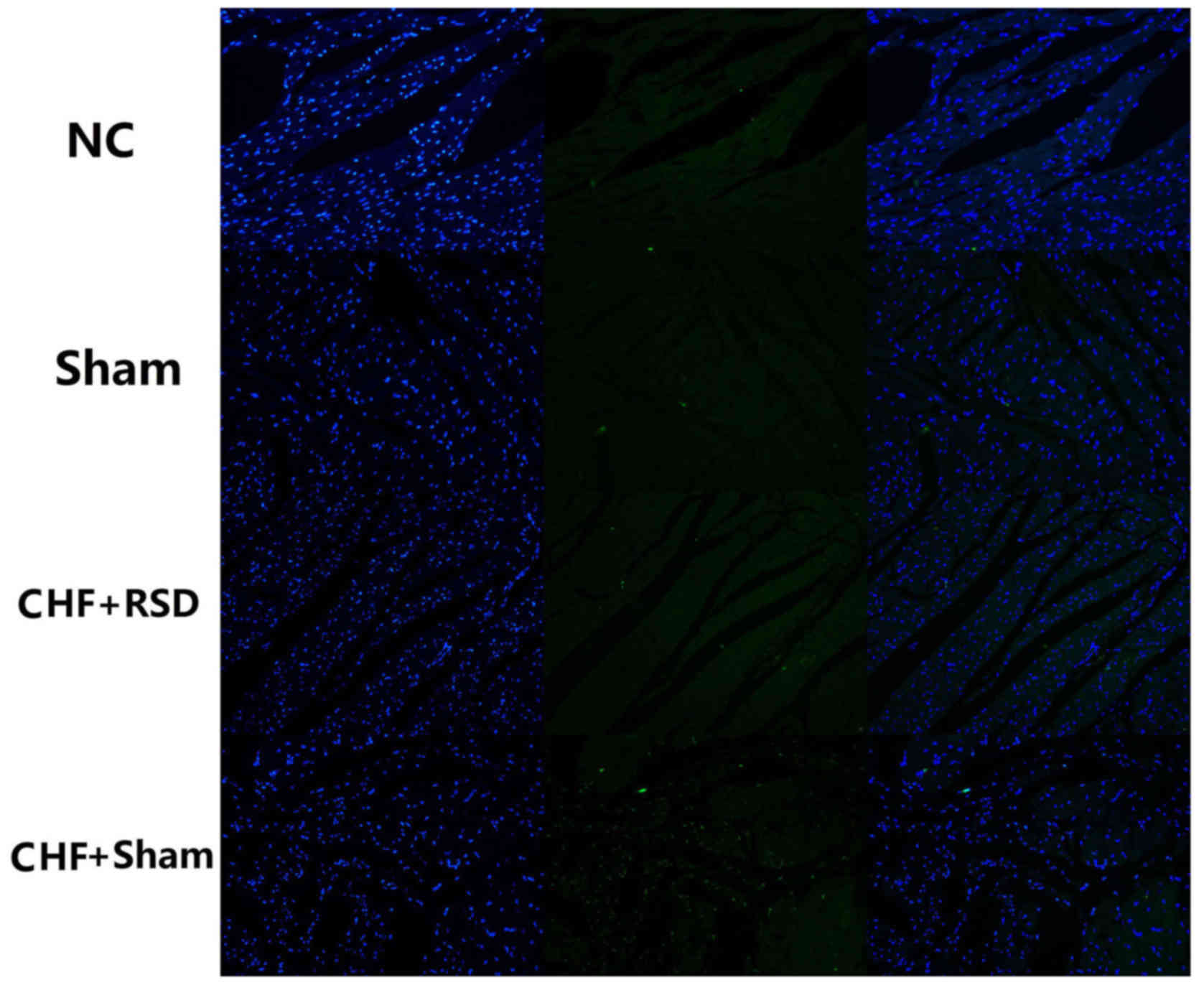
TUNEL assay and laser scanning
confocal microscopy to detect myocardial apoptosis
TUNEL method is commonly used for detecting
apoptosis. In the present study, TUNEL assay (Beckman Coulter,
Inc., Brea, CA, USA) was applied together with laser scanning
confocal microscopy (LSCM) to detect myocardial apoptosis in rat
tissues in the CHF+sham, CHF+RSD, control and sham groups. Briefly,
the rat heart were extracted, fixed in 4% polyformaldehyde
solution, paraffin-embedded and prepared into 5-µm sections. These
sections were dewaxed, treated with 3% H2O2
for 10 min to block endogenous peroxidase activity, rinsed three
times with 1X PBS for 5 min each and digested with fresh proteinase
K working solution (10 mg/l in 10 mmol/l Tris-HCl, pH 7.4–8.0) at
room temperature for 20 min. After rinsing again with 1X PBS three
times for 5 min each, the sections were incubated with TUNEL
solution in the dark at 37°C in a humidified chamber for 60 min,
and rinsed with 1X PBS three times for 5 min each time.
Subsequently, the samples were incubated with horseradish
peroxidase-labeled anti-FITC antibody (cat. no. C1063, Annexin
V-FITC kit; Beyotime Institute of Biotechnology, Haimen, China) at
37°C in a humidified chamber in dark for 30 min, rinsed as before
and stained with DAB for 5–10 min. After further rinsing as before,
the sections were stained with hematoxylin for a few sec, washed
with running water, dehydrated, mounted on slides and sealed with
neutral gum. Negative controls were prepared in a similar manner,
but without TdT in the TUNEL reaction solution. The slides were
subjected to LSCM at a magnification of ×400 to observe the level
of myocardial apoptosis of rats in each group.
Statistical analysis
Data are expressed as the mean ± standard deviation.
SPSS software version l7.0 (SPSS, Inc., Chicago, IL, USA) was used
for statistical analysis. One-way analysis of variance was
conducted to compare experimental groups, followed by Scheffe's
post hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
RSD significantly inhibits the levels
of Ang II and aldosterone in the plasma of rats with heart
failure
The present study demonstrated that, following renal
sympathetic activation, increased norepinephrine and Ang II were
released, while myocardial apoptosis was also increased (Table II). In order to determine whether
RSD was able to inhibit myocardial apoptosis of rats with heart
failure, ELISA was employed to detect the contents of
norepinephrine, Ang II and aldosterone in the plasma of rats in the
CHF+sham, CHF+RSD, control and sham groups. The results revealed
that the expression levels of plasma Ang II and aldosterone
decreased from 491.1±14.8 and 517.3±16.0 in CHF+sham rats to
388.3±13.7 and 408.5±12.3 in the CHF+RSD rats, respectively
(P<0.01; Table II). The
expression level of norepinephrine was also reduced from 264.6±13.6
in rats of the CHF+sham group to 200.7±8.0 in rats of the CHF+RSD
group, although this difference was not statistically significant.
By contrast, the expression levels of norepinephrine, Ang II and
aldosterone demonstrated no significant differences between rats in
the control and sham groups (P>0.05). These experimental results
are consistent with a previous study by DiBona (17) using a canine model established with
rapid pacing. Thus, RSD was confirmed to lower the CHF-induced
release of Ang II and aldosterone induced by CHF.
| Table II.Expression levels of norepinephrine,
Ang II and aldosterone in the rat plasma. |
Table II.
Expression levels of norepinephrine,
Ang II and aldosterone in the rat plasma.
| Group | n | Norepinephrine | Ang II | Aldosterone |
|---|
| Control | 15 | 159.3±11.1 | 208.5±14.3 | 205.3±14.8 |
| Sham | 13 | 168.2±13.7 | 201.6±10.5 | 197.0±9.5 |
| CHF+sham | 12 | 264.6±13.6 |
491.1±14.8a |
517.3±16.0a |
| CHF+RSD | 10 | 200.7±8.0 |
388.3±13.7b |
408.5±12.3b |
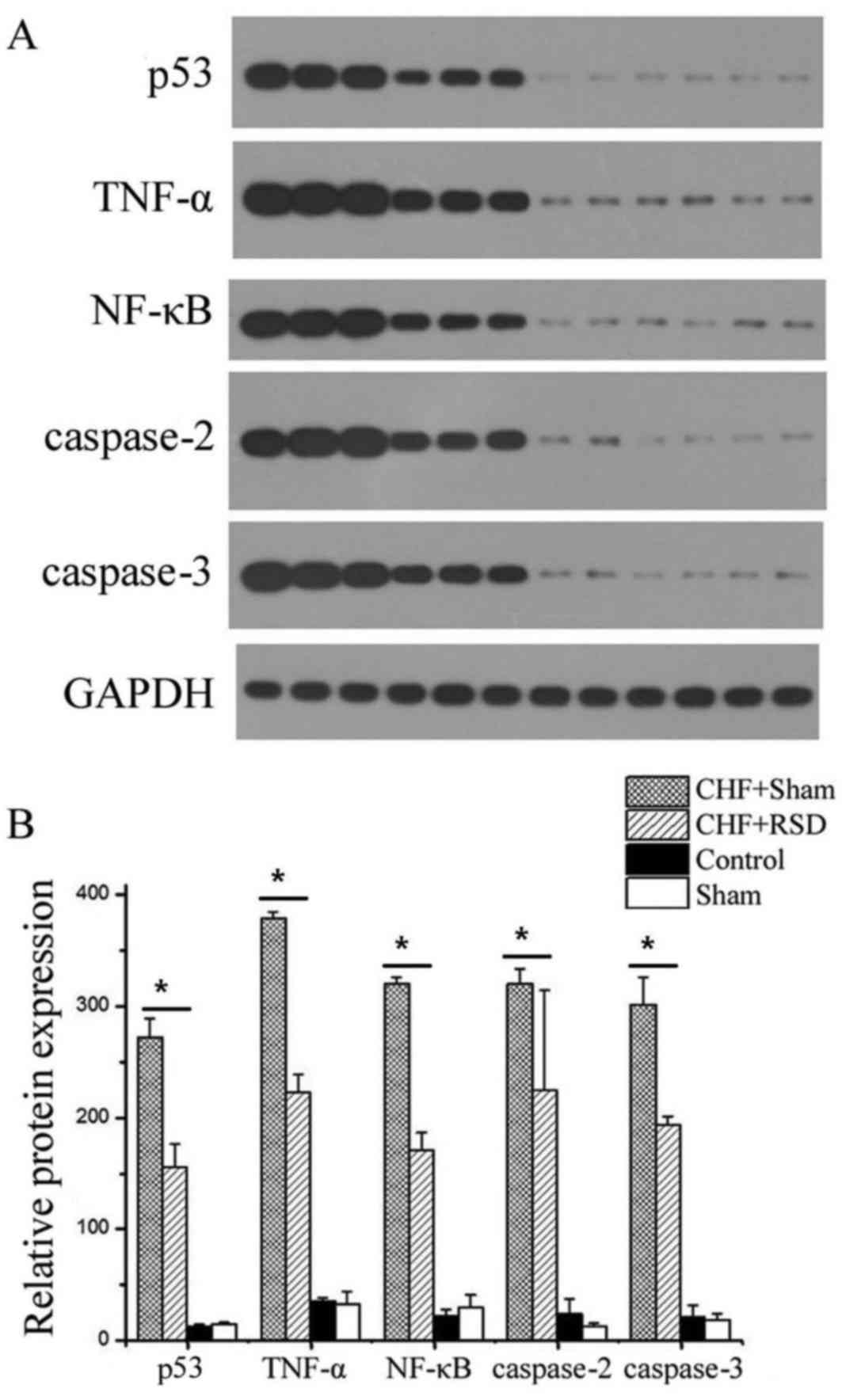
RSD significantly inhibits the
expression of p53, TNF-α, NF-κB, as well as caspases-2 and −3, at
the mRNA and protein levels
To determine the effects of RSD on the myocardial
apoptosis and remodeling of rats with heart failure, RT-qPCR and
western blot analyses were applied to detect the expression levels
of the apoptosis-associated factors p53, TNF-α, NF-κB, caspase-2
and −3 at the transcriptional and translational levels. The results
revealed that RSD significantly inhibited the expression levels of
p53, TNF-α, NF-κB, caspase-2 and −3. In addition, myocardial
tissues were extracted from rats in the CHF+sham, CHF+RSD, control
and sham groups, and used to measure the mRNA expression by qPCR.
The results showed that the mRNA expression levels of the apoptotic
factors p53, TNF-α, NF-κB, caspase-2 and −3 in the myocardial
tissues of rats in the CHF+RSD group were significantly reduced by
71.6, 39.3, 42.3, 59.1 and 60.7%, respectively, compared with those
in the CHF+sham group (Fig. 1;
P=7.45×10−7, 4.56×10−8, 4.33×10−9,
1.41×10−7 and 3.00×10−6, respectively).
Furthermore, western blot analysis revealed that, compared with the
CHF+sham group, the protein levels of p53, TNF-α, NF-κB, caspase-2
and −3 were reduced by 42.6, 41.3, 46.7, 30.0 and 35.8%,
respectively, in the myocardial tissues of CHF+RSD rats (Figs. 2 and 3; P=0.002, 1.07×10−4,
1.13×10−4, 1.39×10−4 and 0.002,
respectively). By contrast, the mRNA and protein levels of p53,
TNFα, NF-κB, caspase-2 and −3 were not significantly different
between rats in the control and sham groups (P>0.05).
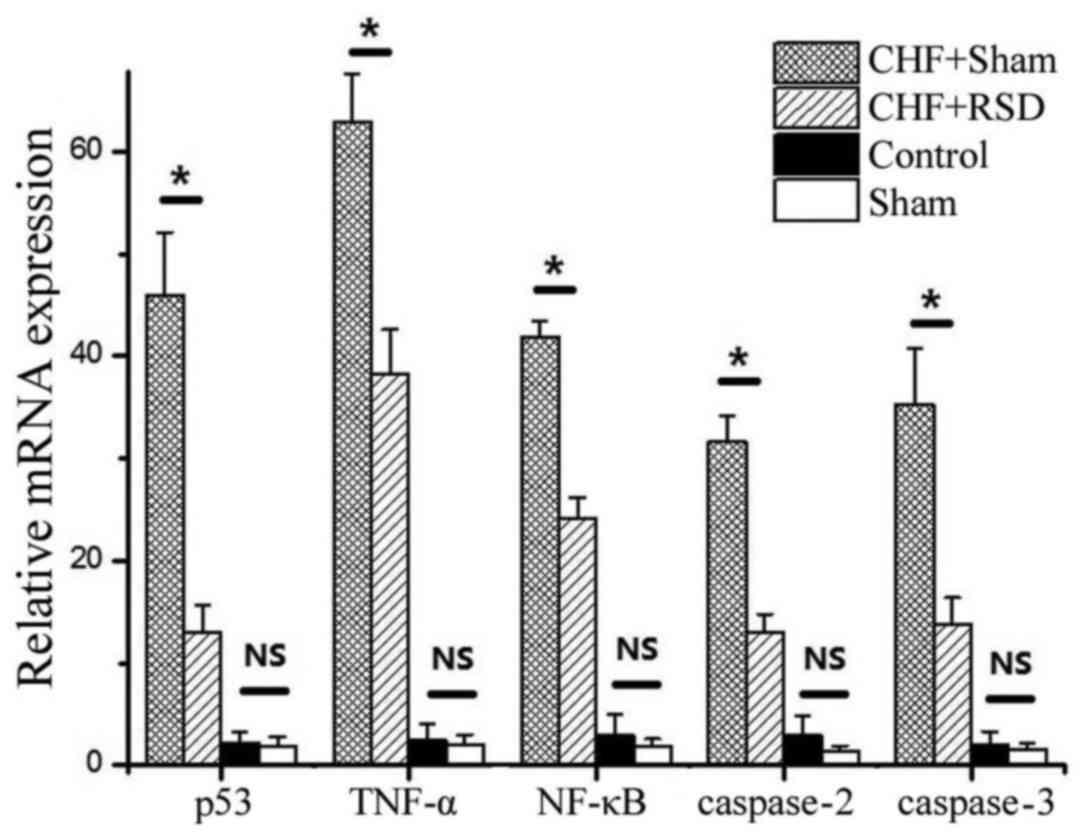 | Figure 1.RT-qPCR analysis of apoptosis factors
in rat myocardial tissues. The mean mRNA levels of P53, TNF-α,
NF-κB, caspase-2 and −3 in myocardial tissues of rats in the
CHF+sham, CHF+RSD, control and sham groups are shown from three
independent experiments. *P<0.05. RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; NS,
non-significant difference (P>0.05). CHF, congestive heart
failure; RSD, renal sympathetic denervation; TNF, tumor necrosis
factor; NF, nuclear factor. |
Overall, the aforementioned results indicated that
RSD was able to significantly inhibit the expression of the
apoptotic factors p53, TNFα, NF-κB, caspase-2 and −3 in rat
myocardial tissues at both the mRNA and protein levels, and these
factors were closely associated with myocardial apoptosis.
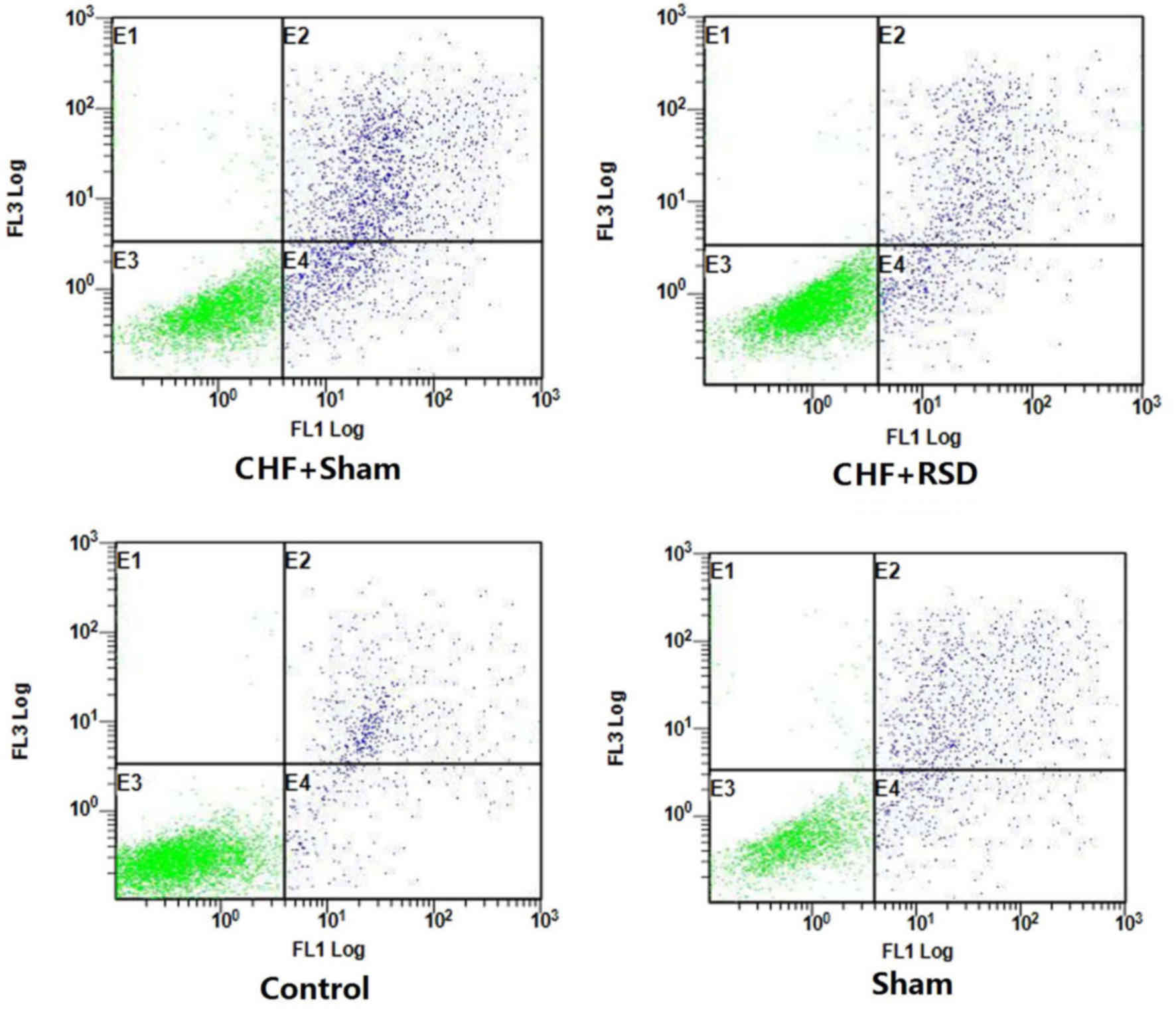
RSD inhibits apoptosis in myocardial
tissues
To further determine the impacts of RSD on the
apoptotic process of rat myocardial tissues, the apoptotic levels
of myocardial tissues of rats in the RSD+sham and CHF+sham groups
were detected and compared using Annexin V-FITC/PI-labeled flow
cytometry and TUNEL staining/LSCM. The results demonstrated that
the apoptosis level of myocardial tissues was decreased from 11.35%
in the CHF+sham group to 3.22% in the CHF+RSD group (Fig. 3; P<0.05). However, there was no
significantly difference in the apoptosis level between rats in the
control and sham groups (Fig. 3;
P>0.05). Consistently, TUNEL staining/LSCM also observed that
the apoptosis level was significantly decreased in myocardial
tissues of rats in the CHF+RSD group compared with that in the
CHF+sham group, but showed no significant difference between rats
in the control and sham groups (Fig.
4).
In conclusion, RSD evidently inhibited myocardial
apoptosis and intervened in the process of heart failure in the rat
tissues.
Discussion
Renal sympathetic activation is the joint
pathophysiologic channel of heart failure and hypertension, whereas
RSNA serves a crucial role in the onset and progression of heart
failure. Subsequent to renal sympathetic activation, renal afferent
nerves may induce activation of cardiac, renal, central, skeletal
muscle and other multi-organ sympathetic nerves (14), resulting in increased norepinephrine
release and RAS system activation, and further promoting the
myocardial remodeling process. A previous study has confirmed that
catheter-based renal sympathetic nerve ablation can safely and
effectively lower the blood pressure of patients with resistant
hypertension (16). The REACH pilot
study (18) proved that RSD
significantly increased the six-minute walking distance of patients
with chronic heart failure and normal blood pressure, but could not
evidently change their blood pressure, indicating that RSD ablation
of renal arteries may significantly improve the cardiac function of
heart failure patients with no adverse effects on the hemodynamics
and renal function. Previous results have also indicated that the
effect of RSD on the prognosis of patients with heart failure is
not mediated by the decrease in blood pressure, which suggests the
potential application of non-pharmacological treatment for patients
with heart failure (18). Recently,
Li et al applied RSD to patients with heart dysfunction,
myocardial fibrosis, and neurological immune response. The authors
observed that RSD significantly improved the myocardial fibrosis
and left ventricular and atrial hypertrophy, while inhibiting the
SNS, RAAS and arginine vasopressin of rats with transverse aortic
constriction (23).
The present study examined the levels of
norepinephrine, Ang II and aldosterone in the plasma of rats in
different groups. Consistent with the aforementioned results, the
present study observed that the expression levels of Ang II and
aldosterone in the plasma of rats in the CHF+RSD group were
significantly decreased compared with rats in the CHF+sham group.
Furthermore, to study the effect of RSD on myocardial apoptosis, a
model of rats with heart failure due to pressure load induced by
isoproterenol was constructed, and changes in the expression of
apoptosis-associated factors were examined. The results revealed
that RSD evidently suppressed the expression of
apoptosis-associated factors at the mRNA and protein levels.
RT-qPCR analysis showed that the mRNA levels of apoptotic factors
p53, TNF-α, NF-κB, caspase-2 and −3 in the myocardial tissues of
CHF+RSD rats were clearly reduced by 1.6, 39.3, 42.3, 59.1 and
60.7%, respectively, when compared with those in the CHF+sham
group. Western blot analysis also observed that the protein levels
of these factors in the myocardial tissues of CHF+RSD rats were
significantly decreased by 42.6, 41.3, 46.7, 30.0 and 35.8%,
respectively, compared with the CHF+sham group. These findings
indicated that RSD can effectively reduce myocardial apoptosis
after heart failure and improve myocardial remodeling of rats with
heart failure. A study by Hu et al investigating rats with
heart failure and myocardial infarction also suggested that RSD had
an important effect on myocardial remodeling (21,24),
which is in a good agreement with the present study results.
TUNEL staining/LSCM was also performed in the
present study, and demonstrated that RSD significantly improved
myocardial apoptosis in rats with heart failure. In addition,
FITC-Annexin V/PI double-labeled flow cytometry found that the
apoptosis levels of myocardial tissues of CHF+RSD rats were lowered
by 3.22% compared with those of CHF+sham rats (P<0.05), while no
significant differences were detected between the control and sham
groups (P>0.05). Thus, the results of the two methods
investigating myocardial apoptosis were consistent.
Currently, the use of RSD for heart failure therapy
is still a novel method. A limited number of inpatients have been
treated with RSD and followed up for long term. Grec et al
pointed out that RSD treatment not only provides an insight on the
treatment of resistant hypertension, but also brings new hope for
patients with cardiovascular metabolic diseases (25). Since norepinephrine and Ang II are
two important factors for myocardial apoptosis, RSD is likely to
achieve its role of inhibiting myocardial apoptosis by suppressing
the release of norepinephrine and Ang II. The present study has
proven the following: i) Renal sympathetic activation results in
increased release of norepinephrine, which further induces
myocardial apoptosis through the release of Ang II, which exerts
the same role by regulating the expression of p53 and NF-кB; ii)
RSD inhibited renal sympathetic activation, thus significantly
attenuating the expression levels of p53, TNF-α, NF-κB, caspase-2
and −3, and improving myocardial apoptosis.
In conclusion, the present study confirmed that, by
suppressing the release of norepinephrine and Ang II, RSD can
significantly inhibit myocardial apoptosis in rats with
norepinephrine-induced heart failure, lower the expression levels
of important myocardial apoptosis factors p53, TNF-α, NF-κB,
caspase-2 and −3, and improve the heart failure conditions of rats
with heart failure at the tissue level. The present study not only
provides data supporting the use of RSD to treat heart failure, but
also provides an insight into the use of non-pharmacological
treatment for heart failure.
Acknowledgements
The present study was supported by a grant from the
Foundation of Health and Family planning Commission of Hubei
Province (no. WJ2015MB293).
References
|
1
|
Gu DF, Huang GY and He J: Investigation on
epidemiology of heart failure in China and the incidence rate. Chin
Cardiovascular Dis Magazine. 31:3–6. 2003.(In Chinese).
|
|
2
|
Yancy CW, Jessup M, Bozkurt B, Butler J,
Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi
JL, et al: 2013 ACCF/AHA guideline for the management of heart
failure: A report of the American College of Cardiology
Foundation/American Heart Association Task Force on Practice
Guidelines. J Am Coll Cardiol. 62:e147–e239. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kerr JF, Wyllie AH and Currie AR:
Apoptosis: A basic biological phenomenon with wide-ranging
implications in tissue kinetics. Br J Cancer. 26:239–257. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Khoynezhad A, Jalali Z and Tortolani AJ: A
synopsis of research in cardiac apoptosis and its application to
congestive heart failure. Tex Heart Inst J. 34:352–359.
2007.PubMed/NCBI
|
|
5
|
Bae S, Yalamarti B and Kang PM: Role of
caspase-independent apoptosis in cardiovascular diseases, Frontiers
in bioscience. A J Virtual Library. 13:2495–2503. 2008. View Article : Google Scholar
|
|
6
|
Long X, Boluyt MO, Hipolito ML, Lundberg
MS, Zheng JS, O'Neill L, Cirielli C, Lakatta EG and Crow MT: p53
and the hypoxia-induced apoptosis of cultured neonatal rat cardiac
myocytes. J Clin Invest. 99:2635–2643. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang EY, Gang H, Aviv Y, Dhingra R,
Margulets V and Kirshenbaum LA: p53 mediates autophagy and cell
death by a mechanism contingent on Bnip3. Hypertension. 62:70–77.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Braam B, Cupples WA, Joles JA and Gaillard
C: Systemic arterial and venous determinants of renal hemodynamics
in congestive heart failure. Heart Fail Rev. 17:161–175. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fu YC, Chi CS, Yin SC, Hwang B, Chiu YT
and Hsu SL: Norepinephrine induces apoptosis in neonatal rat
cardiomyocytes through a reactive oxygen species-TNF alpha-caspase
signaling pathway. Cardiovasc Res. 62:558–567. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fu YC, Chi CS, Yin SC, Hwang B, Chiu YT
and Hsu SL: Norepinephrine induces apoptosis in neonatal rat
endothelial cells via down-regulation of Bcl-2 and activation of
beta-adrenergic and caspase-2 pathways. Cardiovasc Res. 61:143–151.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zelarayan L, Renger A, Noack C, Zafiriou
MP, Gehrke C, van der Nagel R, Dietz R, de Windt L and Bergmann MW:
NF-kappaB activation is required for adaptive cardiac hypertrophy.
Cardiovasc Res. 84:416–424. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Booth LC, Ramchandra R, Calzavacca P and
May CN: Role of prostaglandins in determining the increased cardiac
sympathetic nerve activity in ovine sepsis. Am J Physiol Regul
Integr Comp Physiol. 307:R75–R81. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chatterjee A, Mir SA, Dutta D, Mitra A,
Pathak K and Sarkar S: Analysis of p53 and NF-κB signaling in
modulating the cardiomyocyte fate during hypertrophy. J Cell
Physiol. 226:2543–2554. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hasking GJ, Esler MD, Jennings GL, Burton
D, Johns JA and Korner PI: Norepinephrine spillover to plasma in
patients with congestive heart failure: Evidence of increased
overall and cardiorenal sympathetic nervous activity. Circulation.
73:615–621. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mompeo B, Maranillo E, Garcia-Touchard A,
Larkin T and Sanudo J: The gross anatomy of the renal sympathetic
nerves revisited. Clin Anat. 7:660–664. 2016. View Article : Google Scholar
|
|
16
|
Schmieder RE, Redon J, Grassi G, Kjeldsen
SE, Mancia G, Narkiewicz K, Parati G, Ruilope L, van de Borne P and
Tsioufis C: ESH position paper: Renal denervation-an interventional
therapy of resistant hypertension. J Hypertens. 30:837–841. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
DiBona GF: Sympathetic nervous system and
the kidney in hypertension. Curr Opinion Nephrol Hypertens.
11:197–200. 2002. View Article : Google Scholar
|
|
18
|
Davies JE, Manisty CH, Petraco R, Barron
AJ, Unsworth B, Mayet J, Hamady M, Hughes AD, Sever PS, Sobotka PA
and Francis DP: First-in-man safety evaluation of renal denervation
for chronic systolic heart failure: Primary outcome from
REACH-Pilot study. Int J Cardiol. 162:189–192. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang X, Zhao Q, Huang H, Tang Y, Xiao J,
Dai Z, Yu S and Huang C: Effect of renal sympathetic denervation on
atrial substrate remodeling in ambulatory canines with prolonged
atrial pacing. PLoS One. 8:e646112013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Clayton SC, Haack KK and Zucker IH: Renal
denervation modulates angiotensin receptor expression in the renal
cortex of rabbits with chronic heart failure. Am J Physiol Renal
Physiol. 300:F31–F39. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu J, Ji M, Niu C, Aini A, Zhou Q, Zhang
L, Jiang T, Yan Y and Hou Y: Effects of renal sympathetic
denervation on post-myocardial infarction cardiac remodeling in
rats. PLoS One. 7:e459862012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li ZZ, Jiang H, Chen D, Liu Q, Geng J, Guo
JQ, Sun RH, Zhu GQ and Shan QJ: Renal sympathetic denervation
improves cardiac dysfunction in rats with chronic pressure
overload. Physiol Res. 64:653–662. 2015.PubMed/NCBI
|
|
24
|
Hu J, Yan Y, Zhou Q, Ji M, Niu C, Hou Y
and Ge J: Effects of renal denervation on the development of
post-myocardial infarction heart failure and cardiac autonomic
nervous system in rats. Int J Cardiol. 172:e414–e416. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grec V and Buksa M: Are we on the path to
solve the enigma of resistant hypertension: Renal sympathetic
denervation. Med Arch. 67:454–459. 2013. View Article : Google Scholar : PubMed/NCBI
|