Introduction
Breast cancer is a common malignancy in women, and
has high morbidity and mortality rates. There are ~250,000 new
cases of breast cancer and ~40,000 breast cancer-associated
mortalities annually in the USA (1).
The risk factors for breast cancer remain relatively unclear,
although it has been reported that multiple factors, including
hormone levels, family heredity and obesity, may influence the
initiation and development of breast cancer (2,3). Due to
its high heterogeneity, breast cancer is characterized into several
subgroups according to genome alterations. However, the diagnosis
and treatment for breast cancer requires improvement. To improve
survival rates and reduce recurrence, increased understanding of
the molecular mechanisms underlying breast tumor progression is
necessary.
Angiogenesis serves an essential role in tumor
growth and metastasis (4). In breast
cancer, tumor cells may disseminate to distant organs or tissues,
including the lung and bone, through blood vessels formed from
endothelial cells (5). In addition,
these new blood vessels formed deliver blood and nutrients for
continued tumor growth. Angiogenesis is stimulated by angiogenic
factors secreted by the tumor, including vascular endothelial
growth factor A (VEGFA), platelet-derived growth factor (PDGF),
fibroblast growth factor (FGF) and angiopoietins (6,7). Thus,
these factors could be therapeutic targets for breast cancer.
Triptolide has been widely recognized as an
anti-inflammatory, antioxidant and antiproliferative compound
(8,9). Triptolide is extracted from the herb
Tripterygium wilfordii and has been used as an antibiotic
(9). A previous study reported that
triptolide was beneficial for the treatment of monocytic and
myelocytic leukemia (10).
Triptolide induces tumor cells apoptosis by activating the caspase
cascade and inhibiting Wnt/β-catenin signaling (11). Furthermore, triptolide can covalently
bind to and inhibit the transcription factor II human protein
(12). These observations suggest
that triptolide has potential efficacy for the treatment of breast
cancer. Indeed, Owa et al has demonstrated that triptolide
exhibits cytotoxicity against breast cancer cells in vitro
and in vivo through causing lysosomal-mediated programmed
cell death (13). However, the
function of triptolide in angiogenesis remains unclear.
The present study examined the effects of triptolide
on breast cancer cells, particularly angiogenesis. This revealed
that triptolide could inhibit VEGFA expression and breast cancer
cell proliferation. In vivo injection of triptolide
inhibited the formation of microvessels. In addition, triptolide
inhibited the activation of extracellular signal-related kinase
(ERK)1/2 and the expression of hypoxia inducible factor (HIF)-1α.
The overexpression of HIF1-α partially induced VEGFA expression
despite continuing triptolide treatment. These results indicate
that triptolide has potential for the clinical treatment of breast
cancer.
Materials and methods
Cell culture and reagents
MCF7, MDAMB231, and T47D were obtained from the Type
Culture Collection of the Chinese Academy of Sciences (Shanghai,
China); SKBR3, Hs578T and BT474 human breast cancer cells were
obtained from the American Type Culture Collection (Manassas, VA,
USA). Cells were maintained in RPMI-1640 medium (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.), 10 U/ml
penicillin and 10 mg/ml streptomycin. Cells were cultured in a
humidified incubator at 37°C with 5% CO2. Triptolide was
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA;
cat. no. sc-200122) and dissolved in dimethyl sulfoxide (DMSO).
Cells were treated with indicated concentration of Triptolide and
cultured in 24, 48 and 72 h and treated with DMSO as control.
Plasmid and small interfering (si)RNA
transfection
Human HIF1-α complementary DNA was amplified using
the polymerase chain reaction (PCR) and subcloned into the
expression vector pCDNA3.1. The primers used for PCR were as
follows: Sense' 5′-CGGGATCCATGGAGGGCGCCG′-3′ and antisense'
5′-GCTCTAGACTAAATAATTCCTACT′-3′. siRNA pools targeting HIF1-α
(HSH008831) or VEGFA (HSH018475) were purchased from GeneCopoeia,
Inc. (Guangzhou, China). siRNA transfection were performed using
Lipofectamine® 2000 Transfection reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol, which has been described previously (14). For overexpression of HIF1-α, the
plasmid was transfected in cells using Lipofectamine 2000.
RNA extraction, reverse
transcription-quantitative (RT-q)PCR and semi-quantitative PCR
Total RNA was extracted from cells using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. cDNA was synthesized using the
PrimeScript RT-PCR kit (Takara Biotechnology Co., Ltd., Dalian,
China) according to the manufacturer's protocol and used as a
template for qPCR. qPCR was performed on the Applied Biosystems
7500 Fast Real-Time PCR system (Thermo Fisher Scientific, Inc.)
using SYBR Green methods (SYBR® Premix Ex Taq™ II;
Takara Biotechnology Co., Ltd.) according to the manufacturer's
protocol, which has been described previously (15). The expression levels of the target
genes were normalized to GAPDH using the 2−ΔΔCq method
(16). The primers used for qPCR
were as follows: VEGFA forward' 5′-AGGGCAGAATCATCACGAAGT′-3′ and
reverse' 5′-AGGGTCTCGATTGGATGGC′-3′; and GAPDH forward'
5′-CTGGGCTACACTGAGCAC′-3′ and reverse' 5′-AAGTGGTCGTTGAGGGCAAT′-3′.
The semi-quantitative real-time PCR was performed using Takara Ex
Taq (#RR001A, Takara Biotechnology Co., Ltd.) with VEGFA primers.
The cDNA used was the same. The following thermocycling condition
were used: 95°C for 10 sec, 58°C for 15 sec and 72°C for 30 sec for
30 cycles. The PCR products were loaded in 1% agarose gel, stained
with ethidium bromide and semiquantification was performed by gray
intensity analysis.
Western blotting
Total protein was extracted using
radioimmunoprecipitation assay buffer (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) with added protease inhibitor and phosphatase
inhibitor cocktail (Roche Applied Science, Mannheim, Germany.
Protein concentration was measured using the bicinchoninic acid
assay. For each sample, 60 µg of total protein per lane was
separated via SDS-PAGE on a 10% gel and then transferred to a
polyvinylidene fluoride membrane. The membranes were blocked with
5% bovine serum albumin (cat. no. B600036; Sangon Biotech;
Shanghai, China) for 1 h at room temperature and then incubated
with primary antibodies at 4°C overnight at a 1:1,000 dilution.
Then, the membranes were washed with PBS-Tween-20 and incubated at
room temperature for 45 min with horseradish peroxidase
(HRP)-conjugated goat anti-rabbit immunoglobulin G secondary
antibodies (cat. no. sc-2004; 1:2,000 dilution; Santa Cruz
Biotechnology, Inc.). The results were visualized using enhanced
chemiluminescence with ECL Western Blotting Substrate (cat. no.
32106; Pierce; Thermo Fisher Scientific, Inc.). GAPDH was used as
the loading control. The primary antibodies used were as follows:
Anti-phosphorylated (p)-ERK1/2 (cat. no. 4376), anti-ERK1/2 (cat.
no. 4695,), anti-p-RAC-α serine/threonine-protein kinase (Akt; cat.
no. 4060), anti-Akt (cat. no. 4691) and anti-HIF1-α (cat. no.
14179) (all Cell Signaling Technology, Inc., Danvers, MA, USA) and
anti-GAPDH (cat. no. sc-25778; Santa Cruz Biotechnology, Inc.). The
quantification of western blot bands was performed using ImageJ
software (National Institutes of Health, Bethesda, MD, USA).
Animal experiments
The animal experimental procedures used in the
present study were approved by the Animal Care and Use Committee of
Shandong University and in accordance with guidelines. The mice
were purchased from Shanghai SLAC Laboratory Animal Co., Ltd.
(Shanghai, China). A total of 1×106 MDAMB231 cells were
injected into the mammary fat pads of 12 8-week-old female nude
mice (weight, 20.21±1.18 g) to produce a mouse model of breast
cancer. Mice were housed at 25°C and had free access to food and
water with a 14-h light/10-h dark cycle. These mice were then
divided into two groups (n=6/group), the untreated control group
and the triptolide treatment group. A total of 15 days after
xenotransplantation of the cells, 1 mg/kg−1 triptolide
was injected into the tumors 3 times per week for a total of 4
weeks or when the tumor size reached ~1.5 cm. Then the mice were
sacrificed, and the tumors were extracted and processed for
immunohistochemistry.
Immunohistochemistry
Tumor tissues were collected 8 weeks after the last
triptolide injection, and underwent 4% formalin fixation overnight
at 4°C and paraffin embedding. The specimens were then sliced into
4-µm-thick sections. Then, the sections were deparaffinized in
xylene and rehydrated in alcohol. After antigen retrieval at ~100°C
for 3 min in citrate buffer, the sections were placed in 3%
H2O2 to quench endogenous peroxidase activity
and samples were blocked with 5% BSA (Sangon Biotech) at room
temperature for 30 min. Primary antibodies were incubated with the
sections overnight at 4°C. Subsequently, the sections were washed
with PBS and incubated with HRP-labeled secondary antibodies for 30
min at 37°C, followed by a further wash. The antibody signals were
visualized using 3′-diaminobenzidine and counterstained with
hematoxylin at room temperature for 2 min. Sections were then
observed under a light microscope. At least three sections per
specimen were stained to confirm reproducibility. The primary
antibodies used were as follows: Anti-VEGFA (cat. no. 19003–1-AP;
1:200 dilution), anti-cluster of differentiation (CD)31 (cat. no.
11265-1-AP; 1:200 dilution) and anti-proliferation marker protein
Ki67 (Ki67; cat. no. 19972-1-AP; 1:500 dilution) were purchased
from ProteinTech Group, Inc. (Chicago, IL, USA).
Immunohistochemistry results were quantified by measuring the
positive stained cells. The positive staining was indicated at a
density >50%.
Endothelial tube formation assay
The tube formation assay was performed using human
umbilical vein endothelial cells (HUVECs; Type Culture Collection
of the Chinese Academy of Sciences), which were cultured in
RPMI-1640 supplemented with 20% FBS at 37°C in an atmosphere
containing 5% CO2 as described previously (17). A total of 2×105 cells/well
were seeded into 12-well culture plates precoated with growth
factor-reduced Matrigel® (BD Biosciences, Franklin
Lakes, NJ, USA). Cells were treated with 5 nM triptolide for 48 h
and cells treated with si-VEGF siRNAs were employed as a positive
control. After a 24-h incubation at 37°C, tube formation was imaged
and quantified using an inverted microscope.
Bromodeoxyuridine (BrdU) incorporation
assay
For the BrdU incorporation assay, HUVECs were
pretreated with 5 nM triptolide or 10 nM siRNAs and cultured for 48
h at 37°C. Cells were then processed for BrdU incorporation using a
BrdU cell proliferation assay Kit (cat. no. K306-200; Biovision,
Milpitas, CA, USA) according to the manufacturer's protocol, which
has been described previously (17).
Representative images were then captured using an inverted
microscope and the number of cells were determined cells with
fluorescence in five random fields and compared these with the
control group.
Statistical analysis
Statistical analysis was performed using an unpaired
Student's t-test. SPSS software (version 21.0; SPSS, Inc., Chicago,
IL, USA). Results from in vitro experiments are presented at
the mean ± standard error of the mean from at least three
experiments. In vivo results are presented as the mean ±
standard deviation. P<0.05 was considered to indicate a
statistically significant difference.
Results
Triptolide suppresses VEGFA
expression
It has been reported that triptolide induces
apoptosis and exhibits toxic effects on breast cancer cells
(18); however, the function of
triptolide in angiogenesis remains unclear. The expression of
VEGFA, a key angiogenic activator, at the transcript level in six
breast cancer cell lines was determined in the present study using
semi-quantitative PCR (Fig. 1A).
This revealed that VEGFA was relatively highly expressed in Hs578T
and MDAMB231 cells compared with BT474, SKBR3, MCF7 and T47D cells.
Next, Hs578T and MDAMB231 cells were treated with 5 nM triptolide,
which significantly reduced VEGFA mRNA levels after 48 and 72 h in
a time-dependent manner (all P<0.05; Fig. 1B). Western blotting was employed to
confirm this observation at the protein level. In addition, the two
cell lines were treated with different concentrations of triptolide
ranging from 5–20 nM. Treatment with 10 and 20 nM triptolide
significantly reduced the expression of VEGFA mRNA in a
concentration-dependent manner (all P<0.05; Fig. 1C). This was again confirmed at the
protein level via western blot analysis. These results indicate
that triptolide suppresses VEGFA expression in breast cancer
cells.
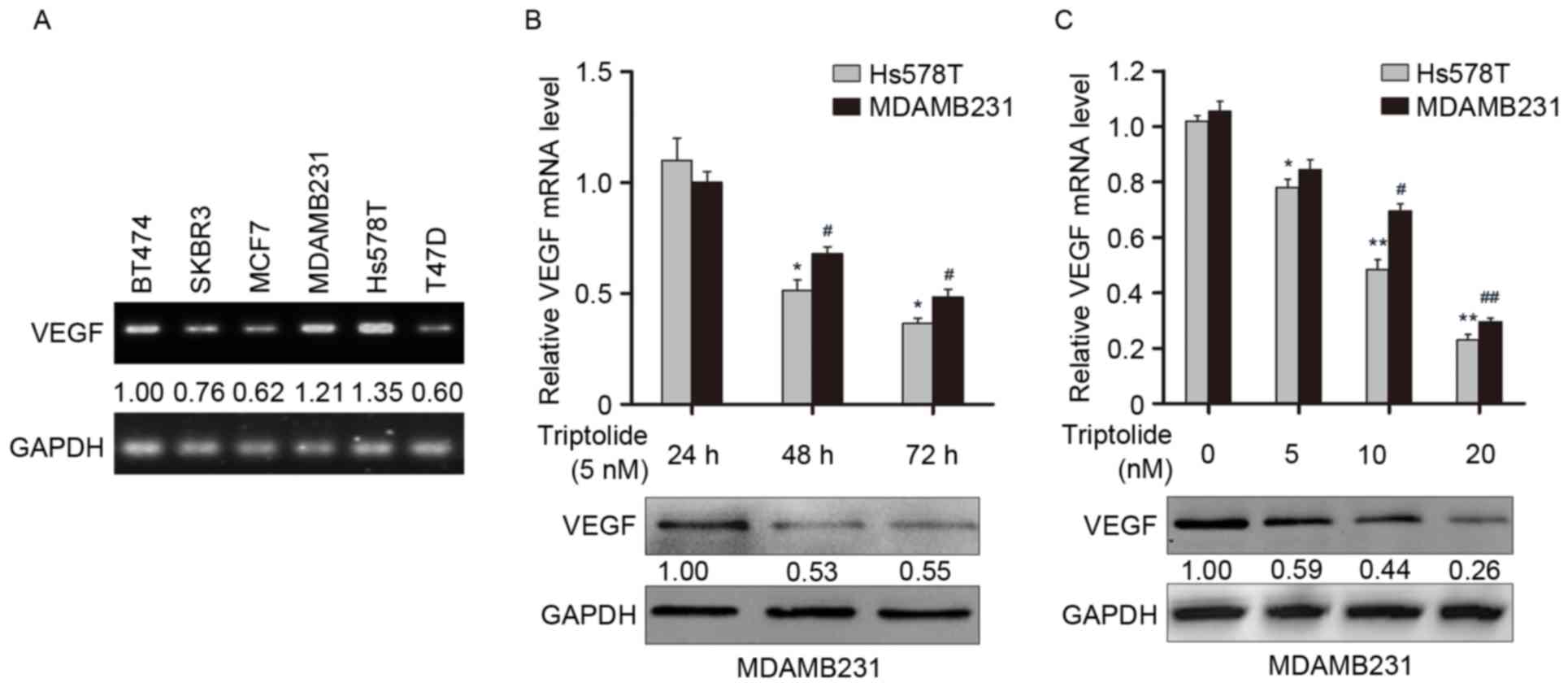 | Figure 1.Triptolide suppresses the expression
of VEGFA in vitro. (A) VEGFA mRNA expression levels in six
breast cancer cell lines were determined using RT-qPCR analysis.
GAPDH was used as the internal control. (B) Hs578T and MDAMB231
were treated with 5 nM triptolide for 24, 48 and 72 h, and the
expression of VEGFA was examined by RT-qPCR (upper panel) and
western blot (lower panel) analysis. (C) Cells were treated with
dimethyl sulfoxide alone or triptolide (5, 10 or 20 nM), and the
expression of VEGFA was examined by RT-PCR (upper panel) and
western blot (lower panel) analysis. *P<0.05, **P<0.01 vs.
Hs578T cells at 24 h or 0 nM triptolide treatment, respectively;
#P<0.05, ##P<0.01 vs. MDAMB231 cells at
24 h 0 nM triptolide treatment, respectively. VEGFA, vascular
endothelial growth factor A; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction. |
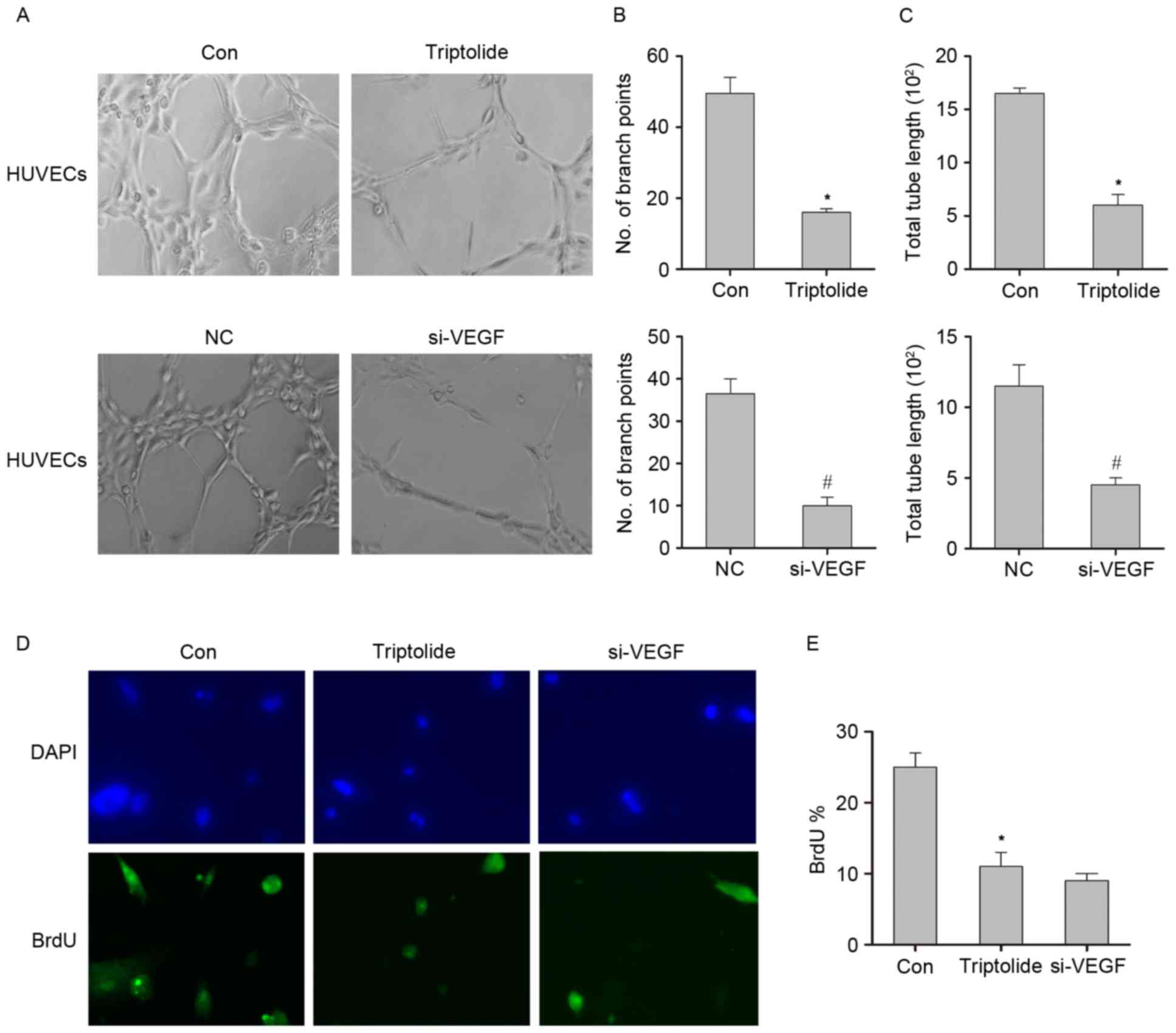
Triptolide inhibits endothelial tube
formation and cellular proliferation
To further address the impacts of triptolide in
regulating VEGFA expression, HUVECs were used to evaluate the
function of triptolide in angiogenesis. A previous study has
indicated that triptolide exhibits toxic effects on these highly
metastatic cells (18). In the
present study, HUVECs were treated with 5 nM triptolide for 48 h,
which induced less apoptosis (data not shown) than described in a
previous study (18). As shown in
Fig. 2A, the tube networks formed by
HUVECs were more extensive in the control group compared with the
triptolide group. Subsequently, siRNA targeting VEGFA was used as a
positive control to directly decrease VEGFA expression, which
revealed that the suppressive effect of triptolide on the tube
networks formed by HUVECs was comparable with that of the HUVECs
treated with the siRNA (Fig. 2A).
These results were quantified by measuring the branch points
(Fig. 2B) and total tube lengths
(Fig. 2C). The data indicated that
triptolide inhibits the tube formation of HUVECs. The effect of
triptolide on endothelial cell proliferation was measured using the
BrdU incorporation assay. Cells treated with triptolide or
VEGFA-targeting siRNA exhibited decreased BrdU incorporation
compared with the control group (Fig. 2D
and E). These data suggest that triptolide inhibits endothelial
cell proliferation and tube formation.
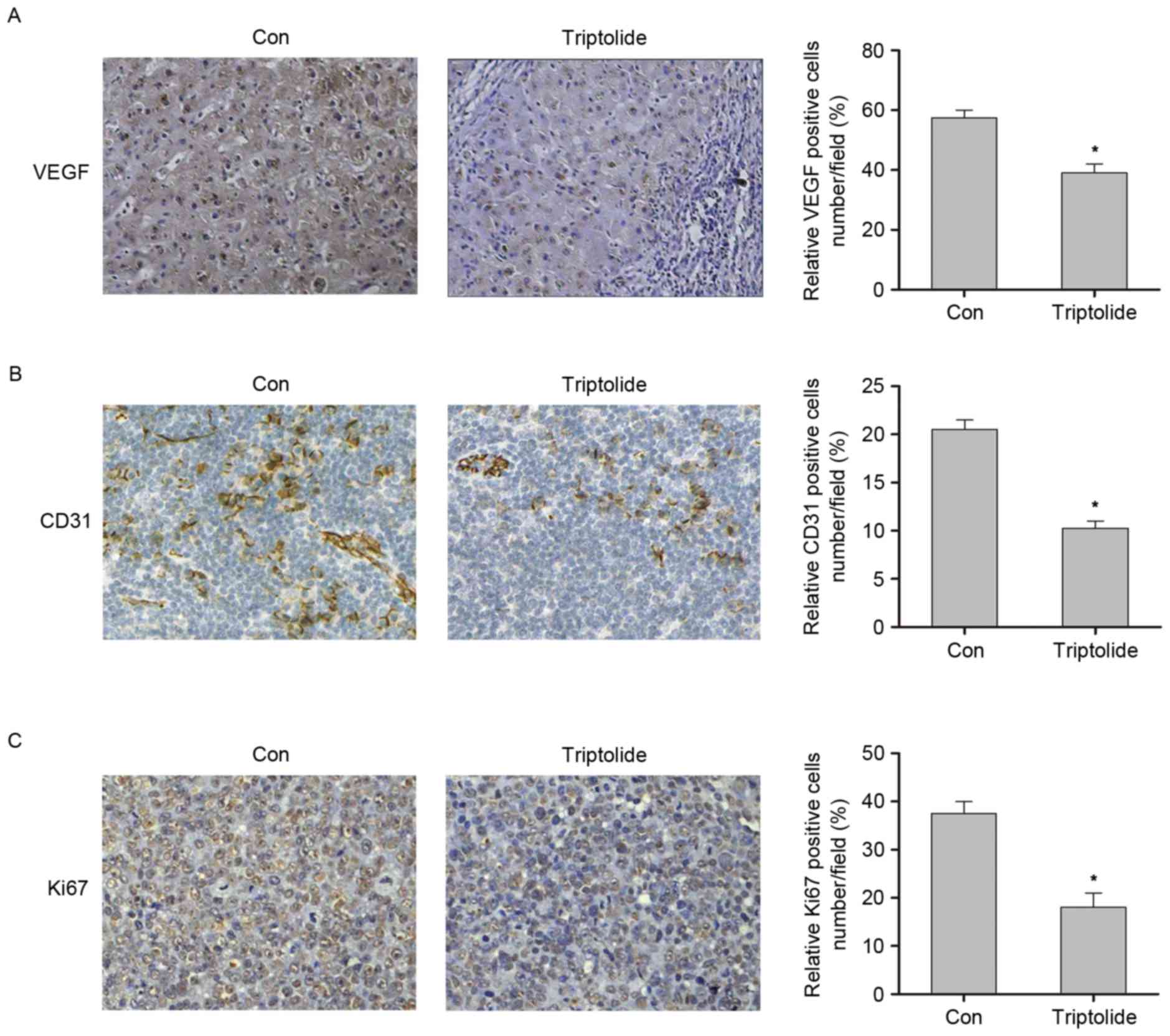
Triptolide inhibits angiogenesis
To analyze the effect of triptolide on breast cancer
angiogenesis in vivo, MDAMB231 cells were injected into the
mammary fat pads of female nude mice, which were subsequently
treated with triptolide. After 6 weeks, the mice were sacrificed
and tumor samples were extracted. Tumor microvasculature formation
in the triptolide treatment group compared with the control group
was investigated via immunohistochemistry. As shown in Fig. 3A, this revealed that the expression
of the proangiogenic factor VEGFA was significantly decreased in
the triptolide group compared with the control group (P<0.05).
Furthermore, immunohistochemical staining for CD31 revealed that
intrametastatic vasculature was significantly decreased in the
triptolide treatment group compared with the control group
(P<0.05; Fig. 3B). In addition, a
significant suppression in tumor cells proliferation was observed
in the triptolide treatment group compared with the control group,
as indicated by Ki67 staining (P<0.05; Fig. 3C). These data indicate that
triptolide inhibits tumor angiogenesis.
Triptolide regulates VEGFA expression
through suppressing HIF1-α expression by inhibiting ERK1/2
activity
To further investigate the mechanism by which
triptolide suppresses VEGFA expression, the Akt and
mitogen-activated protein kinase signaling pathways, which have
been in implicated in regulating VEGFA expression in breast cancer
tumourigenesis (19), were examined.
This demonstrated that triptolide suppressed ERK1/2
phosphorylation, while it had little effect on Akt phosphorylation
(Fig. 4A). HIF1-α expression, which
is regulated by ERK1/2, was also decreased in breast cancer cells
treated with triptolide (Fig. 4A).
As HIF1-α is key transcriptional regulator of VEGFA, knockdown of
HIF1-α significantly suppressed VEGFA expression (P<0.05;
Fig. 4B). To determine whether
HIF1-α was associated with the suppressive effect of triptolide on
VEGFA expression, MDAMB231 and Hs578T cells were treated with
triptolide followed by overexpression of HIF1-α, after which VEGFA
expression was examined. As shown in Fig. 4C, the expression of VEGFA was
significantly upregulated following triptolide treatment and HIF1-α
overexpression compared with triptolide treatment alone
(P<0.05). These data suggest that the regulation of VEGFA
expression by triptolide is partially dependent on the suppression
of HIF1-α expression by inactivation of the ERK1/2 signaling
pathway.
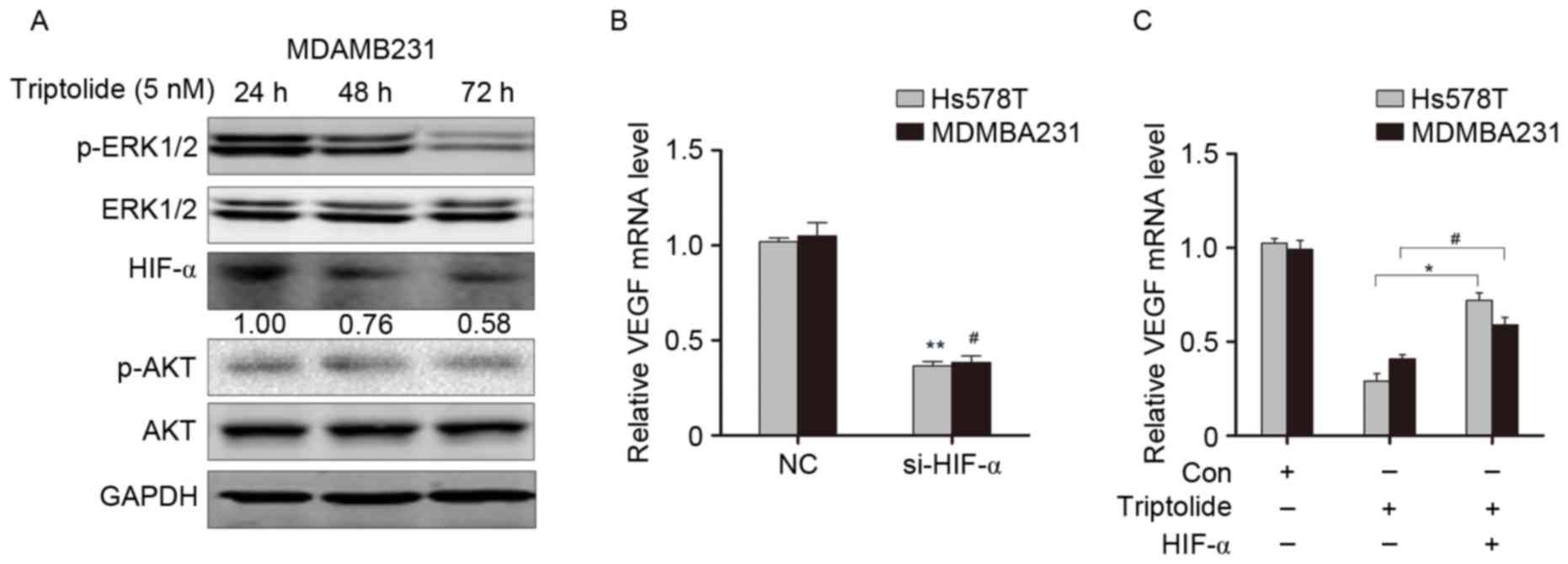 | Figure 4.Triptolide regulates VEGFA expression
through suppressing HIF1-α expression by inhibiting ERK1/2
activity. (A) Western blot analysis of ERK1/2 and Akt
phosphorylation status, and HIF1-α expression, in MDAMB231 beast
cancer cells treated with triptolide. (B) VEGFA expression was
examined by RT-qPCR after knocking down HIF1-α expression. (C)
VEGFA expression was examined by RT-qPCR after overexpression of
HIF1-α and triptolide treatment. *P<0.05, **P<0.01 vs. Hs578T
cells at 24 h; #P<0.05 vs. MDAMB231 cells at 24 h.
VEGFA, vascular endothelial growth factor A; ERK1/2, extracellular
signal-related kinase 1/2; HIF-1α, hypoxia inducible factor 1-α;
Akt, RAC-α serine/threonine-protein kinase; Con, control;
si-HIF-1α, small interfering RNA targeting HIF-1α; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction. |
Discussion
It has been reported that triptolide has multiple
functions, including immunosuppression and anti-inflammation
(20–22). Recently, the in vivo
inhibition of interleukin (IL)-1β by triptolide was examined in a
mouse model of ulcerative colitis (23). In addition, triptolide inhibits the
growth and metastasis of solid tumors, including melanoma, bladder
cancer and gastric cancer (24).
Furthermore, a recent study identified that triptolide induced
breast cancer cell apoptosis and had toxic effects on tumor stem
cells by regulating the Wnt/β-catenin signaling pathway (18). Thus, the present study aimed to
further investigate the function of triptolide on tumor
angiogenesis.
Hs578T and MDAMB231 breast cancer cells were used to
examine the functions of triptolide due to their relatively high
expression of VEGFA. Notably, it has been reported that Hs578T and
MDAMB231 cells are more sensitive to epidermal growth factor (EGF)
stimulation (25). The invasive
abilities of Hs578T and MDAMB231 cells were increased after EGF
treatment compared with MDAMB453 and T47D cells (25). Furthermore, in the present study
triptolide was demonstrated to suppress VEGFA expression in a dose-
and time-dependent manner. In addition, the results of the tube
formation assay revealed that triptolide inhibited HUVEC
proliferation and migration. These results led to the investigation
of the effect of triptolide in breast cancer in vivo. In the
MDAMB231 cell xenograft transplantation mouse model, triptolide
inhibited VEGFA expression, as demonstrated by
immunohistochemistry. Ki67 and CD31 staining of the tumor sections
also suggested decreased cell proliferation and angiogenesis.
The results of western blot analysis in the present
study indicate that triptolide suppresses ERK1/2 activation and
inhibits HIF1-α expression in breast cancer cells. Triptolide has
been demonstrated to regulate several key signaling pathways in
tumor progression, including the
phosphatidylinositol-4,5-bisphosphate
3-kinase/Akt/Serine/threonine-protein kinase mTOR, tumor necrosis
factor (TNF)-related apoptosis-inducing ligand and nuclear factor
(NF)-κB signaling pathways (26,27). The
present study examined Akt phosphorylation following triptolide
treatment and identified a slight decrease in p-Akt, although this
decrease was not significant.
Previous studies have indicated that triptolide can
inhibit the expression of inflammation-associated genes, including
IL-6, TNF-α, and the adhesion molecules intercellular adhesion
molecule 1 and P-selectin, in HUVECs treated with
lipopolysaccharide (LPS) (28).
However, the expression of HIF1-α was increased when LPS and
triptolide were combined (28).
These observations may be due to the fact that the tumor cells used
were transformed epithelial cells that already expressed a higher
level of inflammatory factors and HIF1-α compared with endothelial
cells. Other studies have demonstrated that LPS can induce the
epithelial-mesenchymal transition of tumor cells through regulation
of the NF-κB signaling pathway, and that triptolide reverses this
effect (29–31). In the present study, an siRNA pool
was used to knockdown HIF1-α, which confirmed that VEGFA expression
was decreased (32,33). Then, HIF1-α was overexpressed and the
cells were treated with triptolide. Consistent with the hypothesis
that triptolide regulates VEGFA expression through suppressing
HIF1-α expression by inhibiting ERK1/2 activity, the overexpression
of HIF1-α upregulated VEGFA expression, even with triptolide
treatment.
In conclusion, the results of the present study
indicate that triptolide inhibits breast cancer cell proliferation
and angiogenesis through regulating the ERK1/2-HIF1-α-VEGFA axis.
Thus, triptolide is a potential therapeutic strategy for the
treatment of breast cancer that required further study.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Endogenous Hormones and Breast Cancer
Collaborative Group; Key TJ, Appleby PN, Reeves GK, Travis RC,
Alberg AJ, Barricarte A, Berrino F, Krogh V, Sieri S, et al: Sex
hormones and risk of breast cancer in premenopausal women: A
collaborative reanalysis of individual participant data from seven
prospective studies. Lancet Oncol. 14:1009–1019. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Couch FJ, Hart SN, Sharma P, Toland AE,
Wang X, Miron P, Olson JE, Godwin AK, Pankratz VS, Olswold C, et
al: Inherited mutations in 17 breast cancer susceptibility genes
among a large triple-negative breast cancer cohort unselected for
family history of breast cancer. J Clin Oncol. 33:304–311. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Deryugina EI and Quigley JP: Tumor
angiogenesis: MMP-mediated induction of intravasation- and
metastasis-sustaining neovasculature. Matrix Biol. 44–46:94–112.
2015. View Article : Google Scholar
|
|
5
|
Richert MM, Vaidya KS, Mills CN, Wong D,
Korz W, Hurst DR and Welch DR: Inhibition of CXCR4 by CTCE-9908
inhibits breast cancer metastasis to lung and bone. Oncol Rep.
21:761–767. 2009.PubMed/NCBI
|
|
6
|
Bono F, de Smet F, Herbert C, de Bock K,
Georgiadou M, Fons P, Tjwa M, Alcouffe C, Ny A, Bianciotto M, et
al: Inhibition of tumor angiogenesis and growth by a small-molecule
multi-FGF receptor blocker with allosteric properties. Cancer Cell.
23:477–488. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Seftor RE, Hess AR, Seftor EA, Kirschmann
DA, Hardy KM, Margaryan NV and Hendrix MJ: Tumor cell vasculogenic
mimicry: From controversy to therapeutic promise. Am J Pathol.
181:1115–1125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou J, Xi C, Wang W, Fu X, Jinqiang L,
Qiu Y, Jin J, Xu J and Huang Z: Triptolide-induced oxidative stress
involved with Nrf2 contribute to cardiomyocyte apoptosis through
mitochondrial dependent pathways. Toxicol Lett. 230:454–466. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu J, Jiang Z, Xiao J, Zhang Y, Lin S,
Duan W, Yao J, Liu C, Huang X, Wang T, et al: Effects of triptolide
from Tripterygium wilfordii on ERalpha and p53 expression in two
human breast cancer cell lines. Phytomedicine. 16:1006–1013. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pigneux A, Mahon FX, Uhalde M, Jeanneteau
M, Lacombe F, Milpied N, Reiffers J and Belloc F: Triptolide
cooperates with chemotherapy to induce apoptosis in acute myeloid
leukemia cells. Exp Hematol. 36:1648–1659. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Park SW and Kim YI: Triptolide induces
apoptosis of PMA-treated THP-1 cells through activation of
caspases, inhibition of NF-κB and activation of MAPKs. Int J Oncol.
43:1169–1175. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen F, Gao X and Shilatifard A: Stably
paused genes revealed through inhibition of transcription
initiation by the TFIIH inhibitor triptolide. Genes Dev. 29:39–47.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Owa C, Messina ME Jr and Halaby R:
Triptolide induces lysosomal-mediated programmed cell death in
MCF-7 breast cancer cells. Int J Womens Health. 5:557–569.
2013.PubMed/NCBI
|
|
14
|
Hu C, Huang F, Deng G, Nie W, Huang W and
Zeng X: miR-31 promotes oncogenesis in intrahepatic
cholangiocarcinoma cells via the direct suppression of RASA1. Exp
Ther Med. 6:1265–1270. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li J, Zhao X, Wang D, He W, Zhang S, Cao
W, Huang Y, Wang L, Zhou S and Luo K: Up-regulated expression of
phospholipase C, β1 is associated with tumor cell proliferation and
poor prognosis in hepatocellular carcinoma. Onco Targets Ther.
9:1697–1706. 2016.PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kong W, He L, Richards EJ, Challa S, Xu
CX, Permuth-Wey J, Lancaster JM, Coppola D, Sellers TA, Djeu JY and
Cheng JQ: Upregulation of miRNA-155 promotes tumour angiogenesis by
targeting VHL and is associated with poor prognosis and
triple-negative breast cancer. Oncogene. 33:679–689. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shao H, Ma J, Guo T and Hu R: Triptolide
induces apoptosis of breast cancer cells via a mechanism associated
with the Wnt/β-catenin signaling pathway. Exp Ther Med. 8:505–508.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jiang BH and Liu LZ: PI3K/PTEN signaling
in angiogenesis and tumorigenesis. Adv Cancer Res. 102:19–65. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Qiu D, Zhao G, Aoki Y, Shi L, Uyei A,
Nazarian S, Ng JC and Kao PN: Immunosuppressant PG490 (triptolide)
inhibits T-cell interleukin-2 expression at the level of
purine-box/nuclear factor of activated T-cells and NF-kappaB
transcriptional activation. J Biol Chem. 274:13443–13450. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen X, Murakami T, Oppenheim JJ and
Howard OM: Triptolide, a constituent of immunosuppressive Chinese
herbal medicine, is a potent suppressor of dendritic-cell
maturation and trafficking. Blood. 106:2409–2416. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang X, Zhang L, Duan W, Liu B, Gong P,
Ding Y and Wu X: Anti-inflammatory effects of triptolide by
inhibiting the NF-κB signalling pathway in LPS-induced acute lung
injury in a murine model. Mol Med Rep. 10:447–452. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang H, Gong C, Qu L, Ding X, Cao W, Chen
H, Zhang B and Zhou G: Therapeutic effects of triptolide via the
inhibition of IL-1β expression in a mouse model of ulcerative
colitis. Exp Ther Med. 12:1279–1286. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang S, Chen J, Guo Z, Xu XM, Wang L, Pei
XF, Yang J, Underhill CB and Zhang L: Triptolide inhibits the
growth and metastasis of solid tumors. Mol Cancer Ther. 2:65–72.
2003.PubMed/NCBI
|
|
25
|
Koh MS and Moon A: Activation of H-Ras and
Rac1 correlates with epidermal growth factor-induced invasion in
Hs578T and MDA-MB-231 breast carcinoma cells. Biochem Biophys Res
Commun. 406:25–29. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim SH, Kang JG, Kim CS, Ihm SH, Choi MG,
Yoo HJ and Lee SJ: Synergistic cytotoxicity of BIIB021 with
triptolide through suppression of PI3K/Akt/mTOR and NF-κB signal
pathways in thyroid carcinoma cells. Biomed Pharmacother. 83:22–32.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao X, Zhang Q and Chen L: Triptolide
induces the cell apoptosis of osteosarcoma cells through the TRAIL
pathway. Oncol Rep. 36:1499–1505. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Galvez G, Santander S, Raskin I and
Baldeon M: Effects of triptolide on the expression of inflammatory
markers in lipopolysaccharide-treated human endothelial cells
(HUVEC). FASEB J. 29:789–781. 2015.
|
|
29
|
Liu L, Salnikov AV, Bauer N,
Aleksandrowicz E, Labsch S, Nwaeburu C, Mattern J, Gladkich J,
Schemmer P, Werner J and Herr I: Triptolide reverses
hypoxia-induced epithelial-mesenchymal transition and stem-like
features in pancreatic cancer by NF-κB downregulation. Int J
Cancer. 134:2489–2503. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sarkar TR, Battula VL, Werden SJ, Vijay
GV, Ramirez-Peña EQ, Taube JH, Chang JT, Miura N, Porter W, Sphyris
N, et al: GD3 synthase regulates epithelial-mesenchymal transition
and metastasis in breast cancer. Oncogene. 34:2958–2967. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang T, Chen Z and Fang L: Curcumin
inhibits LPS-induced EMT through downregulation of NF-κB-Snail
signaling in breast cancer cells. Oncol Rep. 29:117–124. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ahluwalia A and Tarnawski AS: Critical
role of hypoxia sensor-HIF-1α in VEGF gene activation. Implications
for angiogenesis and tissue injury healing. Curr Med Chem.
19:90–97. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ke Q and Costa M: Hypoxia-inducible
factor-1 (HIF-1). Mol Pharmacol. 70:1469–1480. 2006. View Article : Google Scholar : PubMed/NCBI
|