Introduction
Chronic cigarette smoke is the most important risk
factor for the development of chronic obstructive pulmonary disease
(COPD) (1). Previous results have
indicated that smoking may induce pulmonary vascular remodeling
(PVR), which leads to increased pressure in the pulmonary artery
(2–5), suggesting that smoking might directly
result in PVR at the initial stage of COPD (5,6).
Increased proliferation of pulmonary arterial smooth muscle cells
(PASMCs) directly caused by cigarette smoke has been reported by
previous studies (2,7), and is considered to be one of the
pathological foundations of PVR and irreversible pulmonary
hypertension. However, the molecular mechanisms underlying this
process remain unclear.
It has previously been reported that cigarette smoke
can exert biological effects in an extracellular signal-regulated
kinase 1 and 2 (ERK1/2)-dependent manner (8). ERK1/2 are members of the large family
of mitogen-activated protein kinases. Activation of ERK1/2
(p-ERK1/2) has been implicated as a regulator in numerous
fundamental cellular activities (9,10).
Studies by the current authors and other researchers have addressed
the role of the ERK1/2 signaling pathway in the proliferation of
airway smooth muscle cells and multiple arterial smooth muscle
cells (11–14).
Cell cycle progression is regulated precisely at
various biological checkpoints by cyclins, cyclin-dependent kinases
(CDK) and CDK inhibitors (15).
Cyclin E1, the major E-type cyclin, is expressed during late G1
phase until the end of S-phase (16). The activity of cyclin E1 regulates
cells passing the restriction point ‘R’, which marks a ‘point of no
return’ in cell cycle transition from a resting state into the
division cycle or from G1 phase into S phase (16). Previous studies have reported that
the expression of cyclin E1 could be upregulated by activated
ERK1/2 (17–21). A previous study by the current
authors indicated that cyclin E1 might participate in ERK-related
PVR (22). However, further
investigation is required to determine whether the ERK1/2-cyclin E1
signaling pathway is involved in PVR induced by cigarette
smoke.
To investigate the role of the ERK1/2-cyclin E1
signaling pathway in PVR induced by cigarette smoke, the expression
of ERK1/2 and cyclin E1 in cultured rat PASMCs (rPASMCs) and rat
pulmonary arteries, as well as the structural changes of pulmonary
vessels, were evaluated in the presence of ERK1/2-small interfering
RNA (siRNA). The results indicated an attenuation of PVR by
ERK1/2-siRNA in cigarette smoke-exposed rats, suggesting that
ERK1/2-siRNA may have therapeutic value in the treatment of
COPD.
Materials and methods
Reagents
All siRNAs were purchased from Guangzhou RiboBio
Co., Ltd. (Guangzhou, China). The siRNA sequences were as follows:
ERK1, 5′-GACCGGAUGUUAACCUUUA-3′; ERK2, 5′-CCAGGAUACAGAUCUUAAA-3′;
and negative control, 5-UUC UCC GAA CGU GUC ACG U-3′. All siRNAs
(final concentration 30 nmol/l) were transfected into rPASMCs with
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). For in vivo studies, each rat was
anesthetized with diethyl ether and then administered intranasally
with ERK1/2-siRNA (15 nmol) or an equivalent dose of the negative
control siRNA twice a week. The anesthetization procedure was
performed as follows: The rat was placed into a glass jar with
1–1.5 ml diethyl ether. After 60–90 sec, the inhalation of the
diethyl ether led to the anesthetization of the rat.
Anesthetization was confirmed by swaying the glass jar.
The antibody against p-ERK (9101) was purchased from
Cell Signaling Technology Inc. (Danvers, MA, USA), the antibody
against cyclin E1 (ab7959) was purchased from Abcam (Cambridge, UK)
and the antibody against ERK1/2 (SC-153) was purchased from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA).
Total RNA was isolated using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. First-strand cDNA synthesis was
performed with PrimeScript RT™ reverse transcriptase
(Takara Biotechnology Co., Ltd., Dalian, China). The primers used
for real-time detection were as follows: ERK1 (243 bp), forward,
5′-AGAATGTCATAGGCATCCGAGA-3′ and reverse,
5′-CGCAGGTGGTGTTGATAAGC-3′; ERK2 (201 bp), forward:
5′-ACCTCAAGCCTTCCAACCTC-3′ and reverse,
5′-AGCCCACAGACCAAATATCAAT-3′; cyclin E1 (176 bp), forward,
5′-GGAAAATCAGACCGCCCAG-3′ and reverse, 5′-CATCAGCCAGTCCAGAAGAAC-3′;
β-actin (110 bp), forward, 5′-CGTTGACATCCGTAAAGACCTC-3′ and
reverse, 5′-TAGGAGCCAGGGCAGTAATCT-3′.
Animals and cigarette smoke
exposure
A total of 24 adult male Wistar rats (age, 8–12
weeks; weight, 200–250 g at the start of this study), obtained from
the Animal Application Center at Tongji Hospital (Wuhan, China),
were bred in a barrier system at room temperature (18–23°C) and
40–70% humidity. The rats were fed with food (5 g/100 g weight) and
water (10 ml/100 g weight) every day in a 12-h light/12-h dark
cycle. Rats were randomly divided into four groups: Control,
smoking, smoking + negative control (NC)-siRNA and smoking +
ERK1/2-siRNA. Rats were exposed to normal air or the smoke of 20
Marlboro cigarettes (Philip Morris USA; Altria Group, Inc.,
Richmond, VA, USA) per day for three months. The cigarette smoke
exposure was carried out with a PAB-S200 Animal Passive Smoking
Exposure System (BioLabs Technology Co., Ltd., Beijing, China).
The study was conducted according to the principles
of the Declaration of Helsinki and approved by the Institutional
Review Board of Tongji Hospital of Tongji Medical College, Huazhong
University of Science and Technology in accordance with its
guidelines for the protection of animal subjects. The Institutional
Review Board approved the animal experiments for this research.
Cell culture
Primary rPASMCs were prepared from explants of
endothelium and adventitia-stripped intra-pulmonary arteries of 6
additional Wistar rats (Male, 6–8 weeks, same source and housing
conditions as above) with an average body weight of 220 g (200–250
g). The rats were sacrificed by the intraperitoneal injection of
sodium pentobarbital (150 mg/kg), and the explants and primary
rPASMCs were obtained as previously described (23). Cells were cultured in Dulbecco's
modified Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.)
with 10% heat-inactivated fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) and identified by phase-contrast microscopy and
immunochemical staining. Cells from passage 3–8 were used for all
experiments.
Immunochemical staining
Cells were fixed with 4% paraformaldehyde for 20 min
at room temperature. Nonspecific binding sites were blocked in 10%
bovine serum in phosphate-buffered saline (PBS) at room temperature
for 1 h and cells were immunostained with anti-α-smooth muscle
actin (SMA) primary antibody (1:200 dilution; sc-32251; Santa Cruz
Biotechnology, Inc.) for 1 h at room temperature, followed by
reaction with biotinylated secondary antibody (biotin-conjugated
affinipure goat anti-mouse IgG; SA00004-1; Proteintech Group, Inc.,
Chicago, IL, USA) for 30 min (1:200 dilution) at room temperature.
Subsequently, cells were incubated with HRP-conjugated streptavidin
(cat. 434323, Thermo Fisher) for 30 min at room temperature (1:200
dilution) and the appropriate substrate solution
(3,3′-diaminobenzidine substrate solution; 34002; Pierce; Thermo
Fisher Scientific, Inc.) was added and treated for 5–15 min. Images
were captured under a fluorescence microscope (Nikon Corporation;
Tokyo, Japan).
Western blotting
Cells were lysed by lysing buffer (50 mM Tris-HCl,
pH 7.5, 300 mM NaCl, 1% Triton-X, 5 mM EDTA and 10% glycerol) and
the whole proteins were quantified using a commercial BCA assay kit
(23225; Pierce; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Cell lysates were loaded (30 µg per
lane) and electrophoresed in 12% SDS-PAGE and transferred onto a
polyvinylidene difluoride membrane. At room temperature, the
membrane was incubated in a 5% non-fat milk- tris-buffered saline
with Tween-20 solution for at least 30 min to block non-specific
binding. The membrane was incubated with the primary antibody
(1:1,000 dilution) for 2 h at room temperature. The following
primary antibodies were used to detect various target proteins: The
antibody against p-ERK (9101; Cell Signaling Technology, Inc.),
against cyclin E1 (ab7959; Abcam), against ERK1/2 (SC-153; Santa
Cruz Biotechnology, Inc.) and the antibody against β-actin
(60008–1-Ig; Proteintech Group, Inc.). Then the membrane was
incubated with the appropriate horseradish peroxidase-conjugated
secondary antibody (1:5,000 dilution; goat anti-rabbit IgG;
SA00001-2; Proteintech Group, Inc.) for 45 min at room temperature.
The membrane was subsequently incubated with enhanced
chemiluminescence substrate (32106; Pierce; Thermo Fisher
Scientific, Inc.) for 5 min, and the result was detected by a
luminescent image analyzer (LAS4000; Fujifilm; Tokyo, Japan).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Whole RNA was extracted from the cells using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, INc.) and reversed
transcribed into cDNA using reverse transcriptase (M-MLV; M170A;
Promega Corporation; Madison, WI, USA) and Oligo dT for 1 h at
42°C. qPCR was performed using the SYBR Green method and the
commercial kit (iQ™ SYBR®-Green Supermix; 1708880;
Bio-Rad Laboratories, Inc., Hercules, CA, USA) on ABI-7500 (Life
Technology). Samples were prepared according to the instructions of
the manufacturer of the iQ™ SYBR® Green Supermix and the
qPCR programmed was set as follows: Denaturation at 95°C for 3 min,
followed by 40 cycles of amplification (denaturation at 95°C for 15
sec, annealing and extension at 60°C for 30 sec; the plate was read
at the end of each extension phase), melt curve analysis was
conducted from 55 to 95°C with continuous plate reading. The result
was automatically calculated by the 2−ΔΔCq method
(24) on ABI-7500 following the
completion of the experiment.
Preparation of cigarette smoke extract
(CSE)
Smoke from two commercial Marlboro cigarettes was
bubbled through 50 ml phosphate-buffered saline (PBS). After the
solution was adjusted to pH 7.4 and filtered through a 0.22-µm
sterile filter, it was then defined as 100% CSE. We previously
determined that 2% CSE had the strongest effect on promoting the
proliferation of rPASMCs (25), so
this concentration of CSE was used for the entire in vitro
study. Cells were starved for 24 h in serum-free medium at 37°C
before they were treated with CSE or an equal amount of PBS. At 24
h, cells were harvested for subsequent experiments.
Cell proliferation assays
Cell proliferation was assessed by cell counting and
5-bromo-2-deoxyuridine (BrdU; Roche Diagnostics GmbH, Mannheim,
Germany) incorporation assay. The rPASMCs were seeded into 24-well
plates at a density of 3,000 cells/well. After the aforementioned
treatment with CSE or PBS, cells were counted using a hemocytometer
(wi81386; Dongxiyi Company, China) by trypan blue exclusion.
The BrdU incorporation assay was performed to detect
active DNA synthesis. Briefly, cells were plated on cover slips and
treated with CSE or PBS. They were then incubated with 50 mmol BrdU
for 4 h at 37°C and fixed with cold acetone. The cells were then
permeabilized and counted under a fluorescence microscope. The
results of the BrdU incorporation assay were presented as the
percentage of BrdU-positive cells. The detailed procedure has been
described previously (25).
Cell cycle analysis by flow
cytometry
After treatment with CSE or PBS, 1×106
cells/ml were immediately digested by EDTA-free trypsin and fixed
overnight at 4°C in 70% ethanol. Then, cells were suspended in PBS
containing propidium iodide (0.5 mg/ml; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) and DNase-free RNase (0.1 mg/ml). Flow
cytometry was performed using a FACSort system with CellQuest
software (version 5.1; BD Biosciences, Franklin Lakes, NJ, USA).
The percentages of cells in different phases of the cell cycle were
determined using ModFit software (version 3.2; Verity Software
House, Inc., Topsham, ME, USA).
Tissue preparation
Rats were provided with fresh air for 24 h after the
last exposure to cigarette smoke and then anesthetized with
pentobarbital (50 mg/kg, intraperitoneal). The rats were sacrificed
by exsanguination, then the lungs of all rats were collected. The
intrapulmonary arteries were isolated from left lungs for western
blot and reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) analysis. The right lungs were fixed in 4%
paraformaldehyde overnight, embedded in paraffin and cut into 5-µm
sections for morphological examination.
Morphometric analysis of pulmonary
vessels
Sections of the right lungs were stained with
hematoxylin and eosin to observe morphological changes in the small
pulmonary arteries. For each rat, five arteries with an external
diameter between 50 and 150 µm were randomly selected and evaluated
using a HPIAS-1000 medical image-analysis system (Wuhan Champion
Image Engineering Co., Wuhan, China). The detailed selection
procedure was as follows: Two technicians each selected five
arteries from five different visual fields with an optical
microscope. Ten arteries were selected in total, then five arteries
were randomly selected from this pool. The wall area and total area
of the arteries were measured. Wall thickness (WT)=(Wall area/total
area) ×100% (26). All morphometric
measurements were performed by two technicians operating in a blind
manner. The difference in measurements between the two observers
was <5%.
Assessment of PVR
Further evaluation of PVR was performed by
immunostaining with an anti-α-SMA antibody (sc-32251; 1:200
dilution; Santa Cruz Biotechnology, Inc.). All small intrapulmonary
arteries (≤200 µm in external diameter) adjacent to the alveolar
ducts were analyzed. The fully muscularized vessels (actin staining
>75% of the circumference) were counted and the result was
presented as the percentage of the total small intrapulmonary
arteries.
Immunostaining of ERK1/2 and cyclin
E1
As previously described (27), sections were deparaffinized,
rehydrated, soaked in an antigen retrieval solution with citrate
buffer, and incubated overnight at 4°C with a rabbit polyclonal
antibody against p-ERK (9101; 1:200 dilution; Cell Signaling
Technology) or cyclin E1 (ab7959; 1:200 dilution; Abcam).
Biotinylated secondary antibody (SA00004-1; 1:200 dilution;
Proteintech Group, Inc.) was then added and followed by incubation
with a streptavidin-peroxidase conjugate for 30 min at room
temperature. Images were captured under a fluorescence microscope
(Nikon Corporation).
Statistical analysis
Data are presented as the mean ± standard deviation.
Statistical software (PASW Statistics 18.0; IBM SPSS, Armonk NY,
USA) was used for analysis and the Student's t-test was performed.
P<0.05 (two-sided) was considered to indicate a statistically
significant result.
Results
ERK1/2-siRNA suppresses the expression
of ERK1/2 and cyclin E1 in CSE-treated rPASMCs
Rat PASMCs were treated with 2% CSE for 24 h, then
the mRNA expression of ERK1/2 and cyclin E1 were evaluated by
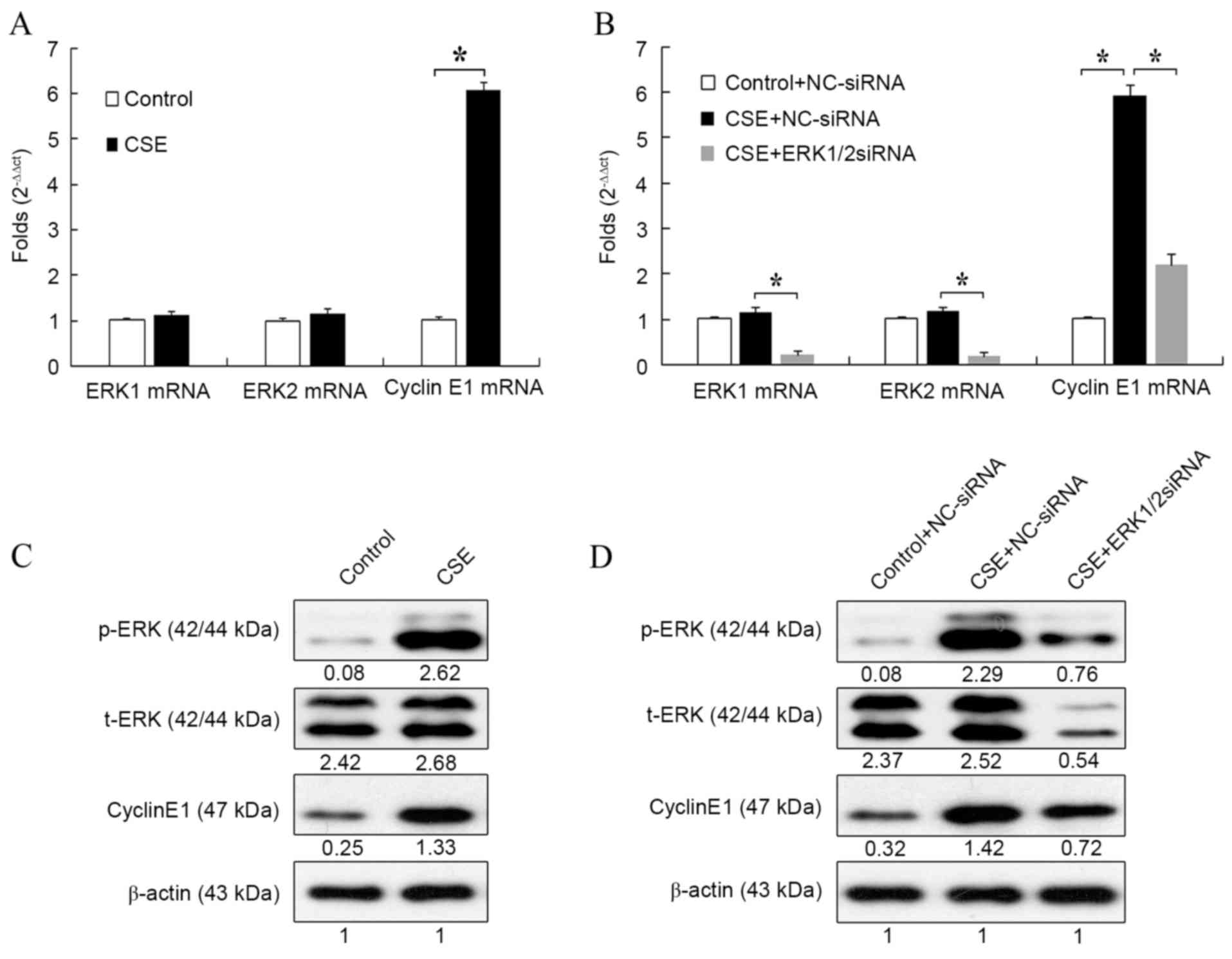
RT-qPCR. Compared with the control, CSE significantly increased the
mRNA expression of cyclin E1 (P<0.05; Fig. 1A). However, it had no influence on
the mRNA expression of ERK1/2.
Next, the effect of ERK1/2-siRNA on the expression
of ERK1/2 and cyclin E1 during CSE treatment was detected. As shown
in Fig. 1B, mRNA expression of
cyclin E1 was significantly increased after CSE treatment compared
with the control (P<0.05). However, this increase was
significantly attenuated by treatment with ERK1/2-siRNA
(P<0.05). This result suggested that ERK1/2-siRNA could block
the effect of CSE on the mRNA expression of cyclin E1. The mRNA
expression of ERK1/2 was also evaluated to verify the function of
ERK1/2-siRNA. For both ERK1 and ERK2, mRNA expression was
significantly reduced in CSE + ERK1/2-siRNA-treated cells compared
with CSE + NC-siRNA-treated cells (both P<0.05).
The effect of CSE on ERK1/2 and cyclin E1 expression
was also evaluated at the protein level. Treatment with CSE
increased the protein level of cyclin E1 (Fig. 1C), which was consistent with the
results at the mRNA level. Also, the expression of p-ERK, but not
t-ERK, was markedly upregulated by CSE treatment, which suggested
that CSE could stimulate the phosphorylation of ERK1/2 and activate
the ERK1/2 signaling pathway (Fig.
1C).
The effect of ERK1/2-siRNA during CSE treatment was
also detected at the protein level. Consistent with the results at
the mRNA level, cyclin E1 expression was increased by CSE +
NC-siRNA treatment and attenuated by CSE + ERK1/2-siRNA treatment
(Fig. 1D). ERK1/2-siRNA also
markedly reduced the protein levels of both t-ERK and p-ERK. The
results of Fig. 1 suggested that CSE
could increase the expression of cyclin E1 through activation of
the ERK1/2 signaling pathway.
Notably, although CSE-induced expression of cyclin
E1 was suppressed by ERK1/2-siRNA at both the mRNA and protein
level, its expression was still higher than that in the control
group (Fig. 1B and D). This was
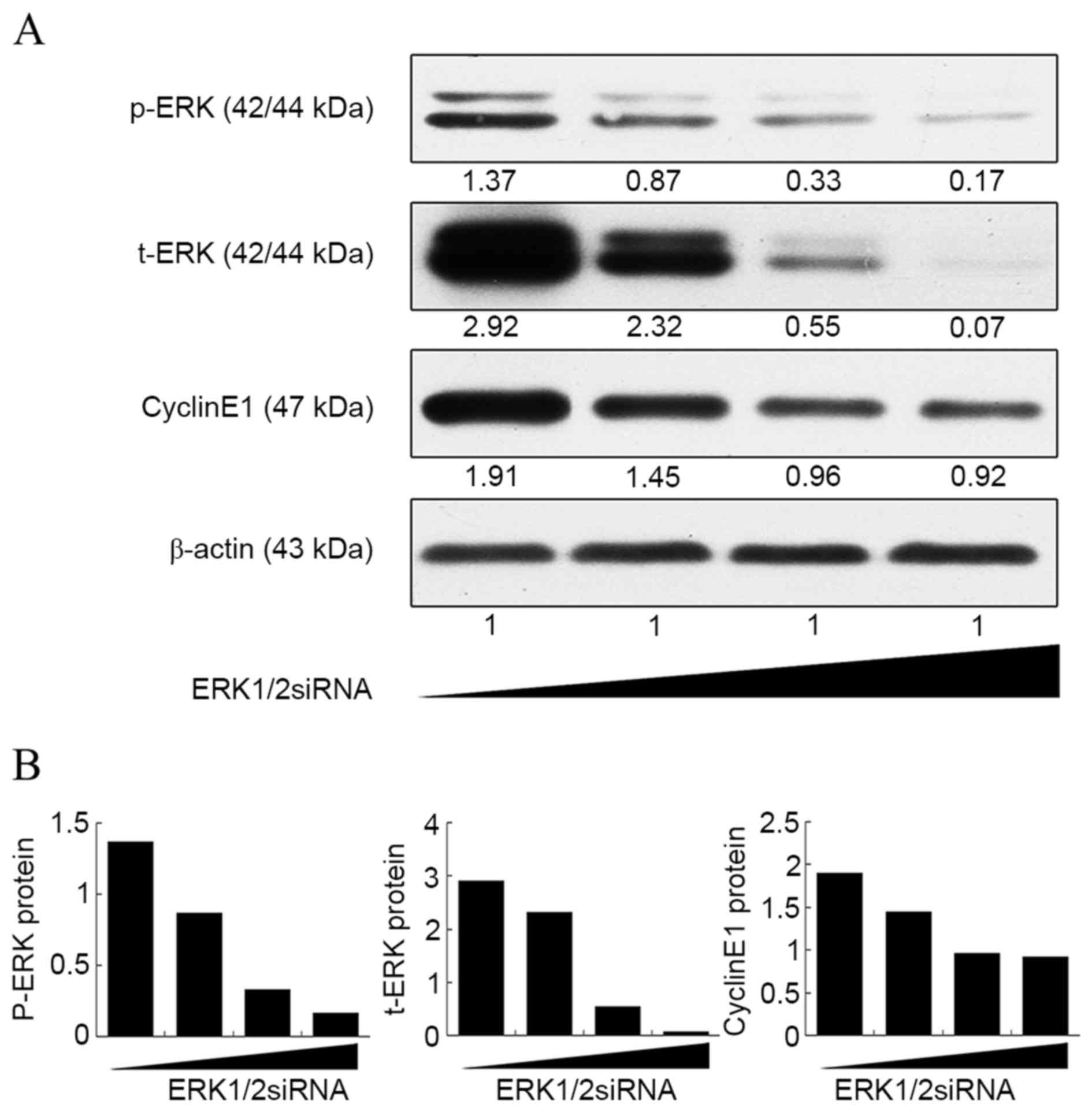
further confirmed by a dose-response curve experiment (Fig. 2). It was found that the expression of
cyclin E1 no longer decreased as the concentration of ERK1/2-siRNA
was increased above 20 nmol/l, whereas the ERK1/2 expression
continued to decrease steadily. This result suggested that the
ERK1/2 signaling pathway is not the only pathway involved in the
upregulation of cyclin E1 by CSE.
ERK1/2-siRNA inhibits progression from
G1 phase to S phase and suppresses rPASMC proliferation induced by
CSE
The distribution of cells in different cell cycle
stages was detected after CSE treatment. A significantly decreased
proportion of cells in G0/G1 phase and a significantly increased
proportion of cells in S phase were observed in the CSE-treated
group compared with the control (P<0.05; Fig. 3A). Furthermore, the effect of
ERK1/2-siRNA during CSE treatment was evaluated. The results
indicated that ERK1/2-siRNA could significantly attenuate the
effects of CSE on the cell cycle (P<0.05; Fig. 3B). Cell proliferation was also
evaluated by cell counting (Fig. 3C and
D) and BrdU incorporation (Fig. 3E
and F). It was found that CSE significantly promoted the
proliferation of rPASMCs compared with the control (P<0.05;
Fig. 3C and E), and this effect was
significantly reduced but not abolished by ERK1/2-siRNA (P<0.05;
Fig. 3D and F).
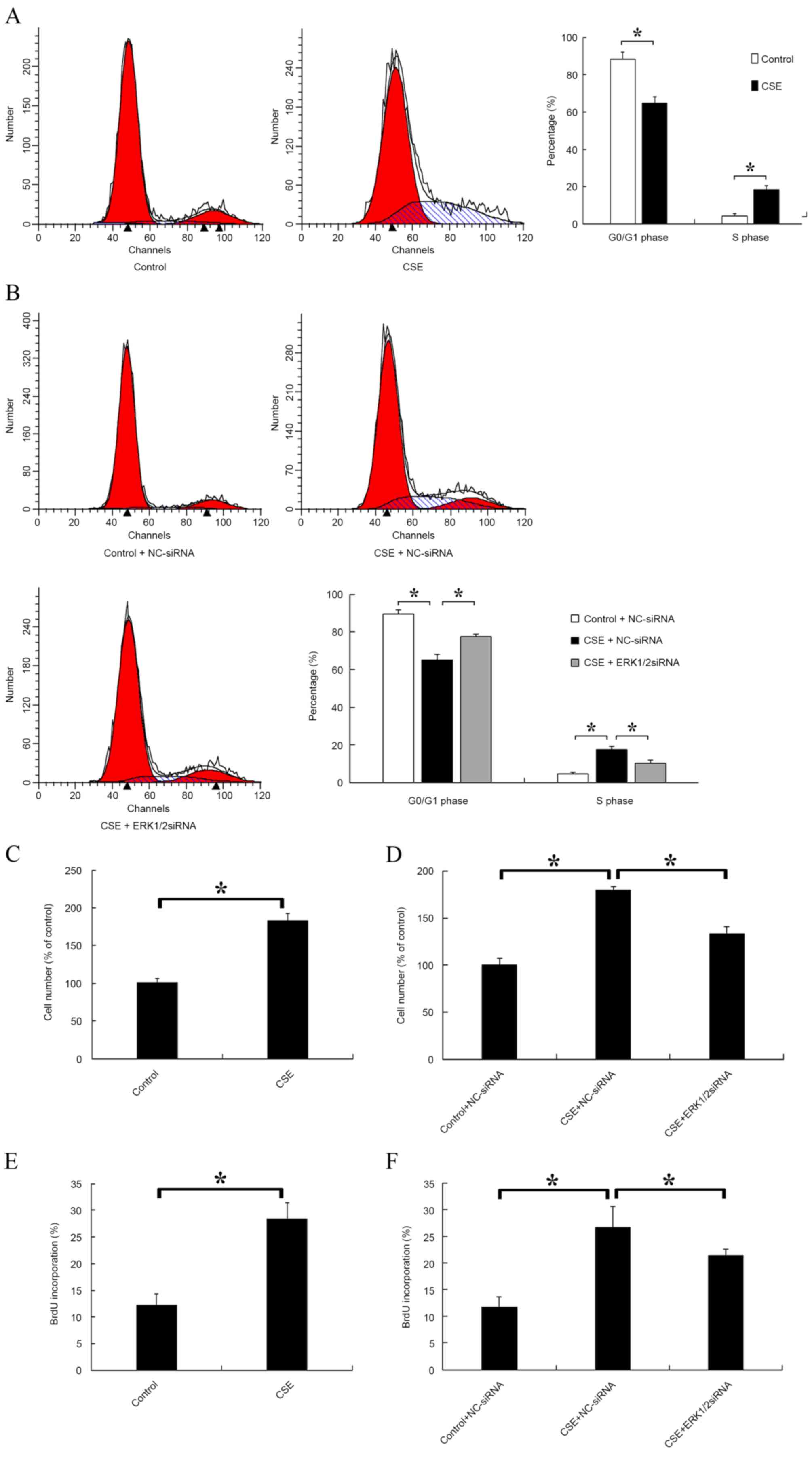 | Figure 3.Effect of CSE and ERK1/2-siRNA on rat
pulmonary artery smooth muscle cell cycle and cell proliferation.
(A) Cells were treated with CSE or the control medium for 24 h,
then the distribution of cell cycle stages was detected. The left
red, right red and blue areas represent G0/G1, G2/M and S phase,
respectively. (B) Cells were transfected with ERK1/2-siRNA or
NC-siRNA at a final concentration of 30 nmol/l for 24 h, then cells
were treated with CSE or control medium for a further 24 h. (C)
Cells were treated with CSE or the control medium for 24 h, then
the cell number of each group was counted. (D) Cells were treated
as in (B), then the cell number of each group was counted. (E)
Cells were treated with CSE or the control medium for 24 h, then
incubated with 50 mmol BrdU for 4 h at 37°C and fixed with cold
acetone. The BrdU-positive cells were counted. (F) Cells were
treated as in (B), then incubated with 50 mmol BrdU for 4 h at
37°C. *P<0.05 vs. control. ERK, extracellular signal-regulated
kinase; siRNA, small interfering RNA; CSE, cigarette smoke extract;
NC, negative control; BrdU, 5-bromo-2-deoxyuridine. |
ERK1/2-siRNA reduces the expression of
ERK1/2 and cyclin E1 in the pulmonary vessels of cigarette
smoke-exposed rats
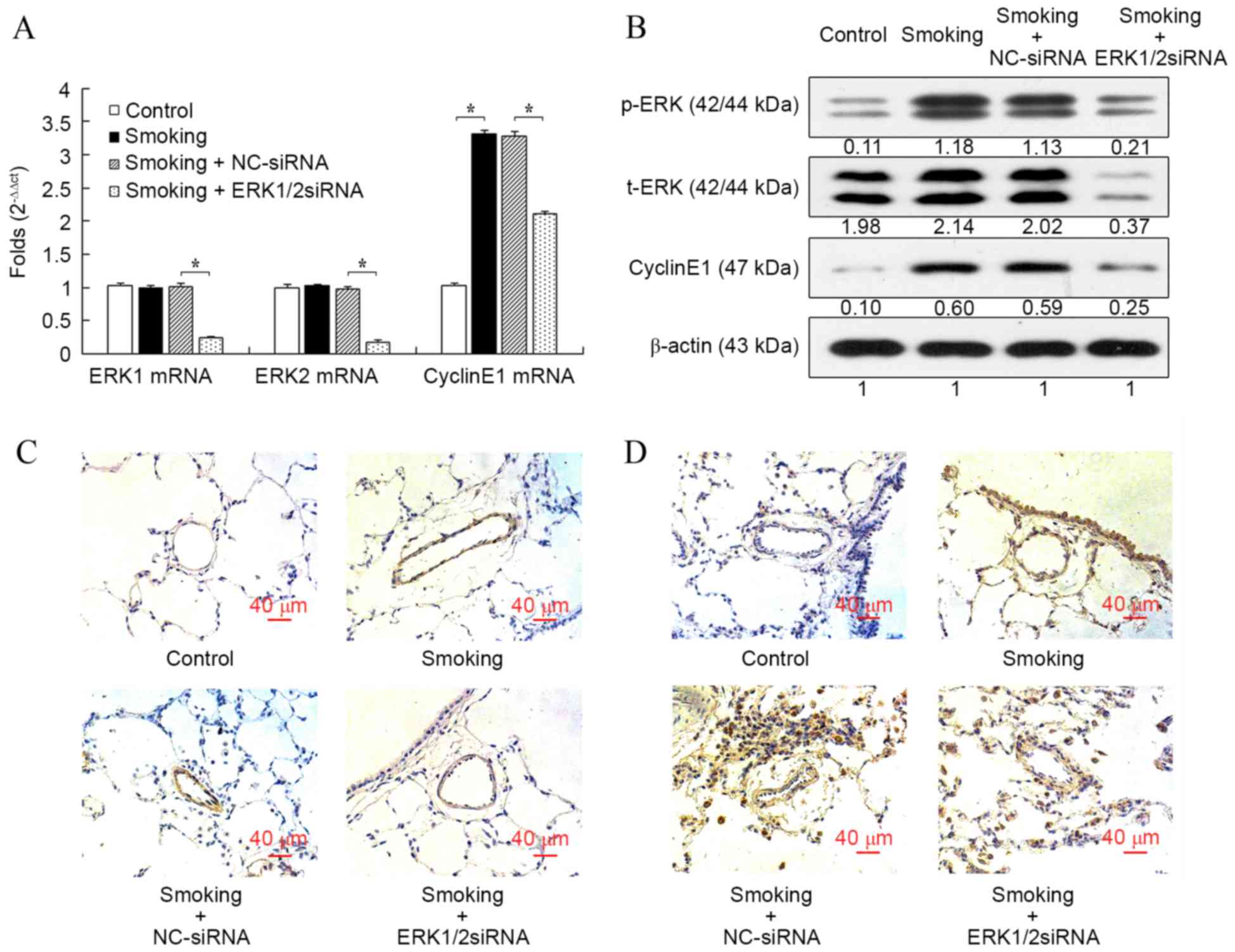
Consistent with the results in vitro,
cigarette smoke exposure significantly increased the mRNA
expression of cyclin E1 in rat pulmonary arteries compared with the
control group (P<0.05; Fig. 4A),
whereas the mRNA expression levels of ERK1 and ERK2 were not
influenced. Furthermore, the mRNA level of cyclin E1 induced by the
cigarette smoke was significantly reduced after the transfection of
ERK1/2-siRNA (P<0.05; Fig. 4A).
The mRNA levels of ERK1 and ERK2 were also significantly reduced
after transfection with ERK1/2-siRNA (both P<0.05), which
confirmed the function of ERK1/2-siRNA. When compared with the
control group, protein expression cyclin E1 and p-ERK, but not
t-ERK, were all markedly increased in the smoking group, and
ERK1/2-siRNA inhibited the increased expression induced by
cigarette smoke (Fig. 4B). However,
the protein expression of cyclin E1 in the ERK1/2-siRNA group was
still slightly increased compared with the control group. The
immunostaining of p-ERK (Fig. 4C)
and cyclin E1 (Fig. 4D) in rat lung
histologic sections were consistent with the results at mRNA and
protein levels.
ERK1/2-siRNA ameliorates rat PVR
induced by cigarette smoke
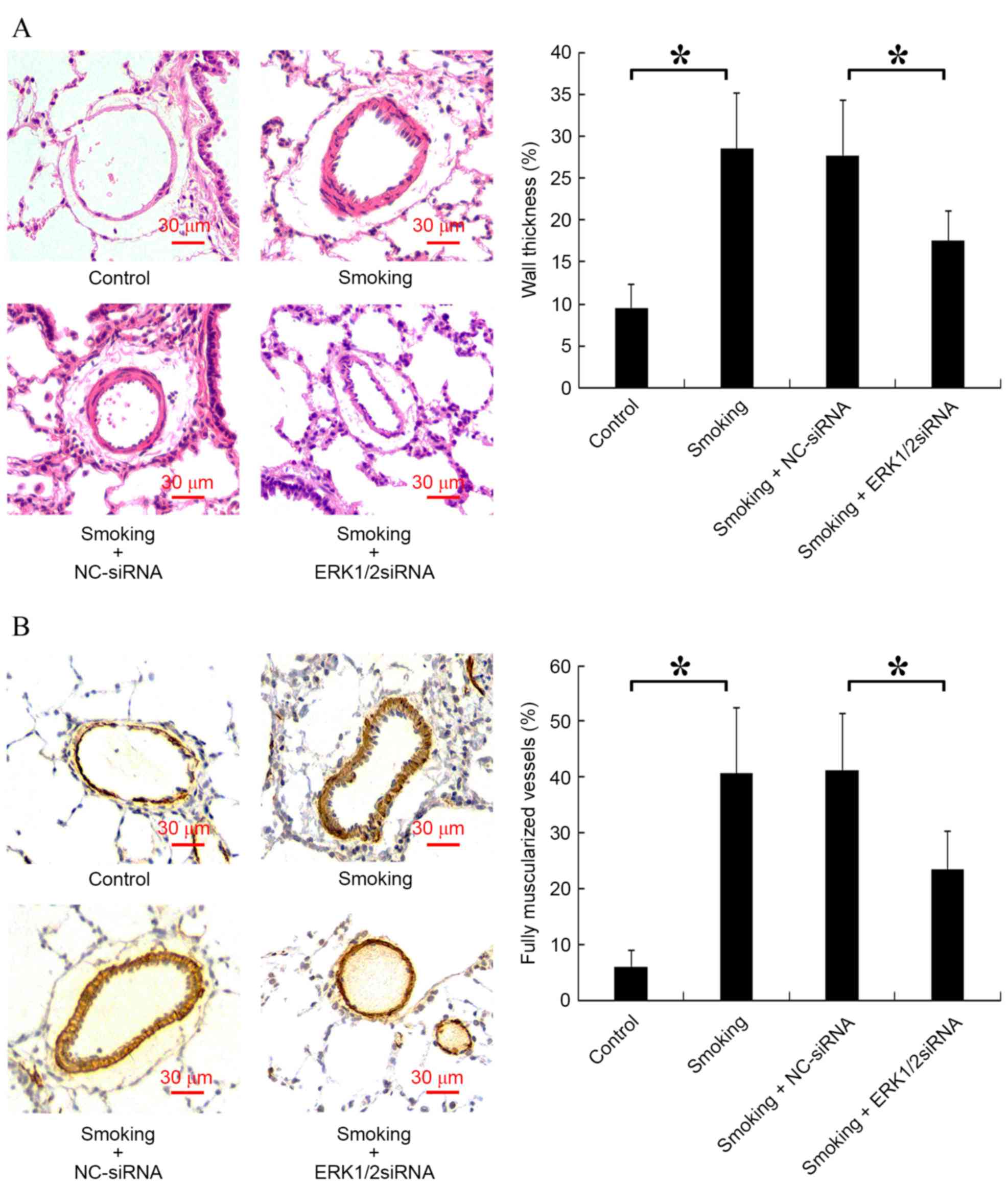
The vessel wall thickness and the number of fully
muscularized vessels are key indicators of PVR (28). Analysis of vessel wall thickness
revealed a significant increase in the smoking group compared with
the control group (P<0.05) and a significant decrease in the
smoking + ERK1/2-siRNA group compared with the smoking + NC-siRNA
group (P<0.05; Fig. 5A).
Furthermore, it was indicated that cigarette smoke significantly
increased the proportion of fully muscularized vessels in rats
compared with the control group, and smoking + ERK1/2-siRNA
significantly reduced this proportion compared with the smoking +
NC-siRNA group (P<0.05; Fig.
5B).
Discussion
The present study indicated that the knockdown of
ERK1/2 expression by siRNA suppressed rPASMC proliferation induced
by CSE and moderated PVR in rats exposed to cigarette smoke. It was
also observed that ERK1/2-siRNA reduced the G1/S transition induced
by CSE via the reduction of cyclin E1 expression. These results
suggested that the ERK1/2 signaling pathway is at least partially
implicated in the abnormal proliferation of rPASMCs (induced by
CSE) and PVR in rats (caused by cigarette smoke exposure), through
the regulation of cyclin E1 expression.
In the present study, ERK1/2-siRNA was used to
silence the gene expression of ERK1/2 in vitro and in
vivo. It was found that t-ERK1/2 was expressed stably in
rPASMCs treated with CSE or pulmonary vessels of cigarette
smoke-exposed rats at both the mRNA and protein level, compared
with control groups. However, p-ERK1/2 was significantly
upregulated in those groups, suggesting that ERK1/2 could be
activated by cigarette smoke. ERK1/2-siRNA reduced p-ERK1/2
expression in vitro and in vivo at both the mRNA and
protein level, by knocking down t-ERK1/2 expression but not the
ratio of p-ERK/t-ERK.
Activated ERK1/2 performs multiple cellular
functions, including cell proliferation, adhesion and migration
(9,10). Previous results have revealed that
cigarette smoke exerts its biological effects via the ERK1/2
signaling pathway (8). Moreover, a
critical role for ERK1/2 activation in the abnormal proliferation
of vascular smooth muscle cells and airway smooth muscle cells has
been suggested during vessel remodeling (11–14). The
present study extended this finding and demonstrated that the
expression of p-ERK1/2 was upregulated by CSE stimulation,
indicating that activated ERK1/2 is involved in the CSE-induced
proliferation of primary rPASMCs. When the effect of ERK1/2 on cell
cycle distribution was analyzed, consistent with a previous study
(29), it was found that activated
ERK1/2 promoted cell cycle progression from G0/G1 to S phase.
Conversely, ERK1/2-siRNA increased the proportion of cell cycle
arrest at the G0/G1 phase.
The molecular mechanism by which ERK1/2 stimulated
rPASMC proliferation was also investigated in the present study.
Previous studies indicated that nicotine, a critical component in
CSE, could upregulate the expression and activity of cyclin E1 in
A549 cells and MKN-45 cells (30,31). In
the current study, ERK1/2-siRNA decreased the mRNA and protein
expression of ERK1/2 and cyclin E1, and inhibited the cell
proliferation of CSE-treated rPASMCs. Combined with our previous
findings by the current authors that cyclin E1 siRNA significantly
decreased rPASMC proliferation (22), these results suggested that CSE could
induce rPASMC proliferation by regulating cyclin E1 expression,
which was mediated by ERK1/2. These results are also consistent
with previous studies in other cell types, which have indicated
that ERK1/2 activates cyclin E1 (17–21). In
short, ERK1/2 may contribute to rPASMC proliferation, as well as
cell cycle progression, and cyclin E1 may be the downstream
mediator of ERK1/2 in this process.
PVR is the major feature of pulmonary hypertension
in COPD patients (32). In the
present in vivo results, increased PVR was observed in
cigarette smoke-exposed rats. A previous study suggested that
ERK1/2 might be involved in this process (33). To elucidate the role of ERK1/2 in PVR
in cigarette smoke-exposed rats, ERK1/2-siRNA was delivered by
intranasal transfection. As discussed above, the expression of
activated ERK1/2 and cyclin E1 were upregulated in the small
intrapulmonary arteries of cigarette smoke-exposed rats, and this
was effectively attenuated by ERK1/2-siRNA. Based on these results,
it was speculated that the mechanism by which ERK1/2 knockdown
improves PVR may involve the inhibition of cigarette smoke-induced
cyclin E1 expression. Measurements of pulmonary vessel wall
thickness and the percentage of fully muscularized arteries
revealed that PVR was reduced in cigarette smoke-exposed rats
treated with ERK1/2-siRNA. The therapeutic efficacy of ERK1/2-siRNA
may be attributed to its inhibitory effect on the proliferation of
pulmonary vascular smooth muscle cells of cigarette smoke-exposed
rats, as observed in the cell culture study.
It is notable that ERK1/2-siRNA did not completely
inhibit the CSE-induced upregulation of cyclin E1 and rPASMC
proliferation in vitro, nor did it completely inhibit the
enhanced expression of cyclin E1 and PVR in cigarette smoke-exposed
rats. These results implied that additional intracellular signaling
molecules might be involved in this process, which may include
Rb-Raf-1 or protein kinase C (31,34).
Indeed, it was also observed that CSE-induced cyclin E1 did not
decrease continuously as the ERK1/2-siRNA increase. Therefore, the
mechanism by which ERK1/2 regulates cyclin E1 expression requires
further clarification.
In conclusion, the present study demonstrated the
role of the ERK1/2-cyclin E1 signaling pathway in rPASMC
proliferation and PVR of cigarette smoke-exposed rats. The results
suggested that cigarette smoke significantly increases rPASMC
proliferation and PVR by upregulating the expression of cyclin E1,
which is mediated by activated ERK1/2. The inhibition of ERK1/2
with siRNA significantly reduces the expression of ERK1/2 and
cyclin E1 both in vitro (CSE-treated rPASMCs) and in
vivo (cigarette smoke-exposed rats), and this inhibitory effect
contributes to the alleviation of PVR. The results of this study
suggest that ERK1/2-siRNA may have therapeutic value in the
treatment of COPD.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant nos. 81070044 and 81370156).
Glossary
Abbreviations
Abbreviations:
|
PVR
|
pulmonary vascular remodeling
|
|
COPD
|
chronic obstructive pulmonary
disease
|
|
rPASMCs
|
rat pulmonary artery smooth muscle
cells
|
|
CSE
|
cigarette smoke extract
|
|
ERK
|
extracellular signal-regulated
kinase
|
References
|
1
|
Churg A, Cosio M and Wright JL: Mechanisms
of cigarette smoke-induced COPD: Insights from animal models. Am J
Physiol Lung Cell Mol Physiol. 294:L612–L631. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Santos S, Peinado VI, Ramírez J, Melgosa
T, Roca J, Rodriguez-Roisin R and Barberà JA: Characterization of
pulmonary vascular remodelling in smokers and patients with mild
COPD. Eur Respir J. 19:632–638. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chaouat A, Naeije R and Weitzenblum E:
Pulmonary hypertension in COPD. Eur Respir J. 32:1371–1385. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wright JL, Levy RD and Churg A: Pulmonary
hypertension in chronic obstructive pulmonary disease: Current
theories of pathogenesis and their implications for treatment.
Thorax. 60:605–609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peinado VI, Pizarro S and Barbera JA:
Pulmonary vascular involvement in COPD. Chest. 134:808–814. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mandegar M, Fung YC, Huang W, Remillard
CV, Rubin LJ and Yuan JX: Cellular and molecular mechanisms of
pulmonary vascular remodeling: Role in the development of pulmonary
hypertension. Microvasc Res. 68:75–103. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ferrer E, Peinado VI, Díez M, Carrasco JL,
Musri MM, Martínez A, Rodríguez-Roisin R and Barberà JA: Effects of
cigarette smoke on endothelial function of pulmonary arteries in
the guinea pig. Respir Res. 10:762009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mercer BA, Kolesnikova N, Sonett J and
D'Armiento J: Extracellular regulated kinase/mitogen activated
protein kinase is up-regulated in pulmonary emphysema and mediates
matrix metalloproteinase-1 induction by cigarette smoke. J Biol
Chem. 279:17690–17696. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mebratu Y and Tesfaigzi Y: How ERK1/2
activation controls cell proliferation and cell death: Is
subcellular localization the answer? Cell Cycle. 8:1168–1175. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ramos JW: The regulation of extracellular
signal-regulated kinase (ERK) in mammalian cells. Int J Biochem
Cell Biol. 40:2707–2719. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xie M, Liu XS, Xu YJ, Zhang ZX, Bai J, Ni
W and Chen SX: ERK1/2 signaling pathway modulates the airway smooth
muscle cell phenotype in the rat model of chronic asthma.
Respiration. 74:680–690. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Preston IR, Hill NS, Warburton RR and
Fanburg BL: Role of 12-lipoxygenase in hypoxia-induced rat
pulmonary artery smooth muscle cell proliferation. Am J Physiol
Lung Cell Mol Physiol. 290:L367–L374. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu B, Ryer EJ, Kundi R, Kamiya K, Itoh H,
Faries PL, Sakakibara K and Kent KC: Protein kinase C-delta
regulates migration and proliferation of vascular smooth muscle
cells through the extracellular signal-regulated kinase 1/2. J Vasc
Surg. 45:160–168. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ding Q, Gros R, Limbird LE, Chorazyczewski
J and Feldman RD: Estradiol-mediated ERK phosphorylation and
apoptosis in vascular smooth muscle cells requires GPR 30. Am J
Physiol Cell Physiol. 297:C1178–C1187. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Morgan DO: Principles of CDK regulation.
Nature. 374:131–134. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Möröy T and Geisen C: Cyclin E. Int J
Biochem Cell Biol. 36:1424–1439. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bessard A, Frémin C, Ezan F, Fautrel A,
Gailhouste L and Baffet G: RNAi-mediated ERK2 knockdown inhibits
growth of tumor cells in vitro and in vivo. Oncogene. 27:5315–5325.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kisielewska J, Philipova R, Huang JY and
Whitaker M: MAP kinase dependent cyclinE/cdk2 activity promotes DNA
replication in early sea urchin embryos. Dev Biol. 334:383–394.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lents NH, Keenan SM, Bellone C and
Baldassare JJ: Stimulation of the Raf/MEK/ERK cascade is necessary
and sufficient for activation and Thr-160 phosphorylation of a
nuclear-targeted CDK2. J Biol Chem. 277:47469–47475. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Keenan SM, Bellone C and Baldassare JJ:
Cyclin-dependent kinase 2 nucleocytoplasmic translocation is
regulated by extracellular regulated kinase. J Biol Chem.
276:22404–22409. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee SH, Park C, Jin CY, Kim GY, Moon SK,
Hyun JW, Lee WH, Choi BT, Kwon TK, Yoo YH and Choi YH: Involvement
of extracellular signal-related kinase signaling in esculetin
induced G1 arrest of human leukemia U937 cells. Biomed
Pharmacother. 62:723–729. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu MQ, Liu XS, Wu HX, Xiang M and Xu YJ:
ERK1/2 promotes cigarette smoke-induced rat pulmonary artery smooth
muscle cells proliferation and pulmonary vascular remodeling via
up-regulating cycline1 expression. J Huazhong Univ Sci Technolog
Med Sci. 33:315–322. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Finder JD, Litz JL, Blaskovich MA, McGuire
TF, Qian Y, Hamilton AD, Davies P and Sebti SM: Inhibition of
protein geranylgeranylation causes a superinduction of nitric-oxide
synthase-2 by interleukin-1beta in vascular smooth muscle cells. J
Biol Chem. 272:13484–13488. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zeng DX, Xu YJ, Liu XS, Wang R and Xiang
M: Cigarette smoke extract induced rat pulmonary artery smooth
muscle cells proliferation via PKCα-mediated cyclin D1 expression.
J Cell Biochem. 112:2082–2088. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carraway MS, Ghio AJ, Suliman HB, Carter
JD, Whorton AR and Piantadosi CA: Carbon monoxide promotes hypoxic
pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol.
282:L693–L702. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou M, Chen HL, Cheng S, Mei L, Zhang HL,
Xie M, Xiong WN and Xu YJ: Effect of dexamethasone on expression of
AGR2 protein in asthmatic mice. J Huazhong Univ Sci Technolog Med
Sci. 33:33–36. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zelko IN, Zhu J, Ritzenthaler JD and Roman
J: Pulmonary hypertension and vascular remodeling in mice exposed
to crystalline silica. Respir Res. 17:1602016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brondello JM, McKenzie FR, Sun H, Tonks NK
and Pouysségur J: Constitutive MAP kinase phosphatase (MKP-1)
expression blocks G1 specific gene transcription and S-phase entry
in fibroblasts. Oncogene. 10:1895–1904. 1995.PubMed/NCBI
|
|
30
|
Shin VY, Jin HC, Ng EK, Sung JJ, Chu KM
and Cho CH: Activation of 5-lipoxygenase is required for nicotine
mediated epithelial-mesenchymal transition and tumor cell growth.
Cancer Lett. 292:237–245. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dasgupta P, Rastogi S, Pillai S,
Ordonez-Ercan D, Morris M, Haura E and Chellappan S: Nicotine
induces cell proliferation by beta-arrestin-mediated activation of
Src and Rb-Raf-1 pathways. J Clin Invest. 116:2208–2217. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sakao S, Voelkel NF and Tatsumi K: The
vascular bed in COPD: Pulmonary hypertension and pulmonary vascular
alterations. Eur Respir Rev. 23:350–355. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hu Y, Dietrich H, Metzler B, Wick G and Xu
Q: Hyperexpression and activation of extracellular signal-regulated
kinases (ERK1/2) in atherosclerotic lesions of cholesterol-fed
rabbits. Arterioscler Thromb Vasc Biol. 20:18–26. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Eder AM, Sui X, Rosen DG, Nolden LK, Cheng
KW, Lahad JP, Kango-Singh M, Lu KH, Warneke CL, Atkinson EN, et al:
Atypical PKCiota contributes to poor prognosis through loss of
apical-basal polarity and cyclin E overexpression in ovarian
cancer. Proc Natl Acad Sci USA. 102:pp. 12519–12524. 2005;
View Article : Google Scholar : PubMed/NCBI
|