Introduction
Despite advances in knowledge and technology
regarding health, myocardial ischemia remains the most common cause
of mortality worldwide and the number of people succumbing to
myocardial ischemia is predicted to increase over the coming
decades (1,2). In the majority of cases, myocardial
infarction is a consequence of coronary occlusion that results in
an insufficient blood supply to the myocardium, leading to
irreversible necrosis (3).
Restoration of blood flow of the ischemic region, known as
reperfusion, is the most effective therapeutic strategy of rescuing
myocardial cells and saving patients' lives. However, as it results
in the abrupt restoration of the oxygen supply, myocardial
reperfusion itself may aggravate myocardial injury, leading to
reperfusion injury (4). During
myocardial ischemia and reperfusion, cardiac cell death, calcium
overload and mitochondrial permeability transition pore (mPTP)
opening occur, resulting in the generation of reactive oxygen
species (RO) (5–7). Various protease enzymes are secreted
during ischemia/reperfusion (I/R) injury. These enzymes are
generated from neighboring cells that are damaged following I/R
injury, leading to increased cell death and the injury of normal
cardiomyocytes surrounding the ischemic area (5–7). In
addition, leukocytes and neutrophils infiltrate the ischemic area
during reperfusion and secrete serine various protease enzymes,
including cathepsin G, elastase and trypsin (8). Necrotic cells also secrete
intracellular protease enzymes, which contribute to heart tissue
damage and ultimately impair cardiac function (5–7).
Therefore, inhibiting protease activity may be a method of
preventing cardiac tissue injury following myocardial ischemia.
Secretory leukocyte protease inhibitor (SLPI) is an
11.7 kDa long cationic non-glycosylated protein that belongs to the
Whey Acidic Protein (WAP) family (9–11). SLPI
inhibits numerous leukocyte serine proteases, including trypsin and
chymotrypsin in pancreatic acinar cells, elastase and cathepsin G
in neutrophils, and chymase in mast cells (10–12).
SLPI is a frontline protein that directly and indirectly defends
against infection by viruses, bacteria and fungi (10). SLPI also acts as anti-inflammatory
mediator, protecting host tissue from the excessive tissue damage
induced by proteolytic enzymes secreted during inflammation
(10). Therefore, SLPI may be a
novel therapeutic strategy to treat myocardial ischemia.
Schneeberger et al (13) reported on the effects of rhSLPI in
cardiac transplantation and demonstrated that supplementation of
recombinant human (rh)SLPI in the cold-preservative solution used
to preserve hearts prior to transplant improves the cardiac score
of these hearts following transplantation. rhSLPI serves a crucial
role in early myocardial performance and post-ischemic inflammation
following cardiac transplantation (13). However, the immediate effects of
rhSLPI on myocardial I/R injury at the cellular and molecular
levels have not yet been investigated. Therefore, rhSLPI may be a
developed as a cardioprotective agent to treat myocardial I/R.
Therefore, the aim of the current study was to
investigate the effect of rhSLPI on isolated adult rat ventricular
myocytes (ARVMs) subjected to simulated I/R and to investigate the
cardioprotective effects of rhSLPI on an ex vivo
ischemia/reperfusion isolated murine heart model.
Materials and methods
Reagents
All basic chemicals were purchased from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). M199 medium was
obtained from Gibco; Thermo Fisher Scientific, Inc. (Waltham, MA,
USA). For SDS-PAGE and western blot analysis, 30% polyacrylamide
solution was purchased from Bio-Rad Laboratories, Inc. (Hercules,
CA, USA) and the polyvinylidenedifluoride (PVDF) membrane was
purchased from GE Healthcare Life Sciences (Little Chalfont, UK).
The antibodies for the dual phosphorylated (p)-Thr180/Tyr182 form
of p38 mitogen-activated protein kinase (MAPK; cat. no.
sc-17852-R), total p38 MAPK (cat. no. sc-728), p-Akt (cat. no.
sc-293125) and total Akt (cat. no. sc-8312) were purchased from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Enhanced
chemiluminescence (ECL) solution was purchased from GE Healthcare
Life Sciences and collagenase type II was purchased from
Worthington Biochemical Corporation (Lakewood, NJ, USA). rhSLPI was
purchased from Sino Biological, Inc. (Beijing, China).
Experimental animals
Adult male Wistar rats (8 weeks old) weighing
200–250 g (n=6) and adult male C57BL/6 mice (8 weeks old) weighing
25–30 g (n=18) were obtained from the National Animal Center,
Salaya Campus, Mahidol University (Bangkok, Thailand). All animals
were maintained under environmentally controlled conditions (a
temperature of 22±1°C, humidity of 45–60% and 12-h light/dark
cycle) at the Center for Animal Research, Naresuan University
(Phitsanulok, Thailand). All protocols used in the current study
were approved by the animal ethics committee of the Center for
Animal Research, Naresuan University (approval no.
NU-AE550732).
Isolation of ARVMs and culture
The Wistar rats were euthanized by intraperitoneal
injection (IP) with pentobarbital sodium (Nembutal®
Sodium Solution CII; 100 mg/kg; Akorn, Inc., Lake Forest, IL, USA)
and lithium heparin (150 U; Government Pharmaceutical Organization,
Bangkok, Thailand). Adult rat ventricular myocytes (ARVMs) were
isolated from the hearts using collagenase-based enzymatic
digestion. Hearts were excised and initially perfused for 10 min
with a modified Krebs solution (solution A) containing 130 mM NaCl,
4.5 mM KCl, 1.4 mM MgCl2, 0.4 mM
NaH2PO4, 0.75 mM CaCl2, 4.2 mM
HEPES, 20 mM taurine, 10 mM creatine and 10 mM glucose at pH 7.3 at
37°C. Hearts were then perfused with a calcium-free solution
containing 100 µM EGTA for 5 min (solution B), followed by
perfusion with solution A containing 100 µM CaCl2 and
0.4 mg/ml Type II collagenase for 30 min at 37°C. Following
enzymatic perfusion, ventricles were cut into small pieces, which
were incubated in 20 ml collagenase solution. Ventricles were
incubated with 100% O2 for a further 7 min at 37°C and
underwent regular gentle triturating. Isolated cardiomyocytes were
separated from undigested ventricular tissue using a cell strainer.
Subsequently, isolated myocytes were allowed to pellet at the
bottom of the tube (sedimentation by gravity) and the supernatant
was removed and replaced with solution A containing 1% bovine serum
albumin (BSA; Sigma-Aldrich; Merck KGaA) and 500 µM
CaCl2. Isolated cardiomyocytes were then allowed again
to sediment at the bottom of the tube (sedimentation by gravity)
and the supernatant was then removed and replaced with 10 ml
solution A containing 1 mM CaCl2. The cell pellet was
washed with M199 culture medium containing 100 IU/ml
penicillin/streptomycin. Myocytes were resuspended in M199
supplemented with 2 mM creatine, 2 mM carnitine and 5 mM taurine,
and then seeded on pre-laminin-coated 6-well plates (15 µg/ml
laminin). Myocytes were allowed to adhere for 2 h in an incubator
containing 5% CO2 at 37°C. The culture medium was
replenished with fresh modified M199 medium, prior to further
experiments.
Simulated ischemia (sI) protocol
sI was performed following a modified protocol
(14). ARVMs were incubated with a
specific basic buffer (137 mM NaCl, 3.8 mM KCl, 0.49 mM
MgCl2, 0.9 mM CaCl2 and 4.0 mM HEPES), 20 mM
2-deoxyglucose, 20 mM sodium lactate and 1 mM sodium dithionite at
pH 6.5, in order to induce sI. The control buffer was composed of
the basic buffer (137 mM NaCl, 3.8 mM KCl, 0.49 mM
MgCl2, 0.9 mM CaCl2 and 4.0 mM HEPES),
supplemented with 20 mM D-glucose and 1 mM sodium pyruvate. ARVMs
subjected to 20 min sI and 2 h reperfusion (sI/R) were treated with
0, 1, 10, 100, 1,000 and 10,000 ng/ml rhSLPI. The treatment was
applied either 2 h prior to sI, during sI or at the onset of
reperfusion. Following reperfusion, the culture medium was
collected to assess lactate dehydrogenase (LDH) activity and cell
viability was determined using the trypan blue dye exclusion assay.
Another set of experiments was performed to determine the most
effective concentration of rhSLPI. Cells were treated with 0, 200,
400, 600, 800 and 1,000 ng/ml rhSLPI prior to or during sI.
Subsequently, cell viability was determined using the trypan blue
dye exclusion assay and cell injury was assessed by determining
released-LDH activity.
Determination of cell viability
The viability of ARVMs was assessed using the trypan
blue dye exclusion method. Following reperfusion, the culture
medium was removed and cells were incubated in 0.4% trypan blue
solution for 1–2 min at room temperature. Following incubation, the
numbers of trypan blue-positive and negative cells were counted
under a light microscope in 5 different microscopic fields. The
percentage of surviving cells from the total amount of cells was
then calculated and the relative percentage of cell viability was
compared with the control group.
Measurement of cellular injury
Released-Lactate Dehydrogenase (LDH) enzyme activity
was measured by the LDH SCE mod. liquiUV kit (cat. no. 12214; HUMAN
Diagnostics Worldwide, Wiesbaden, Germany). A total of 10 µl
culture medium was mixed with 1,000 µl LDH activity assay buffer
and incubated at 37°C for 5 min. Then, 250 µl substrate reagent was
added. The solution was mixed and after 1 min, absorbance was
measured at a wavelength of 340 nm. The mean absorbance change per
minute (ΔA/min) was measured to evaluate released-LDH activity
using the following formula: LDH activity (U/I)=ΔA/min ×
20,000.
Measurement of intracellular ROS level
production
ARVMs were cultured with modified M199 medium in
24-well black plates and maintained at 37°C in an atmosphere
containing 5% CO2 and 95% O2. The culture
medium was removed and the cells were washed with
phosphate-buffered saline (PBS). Subsequently, cells were incubated
with M199 medium containing 25 µM carboxy-H2DCFDA (Sigma-Aldrich;
Merck KGaA) in a dark room for 30 min at 37°C. The medium
containing carboxy-H2DCFDA was discarded and ARVMs were washed once
with PBS. For rhSLPI treatment, 500 µl M199 medium containing
various concentrations of rhSLPI (0, 200, 400, 600, 800 and 1,000
ng/ml) was added and incubated for 1 h at 37°C. Subsequently, cells
were incubated with 250 µM H2O2 for 30 min at
37°C. The control cells were incubated with M199 medium without
treatment. ROS activity was determined by measuring fluorescence
intensity using an EnSpire Multimode Plate Reader (PerkinElmer,
Inc., Waltham, MA, USA) at an excitation wavelength of 498 nm and
emission wavelength of 522 nm.
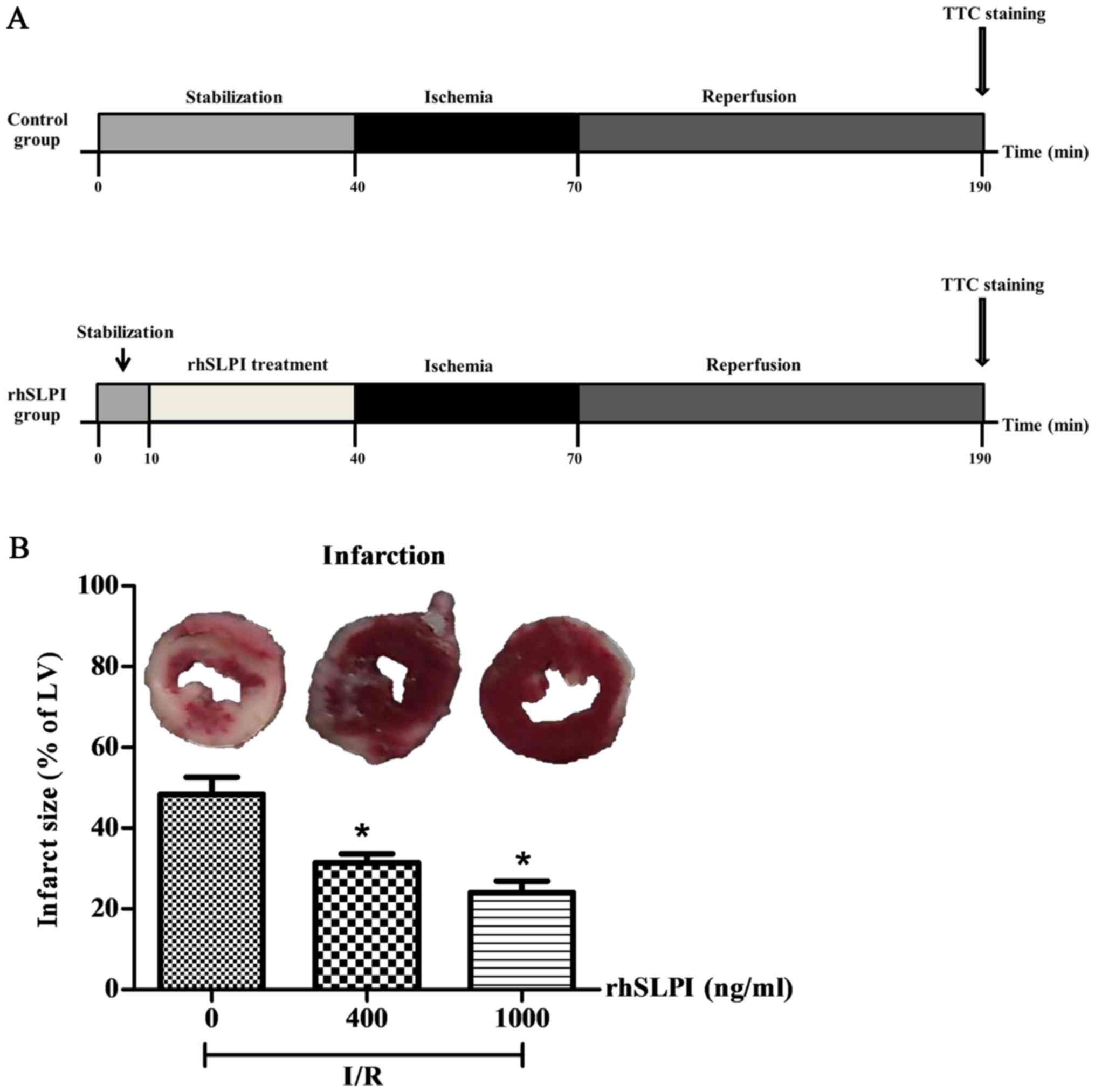
Ex vivo perfusion of isolated mouse
hearts and infarct size measurement
The ex vivo ischemia/reperfusion protocol was
based on a previous study (15). A
total of 18 adult male C57BL/6 mice (25–30 g; n=6 in each group)
were divided into 3 different groups: An I/R group, an I/R + 400
ng/ml rhSLPI group and an I/R + 1,000 ng/ml rhSLPI group. Mice were
euthanized with IP injection of pentobarbital (100 mg/kg) and
heparin (150 U). Hearts were rapidly isolated and placed in
ice-cold modified Krebs-Henseleit buffer (KHB) consisting of 118.5
mM NaCl, 25.0 mM NaHCO3, 4.75 mM KCl, 1.18 mmol
KH2PO4, 1.19 mM MgSO4, 11.0 mM
D-glucose and 1.4 mM CaCl2. The excised heart was
cannulated via the aorta and retrograde perfusion on a Langendorff
system was performed at a constant pressure of 80 mmHg with KHB
equilibrated with 95% O2 and 5% CO2 at 37°C.
Hearts were perfused with KHB for 10 min and were then perfused
with KHB solution containing 400 ng/ml or 1,000 ng/ml rhSLPI for 30
min. Hearts from mice in the I/R group underwent perfusion with KHB
alone. Hearts were then subjected to ischemia by attenuating
perfusion for 30 min (no-flow). Reperfusion was performed with KHB
for a further 2 h. At the end of the protocol, hearts were perfused
with 5 ml 1% triphenyltetrazolium chloride (TTC) in PBS for 1 min
at 37°C and incubated in 1% TTC at 37°C for 10 min. Atria were
removed and hearts were blotted dry, weighed and stored at −20°C
for 1 week. Prior to heart tissue sectioning, the hearts were
thawed, placed in 2.5% glutaraldehyde for 1 min at room temperature
and embedded in 5% agarose. Agarose heart blocks were sectioned
from apex to base in 0.75-mm slices using a vibratome (Agar
Scientific Ltd., Essex, UK). Following sectioning, slices were
fixed in 10% formaldehyde overnight at room temperature prior to
incubation with PBS for 1 day at 4°C. Sections were then compressed
between glass plates (0.75-mm apart) and scanned by a Gel
Doc™ XR+ System (Bio-Rad Laboratories, Inc.). All
analyses of infarct size were performed by two investigators that
were blinded with regards to the group assignments.
Measurement of p38 MAPK and Akt
activation by western blotting
ARVMs were washed twice in ice-cold PBS prior to the
addition of 200 µl 2× SDS-sample buffer containing
β-mercaptoethanol. Cells were scraped and samples were transferred
to pre-cooled micro-centrifuge tubes. For protein extraction,
hearts from the I/R, I/R + 400 ng/ml rhSLPI group and I/R + 1,000
ng/ml rhSLPI groups (each, n=3) were perfused on a Langendorff
perfusion system, subjected to 30 min stabilization, perfused with
KHB containing 400 ng/ml and 1,000 ng/ml rhSLPI or KHB alone as a
vehicle for 30 min prior to 10 min without perfusion (global
ischemia). At the end of the global ischemia, hearts were rapidly
snap frozen. Approximately 50 mg of heart samples were weighed and
homogenized in 500 µl homogenization buffer (20 mM Tris pH 6.8, 1
mM sodium orthovanadate, 5 mM sodium fluoride, and 1
cOmplete™, Mini Protease Inhibitor Cocktail Tablet;
Sigma-Aldrich; Merck KGaA). Heart homogenates were centrifuged at
13,148 × g for 10 min at 4°C. Supernatants were collected and an
equal volume of 2X SDS-PAGE sample buffer containing 10% (v/v)
β-mercaptoethanol and bromophenol blue dye was added. Samples were
boiled for 10 min and stored at −80°C prior to analysis. Extracted
proteins (30 µg/lane) were separated on 12% SDS-polyacrylamide gels
(prepared from a 30% polyacrylamide solution) and transferred to
PVDF membranes that were blocked for 1 h with 5% non-fat milk and
1% BSA in Tris-buffered saline (pH 7.4) containing 0.1% Triton
X-100. Membranes were then probed overnight at 4°C with primary
antibodies against total p38, diphospho-p38, total Akt and p-Akt
(all 1:1,000). Following washing and exposure for 1 h at room
temperature to horseradish peroxidase-conjugated secondary
antibodies, antibody-antigen complexes were visualized using ECL,
revealed as bands corresponding to the detected proteins of
interest. Band densities were detected by Gel DocXR+ Imaging
System, quantified using Image Lab™ software (version
6.0; Bio-Rad Laboratories, Inc., Hercules, CA, USA) and compared,
providing information on the relative abundance of the protein of
interest.
Statistical analysis
All values are presented as the mean ± standard
error of the mean. All comparisons were assessed for significance
using one-way analysis of variance, followed by the Tukey-Kramer
test. Statistical tests were performed using GraphPad Prism version
5 (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
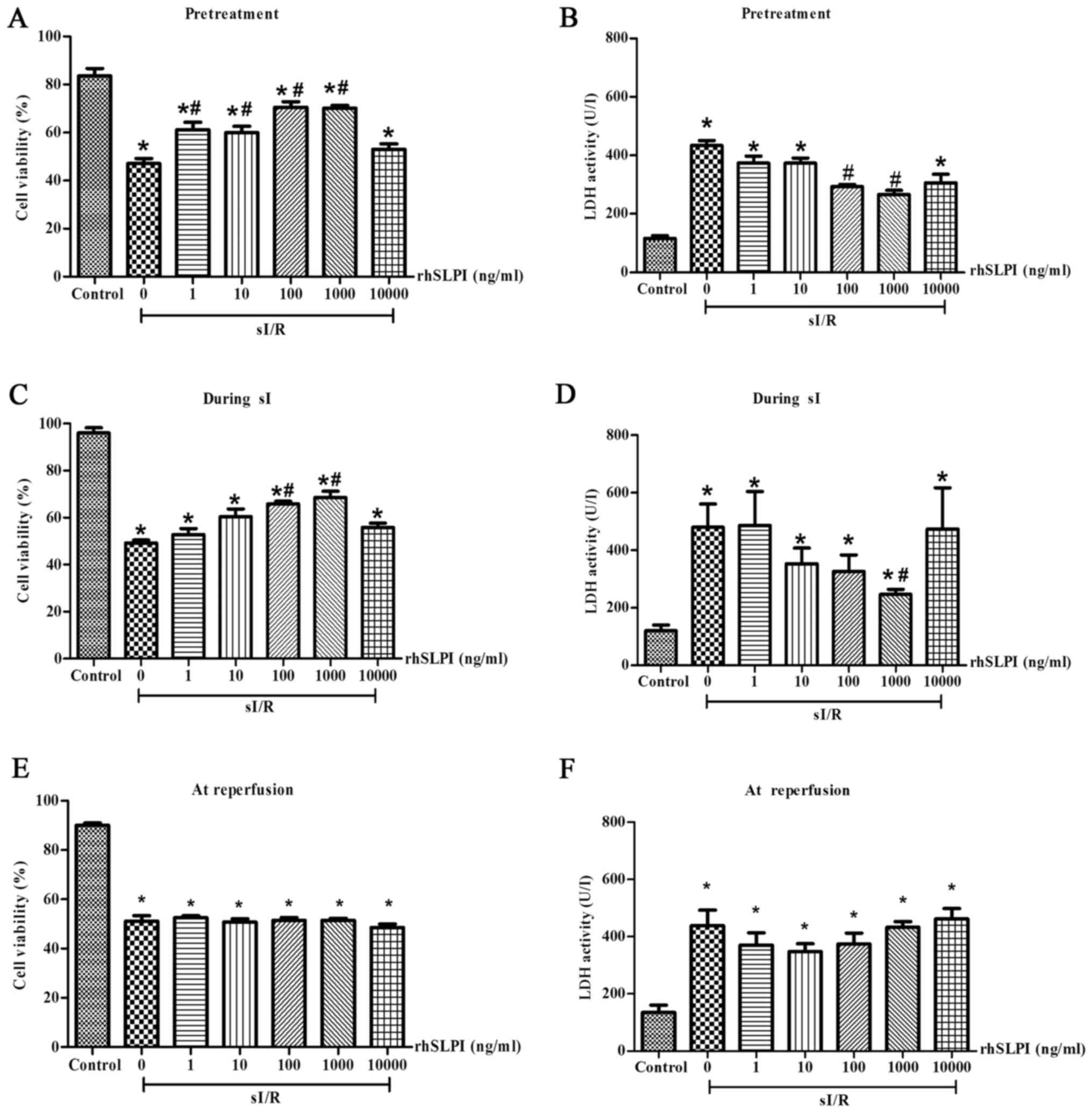
rhSLPI-treatment prior to or during
sI/R attenuates cell death following sI/R injury
The results demonstrated that rhSLPI-treatment
administered 2 h prior to sI or during sI significantly reduced
sI/R-induced cell death. Pretreatment of ARVMs with 1, 10, 100 and
1,000 ng/ml rhSLPI significantly increased cell viability compared
with the untreated sI/R group (P<0.05; Fig. 1A). Furthermore, pre-treatment with
100 and 1,000 ng/ml rhSLPI significantly decreased LDH activity
compared with the untreated sI/R group (P<0.05; Fig. 1B). In addition, treatment of ARVMs
with rhSLPI during sI also significantly decreased the cell death
induced by sI/R. Treatment with 100 ng/ml and 1,000 ng/ml rhSLPI
significantly increased cell viability compared with the untreated
sI/R group (P<0.05; Fig. 1C).
Furthermore, 1,000 ng/ml rhSLPI significantly decreased LDH
activity in the supernatant (P<0.05; Fig. 1D).
By contrast, rhSLPI-treatment at the onset of
reperfusion (Fig. 1E) did not
increase the viability of cardiac cells following sI/R. Similarly,
LDH activity was not significantly reduced when cells were treated
with rhSLPI at the onset of reperfusion (Fig. 1F). This was the case with all doses
of rhSLPI. The results of these indicated that the rhSLPI minimum
concentration that provided maximal cardioprotection in ARVMs was
100 ng/ml in case of pretreatment and 1,000 ng/ml for treatment
during sI.
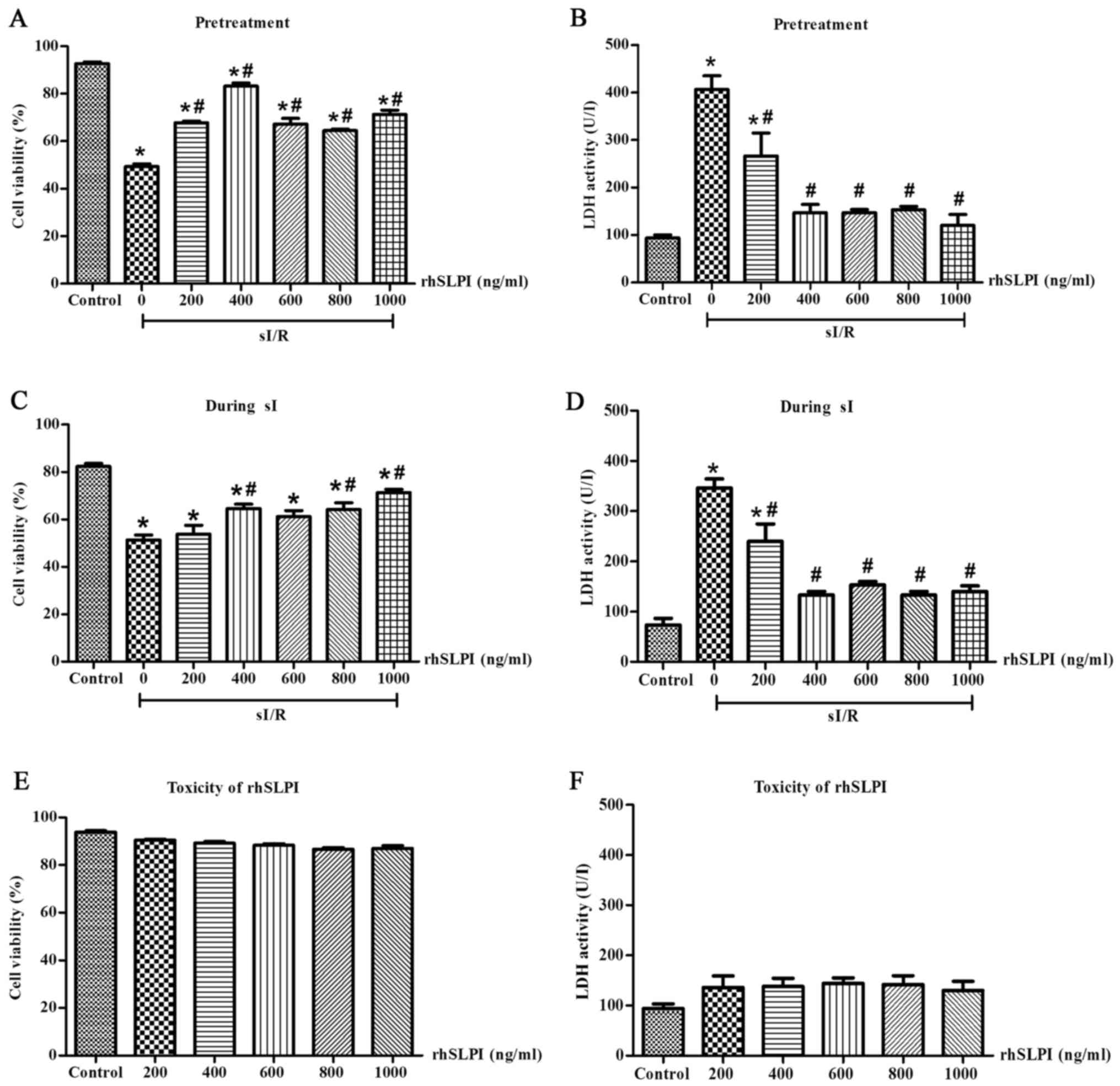
Identification of the maximum rhSLPI
concentration required to provide cardioprotection in ARVMs
subjected to sI/R
To identify the maximum rhSLPI concentration
required to induce cardioprotection in ARVMs without inducing any
toxicity, ARVMs were treated with 0, 200, 400, 600, 800 and 1,000
ng/ml rhSLPI 2 h prior to sI or immediately following the onset of
sI. Following treatment, all groups were subjected to sI for 20 min
followed by 2 h reperfusion. Cell viability and cell injury were
determined.
The results indicated that following 2 h
reperfusion, the percentage of viable cells in the groups treated
with rhSLPI either 2 h prior to sI or at the onset of sI increased
compared with the sI group. Pretreatment of ARVMs with 200, 400,
600, 800 and 1,000 ng/ml rhSLPI significantly increased the
viability of ARVMs exposed to sI/R injury compared with the sI
group (P<0.05; Fig. 2A).
Furthermore, these concentrations significantly decreased LDH
activity in the supernatants compared with the sI group (P<0.05;
Fig. 2B). In addition, treatment of
ARVMs with 400, 800 and 1,000 ng/ml at the onset of sI
significantly increased the viability of ARVMs compared with the sI
group (P<0.05; Fig. 2C) and these
concentrations significantly decreased LDH activity compared with
the sI group (P<0.05; Fig.
2D).
The toxicity of rhSLPI was determined by culturing
ARVMs with 0, 200, 400, 600, 800 and 1,000 ng/ml rhSLPI for 24 h
and then measuring cell viability and cell injury. The results
indicated that treatment with all concentrations of rhSLPI did not
reduce cell viability (Fig. 2E) and
did not increase LDH activity (Fig.
2F). This demonstrates that rhSLPI does not induce toxicity in
ARVMs.
Treatment of ARVMs by rhSLPI decreases
ROS levels
To further investigate the cardio-protective effects
of rhSLPI treatment during sI/R injury, intracellular ROS
generation was measured following H2O2
challenge.
The results demonstrated that exposure to
H2O2 significantly increased intracellular
ROS levels compared with the control group (P<0.05; Fig. 3). However, treatment of ARVMs with
400, 600, 800 and 1,000 ng/ml rhSLPI significantly reduced
intracellular ROS levels compared with untreated ARVMs (P<0.05).
Notably, 400 ng/ml rhSLPI was the lowest concentration of rhSLPI
that significantly reduced ROS production.
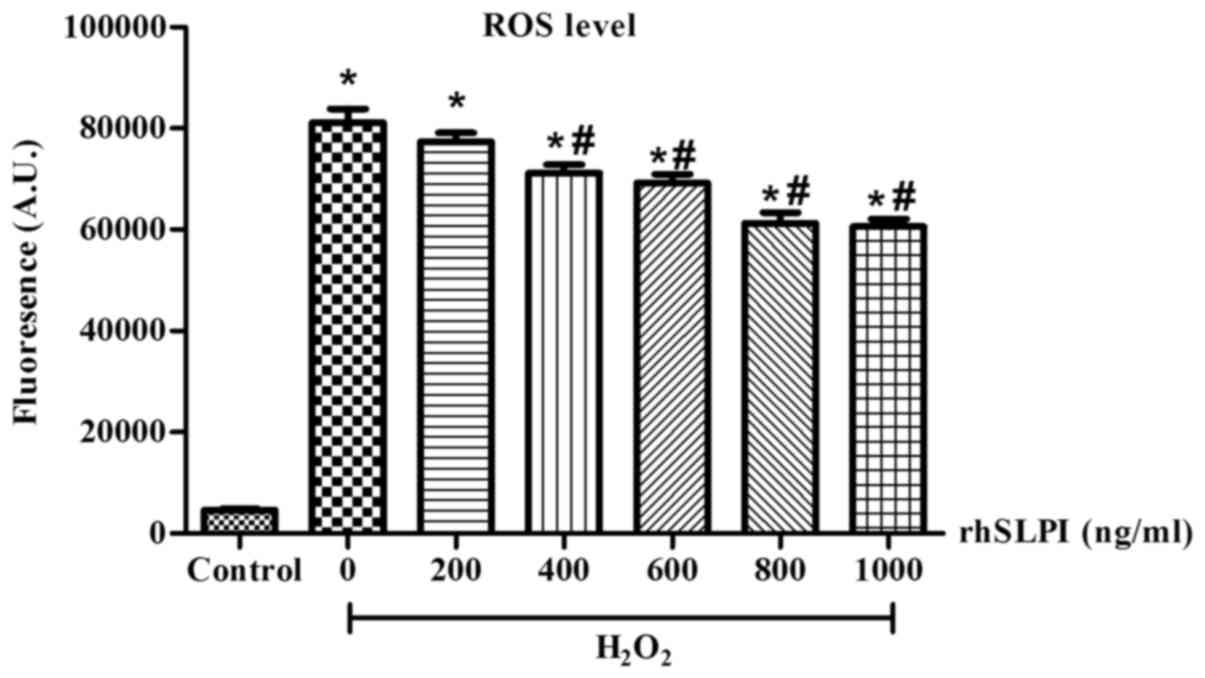 | Figure 3.Cellular ROS levels following
treatment of ARVMs with rhSLPI. ARVMs were incubated with
carboxy-H2DCFDA and treated with 0, 200, 400, 600, 800 and 1,000
ng/ml rhSLPI. Following incubation, H2O2 was
applied to each group and ROS production was determined using the
EnSpire Multimode Plate Reader. Data are presented as the mean ±
standard error of the mean (n=3). *P<0.05 vs. control group;
#P<0.05 vs. H2O2 treated group.
ROS, reactive oxygen species; ARVM, adult rat ventricular myocytes;
rhSLPI, recombinant human secretory leukocyte protease inhibitor;
A.U., arbitrary unit. |
Treatment of ARVMs by rhSLPI
attenuates p38 MAPK phosphorylation and increases Akt activation
following sI
To determine the cellular signaling in response to
rhSLPI treatment in ARVMs during sI/R injury, western blot analysis
was performed to determine the phosphorylation of p38 MAPK and
Akt.
The results demonstrated that the phosphorylation of
p38 MAPK was significantly increased following sI (P<0.05;
Fig. 4A). However, pretreatment with
400 and 600 ng/ml rhSLPI significantly reduced p38 MAPK
phosphorylation (P<0.05). In addition, the results demonstrated
that treatment with 200–1,000 ng/ml rhSLPI prior to sI
significantly increased Akt phosphorylation compared with the sI
group (P<0.05; Fig. 4B).
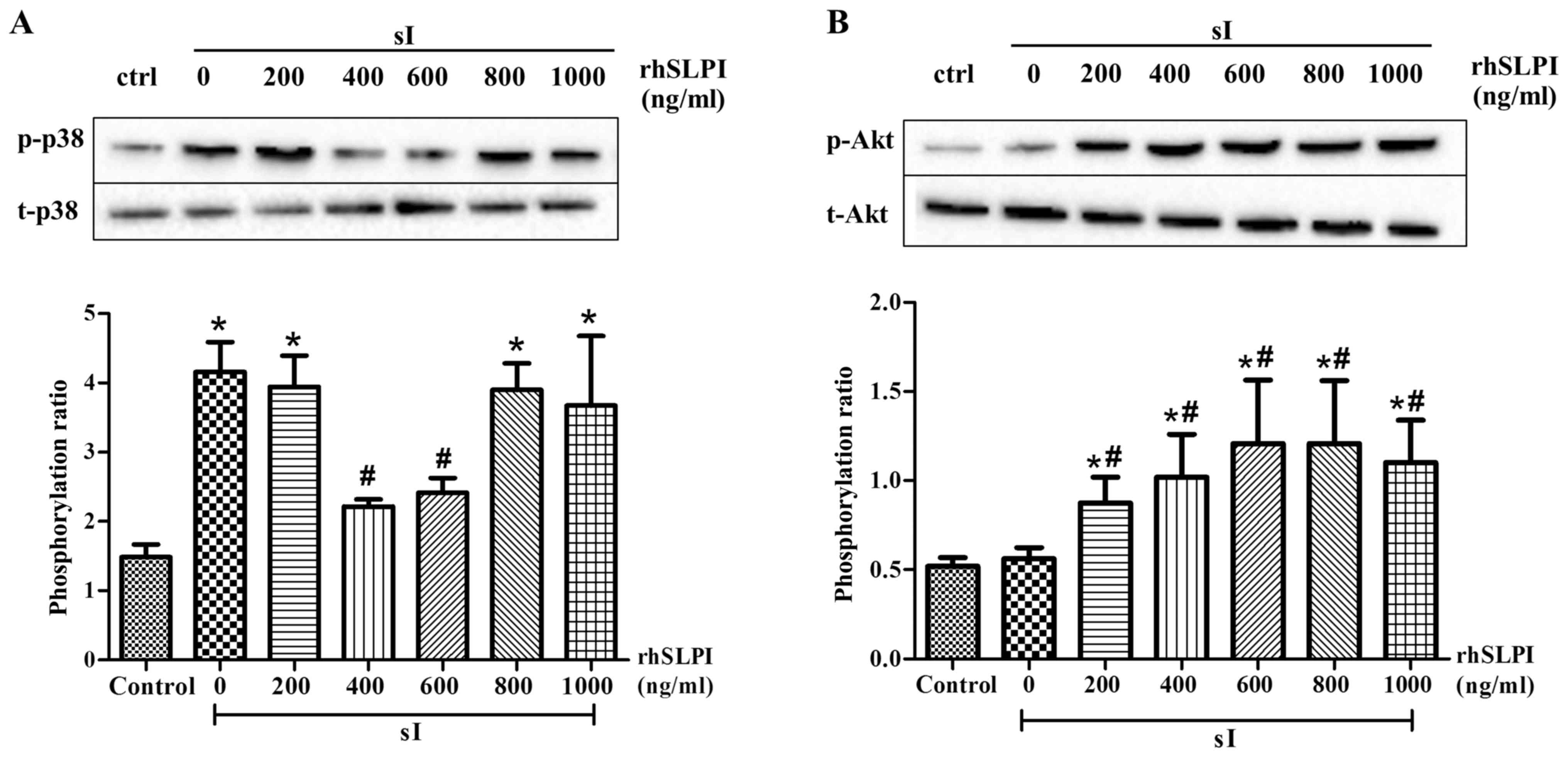 | Figure 4.Cellular signaling in response to
pretreatment of ARVMs with rhSLPI, subjected to
ischemia/reperfusion. ARVMs were treated with 200, 400, 600, 800
and 1,000 ng/ml rhSLPI for 2 h prior to sI. The phosphorylation of
(A) p38 MAPK and (B) Akt was determined by western blot analysis.
Each bar graph represents the phosphorylation ratio (p-/t-) of p38
MAPK and Akt. *P<0.05 vs. control group; #P<0.05
vs. sI group. ARVM, adult rat ventricular myocytes; rhSLPI,
recombinant human secretory leukocyte protease inhibitor; sI,
stimulated ischemia; MAPK, mitogen-activated protein kinase;
p-phosphorylated; t-, total. |
In addition, treatment of ARVMs with rhSLPI at the
onset of sI reduced the phosphorylation of p38 MAPK in a
dose-dependent manner (Fig. 5A).
Treatment of ARVMs with 1,000 ng/ml rhSLPI at the onset of sI
significantly reduced p38 MAPK activation compared with the sI
group (P<0.05). The results also indicated that treatment of
ARVMs with 200–1,000 ng/ml rhSLPI at the onset of sI significantly
increased the phosphorylation of Akt compared with the sI group
(P<0.05; Fig. 5B). This indicates
that treatment of ARVMs with rhSLPI increases Akt phosphorylation
and attenuates the activation of p38 MAPK.
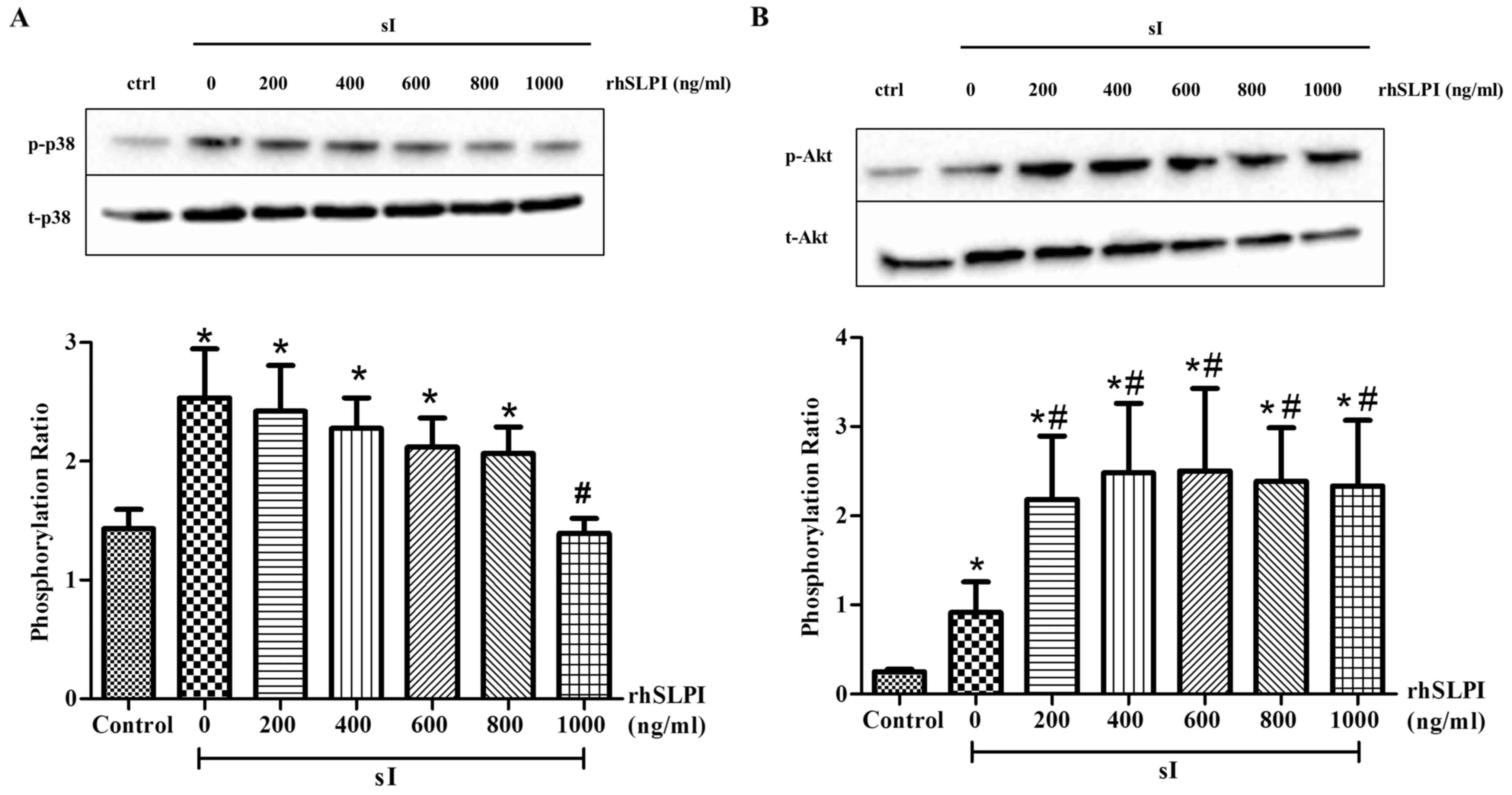 | Figure 5.Cellular signaling following
treatment of ARVMs with rhSLPI during sI. ARVMs were treated with
200, 400, 600, 800 and 1,000 ng/ml rhSLPI at the onset of sI. The
activation of (A) p38 MAPK and (B) Akt was determined by western
blot analysis. Each bar graph represents the phosphorylation ratio
(p-/t-) of p38 MAPK and Akt. *P<0.05 vs. control group;
#P<0.05 vs. sI group. ARVM, adult rat ventricular
myocytes; rhSLPI, recombinant human secretory leukocyte protease
inhibitor; sI, stimulated ischemia; MAPK, mitogen-activated protein
kinase; p-, phosphorylated; t-, total. |
Evaluation of the cardioprotective
effects of rhSLPI in an ex vivo murine model of myocardial I/R
Hearts isolated from adult male C57BL/6 mice were
perfused on a Langendorff system with buffered solutions containing
400 or 1,000 ng/ml rhSLPI during I/R injury (Fig. 6A).
The results indicate that following 30 min global
ischemia and 2 h reperfusion, the infarct size was 48.40±4.23%
(Fig. 6B). However, pretreatment
with either 400 or 1,000 ng/ml rhSLPI significantly reduced the
infarct size compared with the control (P<0.05; Fig. 6B).
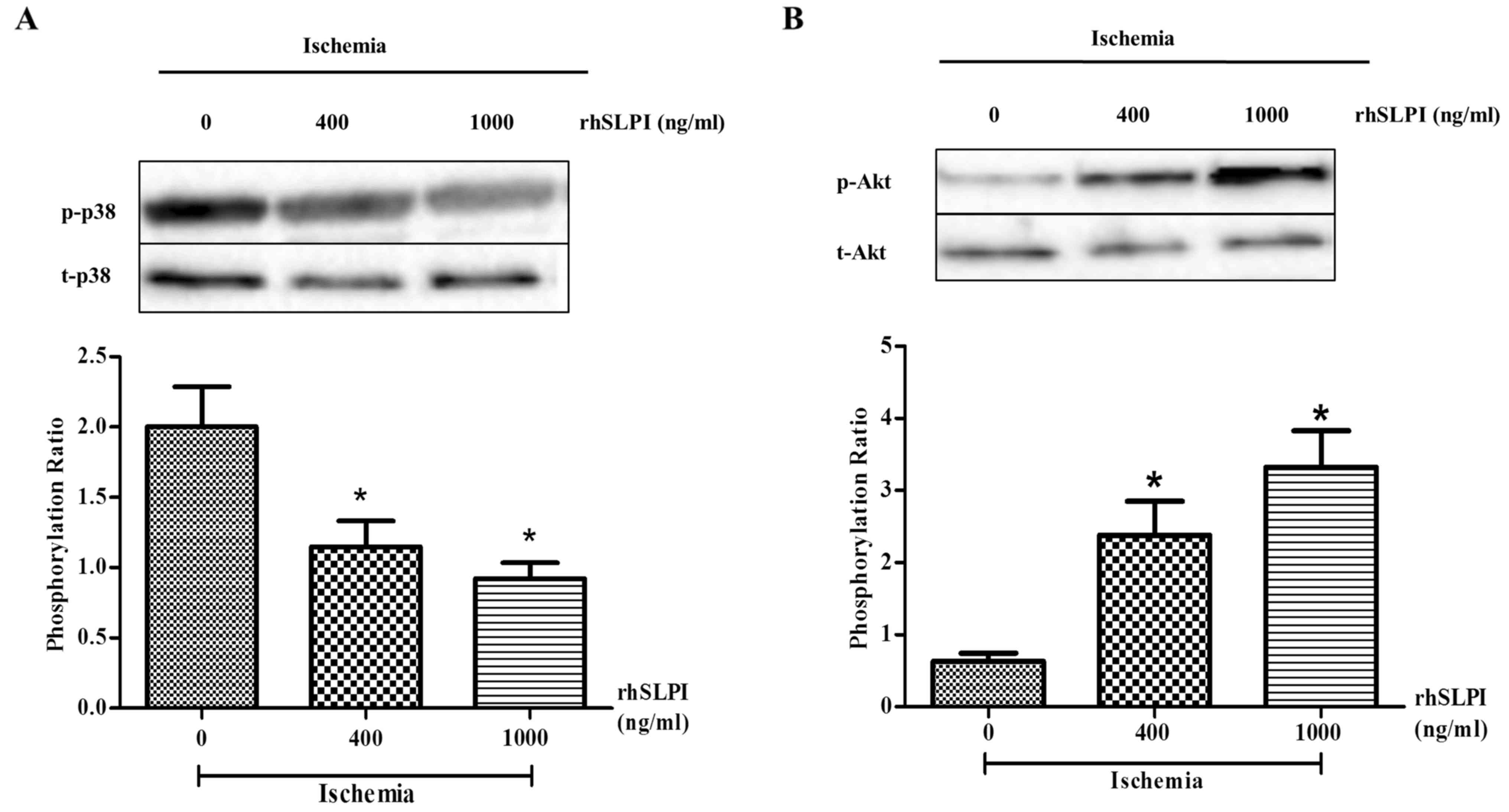
Pretreatment with rhSLPI attenuates
p38 MAPK activation and increases Akt activation in ex vivo I/R
hearts
To further investigate the cardioprotective effects
of rhSLPI pretreatment on ex vivo mouse hearts, cellular
signaling in response to rhSLPI treatment following I/R injury was
investigated. Hearts were perfused with 400 or 1,000 ng/ml rhSLPI
for 30 min prior to global ischemia and 10 min no-flow. p38 MAPK
and Akt phosphorylation ratios were determined by western blotting.
The results demonstrated that, following 10 min global ischemia,
p38 MAPK was highly phosphorylated in the I/R control group
(Fig. 7A). However, pre-treatment
with 400 or 1,000 ng/ml rhSLPI significantly decreased p38 MAPK
activation in a dose-dependent manner (P<0.05; Fig. 7A). By contrast, pretreatment with 400
or 1,000 ng/ml rhSLPI significantly increased Akt phosphorylation
compared with the I/R non-treated control group (P<0.05;
Fig. 7B). These results indicate
that pretreatment with rhSLPI increases Akt phosphorylation and
attenuates p38 MAPK activation in an ex vivo I/R murine
model.
Discussion
The present study demonstrated that rhSLPI exhibits
a protective effects on in vitro and ex vivo sI/R
murine models and determined the mechanism by which treatment with
rhSLPI inhibits the phosphorylation of p38 MAPK and activates the
Akt cell survival pathway in ARVMs and the ex vivo heart.
The results of the current study indicated that in the case of
ARVMs that underwent sI/R injury, the administration of exogenous
rhSLPI, prior to or at the onset of sI significantly reduced
cardiac cell death and cell injury.
During myocardial I/R injury, levels of
pro-inflammatory cytokines, including tumor necrosis factor α
(TNF-α), interleukin (IL) 6, IL-8, and mitochondrial pyruvate
carrier-1 (MPC1), are upregulated and secreted from adjacent
cardiomyocytes, resulting in leukocyte infiltration (5–7).
Infiltrated leukocytes, particularly neutrophil, secrete numerous
serine protease enzymes, including cathepsin G, elastase and
trypsin, which contribute to heart tissue damage, myocardial
necrosis and functional impairment (5,7).
Therefore, inhibiting protease activity, as well as attenuating
inflammatory responses, may be a promising therapeutic strategy to
prevent and treat cardiac tissue injury.
Previous studies have determined the effects of
protease inhibitors on ischemic injury. In a rabbit model of
heterotopic cardiac transplantation, the serine elastase inhibitor
elafin decreased post-cardiac transplant coronary arteriopathy and
decreased myocardial necrosis following transplantation (16). Treatment with two specific elastase
inhibitors, elafin and ICI 200880, in in situ-perfused rat
heart models of repetitive ischemia and myocardial infarction
improved regional myocardial function and reduced infarct size
(17). Furthermore, inhibition of
the serine protease enzymes secreted by neutrophils exhibited
cardioprotective effects against myocardial I/R injury (18,19).
Treatment with PR-39, a potent neutrophil inhibitor, in a murine
model of myocardial I/R injury significantly inhibited the
recruitment of leukocytes into inflamed tissue, thus decreasing
infarct size (18). In a canine
model of heart transplantation, treatment with the neutrophil
elastase inhibitor ONO-5046 Na reduced I/R injury and inhibited
neutrophil elastase and inflammatory cytokine release (19). However, SLPI may be the broadest and
most promising method of treating myocardial I/R injury compared
with other protease inhibitors because SLPI can inhibit various
protease enzyme and exhibits anti-oxidant functions (20). SLPI attenuated cell injury in a
murine hepatic I/R model by inhibiting neutrophil accumulation
(21). Furthermore, SLPI decreased
serum levels of TNF-α, the CXC chemokine macrophage inflammatory
protein, and suppressed activation of the transcription factor
nuclear factor (NF)-κB in the liver (21). Exogenous rhSLPI added to
cold-preservative buffer improved the cardiac score of hearts (a
system that grades myocardial contraction) following
transplantation and reduced the expression of protease enzymes and
pro-inflammatory cytokines in mice undergoing heart transplants
(13).
Studies have investigated the effects of SLPI on
inflammation. SLPI is a protein involved in downregulating
macrophage responses against bacterial lipopolysaccharides (LPS) by
inhibiting NF-κB activation (22).
Ding et al (23) demonstrated
that the anti-inflammatory effects of SLPI on macrophages may be
due to the inhibition of cluster of differentiation (CD)14 on the
macrophage membrane or by inhibiting the formation of LPS-soluble
CD14 complexes. It has been demonstrated that exogenous SLPI is
internalized into monocytes and is distributed throughout the
nucleus and cytoplasm. It then inhibits NF-κB activation by
blocking the degradation of the inhibitor of NF-κB (24). Interestingly, Taggart et al
(24) demonstrated that SLPI
competes with the NF-κB subunit p65 to bind to the promoter of
NF-κB, resulting in the decreased production of pro-inflammatory
cytokines. Furthermore, the spraying of SLPI aerosols suppresses
respiratory epithelial neutrophil elastase (NE) levels and
attenuates the inflammation of the cystic fibrosis epithelial
surface by reducing IL-8 levels (25). Adapala et al (26) reported that SLPI expression in the
adipose tissue is upregulated in obesity and that the pretreatment
of adipocytes with exogenous SLPI results in the downregulation of
LPS-induced IL-6 gene expression and protein secretion in
adipocytes. In addition, exogenous SLPI increased the proliferation
and differentiation of adult neural stem cells via the upregulation
of cyclin D1 and suppression of the cell differentiation regulator
HES1 (27). The treatment of
monocytes with exogenous SLPI inhibited the TNF-α-induced caspase-3
activation and the DNA degradation associated with apoptosis in
monocytes (28). The results of
other studies investigating different types of cells suggest that
treatment with rhSLPI induces similar anti-inflammatory and
anti-apoptotic effects (29–31).
The excessive formation of ROS during I/R injury
induces cardiac cell death directly by inducing cell membrane and
protein damage (32,33) or indirectly by activating
pro-apoptotic pathways, as well as recruiting inflammatory cells
(32,34–38). It
has been suggested that SLPI reduces cellular ROS generation. The
administration of rhSLPI suppressed NE and glutathione levels in
the respiratory epithelial lining fluid of the lung and caused a
concomitant increase in anti-H2O2 capacity
(39). Furthermore, the
overexpression of rhSLPI and glutathione peroxidase-3 reduced
oxidant-induced lung injury and inflammation (40). This indicates that administration of
rhSLPI may be used to treat diseases similar to myocardial I/R
injury that are characterized by an excess of serine proteases and
oxidative stress. The results of the current study demonstrate that
treatment with ARVMs by rhSLPI decrease cellular ROS generation
during H2O2 challenge. To the best of our
knowledge, the current study is the first to demonstrate that the
treatment of ARVMs with rhSLPI reduces cellular ROS generation and
may therefore be an effective method of treating ARVMs in sI/R.
However, the reduction of ROS production and cardiac cell death in
ARVMs following treatment with rhSLPI may be due to increases in
glutathione levels in cardiac cells rather than a direct effect on
ROS scavenging. Therefore, further research investigating the
mechanisms by which rhSLPI reduces intracellular ROS levels in I/R
is warranted.
Myocardial I/R injury is mediated by numerous
signaling pathways, including p38 MAPK. Myocardial I/R injury
activates p38 MAPK, which induces cardiac cell death (15,41–45). The
pre-clinical investigation indicated that the reduction of p38 MAPK
activation reduces myocardial injury (44). Given that the treatment of ARVMs with
rhSLPI reduces cell death following I/R injury, it was hypothesized
that the decrease in cardiac cell death following treatment of
ARVMs with rhSLPI is caused by the attenuation of p38 MAPK
activation. The results of the current study indicated that the
administration of exogenous rhSLPI prior to or at the onset of sI
significantly attenuated the phosphorylation of p38 MAPK. These
results indicate that rhSLPI administration prior to or at the
onset of ischemic insult may attenuate the activation of p38 MAPK
and may therefore be an effective method of protecting cardiac
cells against I/R.
Akt is a serine/threonine protein kinase and serves
an important role in cell survival (46–49). The
activation of Akt signaling protects cardiac cells from apoptosis,
resulting in the attenuation of myocardial I/R injury (46–49). It
was therefore hypothesized that the treatment of ARVMs with rhSLPI
may stimulate Akt phosphorylation during sI/R injury. The results
demonstrated that treatment of ARVMs with rhSLPI prior to or during
sI significantly activated Akt phosphorylation. This suggests that
the administration of rhSLPI not only protects the cell by
inhibiting the activation of p38 MAPK but also increases cell
survival by activating the Akt pathway.
Although it has been demonstrated that various
treatments/interventions administered during reperfusion are
effective at reducing infarct size, the results of the current
study indicated that administering rhSLPI at the onset of
reperfusion failed to protect ARVMs from sI/R induced cell death
and cell injury. These results were similar to those of previous
studies investigating the effects of treatments or interventions
administered prior to ischemia, which may protect the heart from
I/R injury (13,15). Indeed pre-treatment with the p38 MAPK
inhibitors SB203580 and BIRB796 for 30 min prior to ischemia may
significantly reduce infarct size (15). The results of a previous study
conducted in an ischemic rat heart model indicated that SB203580
administered either prior to or during ischemia decreased the
incidence of ventricular tachycardia/ventricular fibrillation in
the ischemic heart, decreased the phosphorylation of heat shock
protein 27 and increased the phosphorylation of connexin 43
(50) However, treatment with
SB203580 at the onset of reperfusion failed to protect the heart
from ischemic insult (50). This may
be due to the fact that, when administered at reperfusion, SB203580
does not inhibit the activation of p38 MAPK and the pro-apoptotic
protein Bax, and does not protect the cardiac mitochondrial
membrane potential (45). In
addition, previous studies demonstrated that pretreatment with
various types of compounds (including amifostine, barbaloin,
simvastatin, lysophosphatidic acid and icariin) may also exhibit
cardioprotective effects against I/R injury (51–55),
similar to the results of the current study. Therefore, the results
of the current study suggest that treatment with rhSLPI (400–1,000
ng/ml) prior to or at the onset of ischemia is most effective at
protecting cardiac cells and reducing cardiac cell injury following
I/R. Pre-treatment with 1,000 ng/ml rhSLPI in ex vivo murine
hearts significantly reduced the infarct size. These results
indicate that timing SLPI treatment with respect to the onset of
ischemia is an important determinant of its therapeutic
efficacy.
There were a number of limitations of the current
study. Treatment of ARVMs by rhSLPI or treatment of ex vivo
hearts with rhSLPI may not be representative of real-world
physiological settings. The ex vivo model is useful in
determining the actual effect of drugs or substance testing, as the
heart undergoes direct perfusion with the test substance and the
effect serum proteins binding to the test substance is avoided. In
addition, the global ischemia protocol used in the current study is
well-suited to study the effects of ischemia and hypoxia and
determine the release of cellular constituents from coronary
effluent, including enzymes and proteins, as well as markers of
cardiac metabolism (56). The
protocols used in the current study, including perfusion setting,
fixation of the heart tissue by glutaraldehyde or formalin and
determination of the infarct size are standard methods that have
been used in previous studies (14,15,50,57,58). As
aforementioned, an ex vivo model may not be representative
of real clinical settings; therefore other relevant models,
including treatment of intact hearts in an in vivo model of
I/R may produce more relevant functional data that are closely
associated with real physiological events in the heart, allowing a
more reliable interpretation of the results. In addition, the
number of animals used in each experiment in the current study was
small; therefore a larger sample size should be used in future
in vivo or clinical studies.
In conclusion, the results of the current study
indicate that treatment with rhSLPI exhibits cardioprotective
effects against myocardial I/R injury in in vitro and ex
vivo models by reducing intracellular ROS levels and infarct
size, attenuating p38 MAPK phosphorylation and activating Akt
phosphorylation. The results suggest that rhSLPI may be developed
as a potential therapeutic strategy to treat patients with ischemic
heart disease.
Acknowledgements
The authors wish to acknowledge the support from the
Franco-Thai Scholarship program in 2015 and-2016 for authors SK and
SBL. The authors would also like to thank the Newton Fund in
cooperation with The Royal Golden Jubilee PhD Program for providing
a PhD placement scholarship for EP, SK and MM. The authors are
grateful to the Center for Animal Research, Naresuan University for
their excellent technical assistance.
Funding
The present study was supported by The Royal Golden
Jubilee PhD Program (grant no. PHD/0043/2555) - joint funding
between the Thailand Research Fund, Naresuan University and
National Research Council of Thailand (NRCT)-Naresuan University
(grant no. R2558B06).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Ethics approval and consent to
participate
The present study was approved by the animal ethics
committee of the Center for Animal Research, Naresuan University
(approval no. NU AE550732).
Authors' contributions
EP and SK conceived and designed the experiments.
EP, JS, SBL, JN, HN, MM and SK performed the experiments and
analyzed the data. EP, MM and SK wrote, re-checked, and proofread
the paper.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
World Health Organization: World Health
Statistics 2008. Geneva: 2008
|
|
2
|
Writing Group Members, . Mozaffarian D,
Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de
Ferranti S, Després JP, et al: Heart disease and stroke
statistics-2016 update: A report from the american heart
association. Circulation. 133:e38–e360. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jennings RB and Reimer KA: The cell
biology of acute myocardial ischemia. Annu Rev Med. 42:225–246.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Epelman S, Liu PP and Mann DL: Role of
innate and adaptive immune mechanisms in cardiac injury and repair.
Nat Rev Immunol. 15:117–129. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Boudoulas KD and Hatzopoulos AK: Cardiac
repair and regeneration: The Rubik's cube of cell therapy for heart
disease. Dis Model Mech. 2:344–358. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jordan JE, Zhao ZQ and Vinten-Johansen J:
The role of neutrophils in myocardial ischemia-reperfusion injury.
Cardiovasc Re. 43:860–878. 1999. View Article : Google Scholar
|
|
8
|
Kuckleburg CJ and Newman PJ: Neutrophil
proteinase 3 acts on protease-activated receptor-2 to enhance
vascular endothelial cell barrier function. Arterioscler Thromb
Vasc Biol. 33:275–284. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bouchard D, Morisset D, Bourbonnais Y and
Tremblay GM: Proteins with whey-acidic-protein motifs and cancer.
Lancet Oncol. 7:167–174. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Majchrzak-Gorecka M, Majewski P, Grygier
B, Murzyn K and Cichy J: Secretory leukocyte protease inhibitor
(SLPI), a multifunctional protein in the host defense response.
Cytokine Growth Factor Rev. 28:79–93. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Moreau T, Baranger K, Dadé S,
Dallet-Choisy S, Guyot N and Zani ML: Multifaceted roles of human
elafin and secretory leukocyte proteinase inhibitor (SLPI), two
serine protease inhibitors of the chelonianin family. Biochimie.
90:284–295. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Doumas S, Kolokotronis A and Stefanopoulos
P: Anti-inflammatory and antimicrobial roles of secretory leukocyte
protease inhibitor. Infect Immun. 73:1271–1274. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schneeberger S, Hautz T, Wahl SM,
Brandacher G, Sucher R, Steinmassl O, Steinmassl P, Wright CD,
Obrist P, Werner ER, et al: The effect of secretory leukocyte
protease inhibitor (SLPI) on ischemia/reperfusion injury in cardiac
transplantation. Am J Transplant. 8:773–782. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jacquet S, Nishino Y, Kumphune S, Sicard
P, Clark JE, Kobayashi KS, Flavell RA, Eickhoff J, cotton M and
Marber MS: The role of RIP2 in p38 MAPK activation in the stressed
heart. J Biol Chem. 283:11964–11971. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kumphune S, Bassi R, Jacquet S, Sicard P,
Clark JE, Verma S, Avkiran M, O'Keefe SJ and Marber MS: A chemical
genetic approach reveals that p38alpha MAPK activation by
diphosphorylation aggravates myocardial infarction and is prevented
by the direct binding of SB203580. J Biol Chem. 285:2968–2975.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cowan B, Baron O, Crack J, Coulber C,
Wilson GJ and Rabinovitch M: Elafin, a serine elastase inhibitor,
attenuates post-cardiac transplant coronary arteriopathy and
reduces myocardial necrosis in rabbits afer heterotopic cardiac
transplantation. J Clin Invest. 97:2452–2468. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tiefenbacher CP, Ebert M, Niroomand F,
Batkai S, Tillmanns H, Zimmermann R and Kübler W: Inhibition of
elastase improves myocardial function after repetitive ischaemia
and myocardial infarction in the rat heart. Pflugers Arch.
433:563–570. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hoffmeyer MR, Scalia R, Ross CR, Jones SP
and Lefer DJ: PR-39, a potent neutrophil inhibitor, attenuates
myocardial ischemia-reperfusion injury in mice. Am J Physiol Heart
Circ Physiol. 279:H2824–H2828. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ueno M, Moriyama Y, Toda R, Yotsumoto G,
Yamamoto H, Fukumoto Y, Sakasegawa K, Nakamura K and Sakata R:
Effect of a neutrophil elastase inhibitor (ONO-5046 Na) on
ischemia/reperfusion injury using the left-sided heterotopic canine
heart transplantation model. J Heart Lung Transplant. 20:889–896.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schneeberger S, Brandacher G, Mark W,
Amberger A and Margreiter R: Protease inhibitors as a potential
target in modulation of postischemic inflammation. Drug News
Perspect. 15:568–574. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lentsch AB, Yoshidome H, Warner RL, Ward
PA and Edwards MJ: Secretory leukocyte protease inhibitor in mice
regulates local and remote organ inflammatory injury induced by
hepatic ischemia/reperfusion. Gastroenterology. 117:953–961. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McKiernan PJ, McElvaney NG and Greene CM:
SLPI and inflammatory lung disease in females. Biochem Soc Trans.
39:1421–1426. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ding A, Thieblemont N, Zhu J, Jin F, Zhang
J and Wright S: Secretory leukocyte protease inhibitor interferes
with uptake of lipopolysaccharide by macrophages. Infect Immun.
67:4485–4489. 1999.PubMed/NCBI
|
|
24
|
Taggart CC, Cryan SA, Weldon S, Gibbons A,
Greene CM, Kelly E, Low TB, O'neill SJ and McElvaney NG: Secretory
leucoprotease inhibitor binds to NF-kappaB binding sites in
monocytes and inhibits p65 binding. J Exp Med. 202:1659–1668. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McElvaney NG, Nakamura H, Birrer P, Hébert
CA, Wong WL, Alphonso M, Baker JB, Catalano MA and Crystal RG:
Modulation of airway inflammation in cystic fibrosis. In vivo
suppression of interleukin-8 levels on the respiratory epithelial
surface by aerosolization of recombinant secretory leukoprotease
inhibitor. J Clin Invest. 90:1296–1301. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Adapala VJ, Buhman KK and Ajuwon KM: Novel
anti-inflammatory role of SLPI in adipose tissue and its regulation
by high fat diet. J Inflamm (Lond). 8:52011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mueller AM, Pedré X, Stempfl T, Kleiter I,
Couillard-Despres S, Aigner L, Giegerich G and Steinbrecher A:
Novel role for SLPI in MOG-induced EAE revealed by spinal cord
expression analysis. J Neuroinflammation. 5:202008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McGarry N, Greene CM, McElvaney NG, Weldon
S and Taggart CC: The ability of secretory leukocyte protease
inhibitor to inhibit apoptosis in monocytes is independent of its
antiprotease activity. J Immunol Res. 2015:5073152015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Subramaniyam D, Hollander C, Westin U,
Erjefält J, Stevens T and Janciauskiene S: Secretory leukocyte
protease inhibitor inhibits neutrophil apoptosis. Respirology.
16:300–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Seto T, Takai T, Ebihara N, Matsuoka H,
Wang XL, Ishii A, Ogawa H, Murakami A and Okumura K: SLPI prevents
cytokine release in mite protease-exposed conjunctival epithelial
cells. Biochem Biophys Res Commun. 379:681–685. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee SY, Nho TH, Choi BD, Jeong SJ, Lim DS
and Jeong MJ: Secretory leukocyte protease inhibitor reduces
inflammation and alveolar bone resorption in LPS-induced
periodontitis in rats and in MC3T3-E1 preosteoblasts. Animal Cells
Systems. 20:344–352. 2016. View Article : Google Scholar
|
|
32
|
Hoffman JW Jr, Gilbert TB, Poston RS and
Silldorff EP: Myocardial reperfusion injury: Etiology, mechanisms,
and therapies. J Extra Corpor Technol. 36:391–411. 2004.PubMed/NCBI
|
|
33
|
Raedschelders K, Ansley DM and Chen DD:
The cellular and molecular origin of reactive oxygen species
generation during myocardial ischemia and reperfusion. Pharmacol
Ther. 133:230–255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gottlieb RA: Cell death pathways in acute
ischemia/reperfusion injury. J Cardiovasc Pharmacol Ther.
16:233–238. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lucchesi BR: Myocardial ischemia,
reperfusion and free radical injury. Am J Cardiol. 65:14I–23I.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Venardos KM, Perkins A, Headrick J and
Kaye DM: Myocardial ischemia-reperfusion injury, antioxidant enzyme
systems, and selenium: A review. Curr Med Chem. 14:1539–1549. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ferrari R, Agnoletti L, Comini L, Gaia G,
Bachetti T, Cargnoni A, Ceconi C, Curello S and Visioli O:
Oxidative stress during myocardial ischaemia and heart failure. Eur
Heart J. 19 Suppl B:B2–B11. 1998.PubMed/NCBI
|
|
39
|
Gillissen A, Birrer P and McElvaney NG:
Recombinant secretory leukoprotease inhibitor augments glutathione
levels in lung epithelial lining fluid. J Appl Physiol (1985).
75:825–832. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Masterson CH, O'Toole DP and Laffey JP:
Over expression of secretory leukocyte protease inhibitor (SLPI)
and glutathione peroxidase-3 (GPX3) attenuate inflammation and
oxidant-mediated pulmonary epithelial injuryAmerican Thoracic
Society 2012 International Conference. San Francisco, California:
2012
|
|
41
|
Kaiser RA, Lyons JM, Duffy JY, Wagner CJ,
McLean KM, O'Neill TP, Pearl JM and Molkentin JD: Inhibition of p38
reduces myocardial infarction injury in the mouse but not pig after
ischemia-reperfusion. Am J Physiol Heart Circ Physiol.
289:H2747–H2751. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gorog DA, Tanno M, Cao X, Bellahcene M,
Bassi R, Kabir AM, Dighe K, Quinlan RA and Marber MS: Inhibition of
p38 MAPK activity fails to attenuate contractile dysfunction in a
mouse model of low-flow ischemia. Cardiovasc Res. 61:123–131. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ma XL, Kumar S, Gao F, Louden CS, Lopez
BL, Christopher TA, Wang C, Lee JC, Feuerstein GZ and Yue TL:
Inhibition of p38 mitogen-activated protein kinase decreases
cardiomyocyte apoptosis and improves cardiac function after
myocardial ischemia and reperfusion. Circulation. 99:1685–1691.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kumphune S, Chattipakorn S and
Chattipakorn N: Role of p38 inhibition in cardiac
ischemia/reperfusion injury. Eur J Clin Pharmacol. 68:513–524.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kumphune S, Surinkaew S, Chattipakorn SC
and Chattipakorn N: Inhibition of p38 MAPK activation protects
cardiac mitochondria from ischemia/reperfusion injury. Pharm Biol.
53:1831–1841. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yu L, Li F, Zhao G, Yang Y, Jin Z, Zhai M,
Yu W, Zhao L, Chen W, Duan W and Yu S: Protective effect of
berberine against myocardial ischemia reperfusion injury: Role of
Notch1/Hes1-PTEN/Akt signaling. Apoptosis. 20:796–810. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mullonkal CJ and Toledo-Pereyra LH: Akt in
ischemia and reperfusion. J Invest Surg. 20:195–203. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fujio Y, Nguyen T, Wencker D, Kitsis RN
and Walsh K: Akt promotes survival of cardiomyocytes in vitro and
protects against ischemia-reperfusion injury in mouse heart.
Circulation. 101:660–667. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Armstrong SC: Protein kinase activation
and myocardial ischemia/reperfusion injury. Cardiovasc Res.
61:427–436. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Surinkaew S, Kumphune S, Chattipakorn S
and Chattipakorn N: Inhibition of p38 MAPK during ischemia, but not
reperfusion, effectively attenuates fatal arrhythmia in
ischemia/reperfusion heart. J Cardiovasc Pharmacol. 61:133–141.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wu SZ, Tao LY, Wang JN, Xu ZQ, Wang J, Xue
YJ, Huang KY, Lin JF, Li L and Ji KT: Amifostine pretreatment
attenuates myocardial ischemia/reperfusion injury by inhibiting
apoptosis and oxidative stress. Oxid Med Cell Longev.
2017:41308242017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang P, Liu X, Huang G, Bai C, Zhang Z
and Li H: Barbaloin pretreatment attenuates myocardial
ischemia-reperfusion injury via activation of AMPK. Biochem Biophys
Res Commun. 490:1215–1220. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Jones SP, Trocha SD and Lefer DJ:
Pretreatment with simvastatin attenuates myocardial dysfunction
after ischemia and chronic reperfusion. Arterioscler Thromb Vasc
Biol. 21:2059–2064. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chen H, Liu S, Liu X, Yang J, Wang F, Cong
X and Chen X: lysophosphatidic acid pretreatment attenuates
myocardial ischemia/reperfusion injury in the immature hearts of
rats. Front Physiol. 8:1532017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ke Z, Liu J, Xu P, Gao A, Wang L and Ji L:
The cardioprotective effect of icariin on ischemia-reperfusion
injury in isolated rat heart: Potential involvement of the PI3K-Akt
signaling pathway. Cardiovasc Ther. 33:134–140. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Skrzypiec-Spring M, Grotthus B, Szelag A
and Schulz R: Isolated heart perfusion according to
Langendorff-still viable in the new millennium. J Pharmacol Toxicol
Methods. 55:113–126. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Herr DJ, Aune SE and Menick DR: Induction
and assessment of ischemia-reperfusion injury in
langendorff-perfused rat hearts. J Vis Exp. 27:e529082015.
|
|
58
|
Csonka C, Kupai K, Kocsis GF, Novák G,
Fekete V, Bencsik P, Csont T and Ferdinandy P: Measurement of
myocardial infarct size in preclinical studies. J Pharmacol Toxicol
Methods. 61:163–170. 2010. View Article : Google Scholar : PubMed/NCBI
|