Introduction
Osteoarthritis (OA), the most common type of
arthritis, reduces the quality of life of affected individuals and
is most common in the population aged >65 years (1). Although previous studies made efforts
to develop therapeutic methods and explore the underlying
mechanisms of OA, they remain far from being completely elucidated;
however, it has been revealed that inflammation is involved in the
development and progression of OA (2). Protein-protein interaction (PPI)
network analysis is a powerful tool to explore the pathological
mechanisms of human diseases (3).
Furthermore, microarray analysis has been widely applied to monitor
gene expression in OA. Thus, combining PPI and public microarray
data will help to identify novel pathways regulating OA
progression.
Long non-coding RNAs (lncRNAs) are a major class of
ncRNAs with a length of >200 nucleotides (4). lncRNAs have important roles in
regulating a vast variety of biological processes at the
transcriptional, post-transcriptional and post-translational level.
Recently, several studies have indicated that lncRNAs may act as
contributors to OA. Certain lncRNAs, including ubiquitin-fold
modifier conjugating enzyme 1 (UFC1) (5), thymosin-β-4 (6), growth arrest-specific 5 (GAS5)
(7) act as natural microRNA sponges
in human OA. A well-known lncRNA, HOX transcript antisense
intergenic RNA (HOTAIR) (8), was
reported to participate in interleukin-1β-induced matrix
metalloproteinase overexpression and chondrocyte apoptosis in
temporomandibular joint OA. However, the functional roles of most
lncRNAs in OA have remained largely elusive.
In the present study, two public datasets were
analyzed to identify differentially expressed lncRNAs and mRNAs in
OA. Next, gene ontology (GO) and Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathway analysis were performed to explore the
potential roles of differentially expressed genes (DEGs). A series
of lncRNAs that were differently expressed in OA were identified.
To provide novel information on the molecular mechanisms and
functional roles of lncRNAs in OA, a co-expression analysis was
also performed. The present study provided useful information for
exploring potential candidate biomarkers for the diagnosis and
prognosis of RA, as well as drug targets.
Materials and methods
Microarray data and data
pre-processing
The microarray datasets GSE48556 (9) and GSE82107 (10) were downloaded from the National
Centre for Biotechnology Information (NCBI) Gene Expression Omnibus
database (http://www.ncbi.nlm.nih.gov/geo/). A total of 139
samples were included in GSE48556. Of these, 106 biopsies were
obtained from OA patients and 33 biopsies were obtained from
healthy controls. A total of 17 samples (10 from OA patients and 7
from healthy controls) were included in the GSE82107 dataset. mRNAs
with fold changes ≥2 and P<0.05 were considered to be
significantly differentially expressed.
GO and KEGG pathway analysis
To identify the functions of DEGs in OA, a GO
function enrichment analysis was performed. KEGG pathway enrichment
analysis was also performed to identify pathways enriched in OA
using the Molecule Annotation System (http://bioinfo.capitalbio.com/mas3/). The P-value was
calculated by hypergeometric distribution and a pathway with
P<0.05 was considered as significant.
LncRNA classification pipeline
LncRNA expression patterns were assessed in the
microarray dataset GSE82107. The following criteria were used to
identify the unique probe sets for lncRNAs from the Affymetrix
array. Refseq IDs labelled as ‘NR_’ which is indicative of ncRNA in
the Refseq database, were retained (11). Finally, 2,448 annotated lncRNA
transcripts with corresponding Affymetrix probe IDs were obtained.
lncRNAs with fold changes ≥2 and P<0.05 were selected as
significantly differentially expressed lncRNAs.
Identification of lncRNA-associated
PPI modules
The Search Tool for the Retrieval of Interacting
Genes/ Proteins (STRING) online software (https://string-db.org) was used to assess the
interactions. The interactions of the proteins encoded by the
differentially expressed genes were searched using STRING online
software, and the combined score of >0.4 was used as the cut-off
criterion. The PPI network was visualized using Cytoscape software
(http://www.cytoscape.org).
Co-expression network construction and
analysis
In the present study, the Pearson correlation
coefficient of DEG-lncRNA pairs was calculated according to their
expression values. The co-expressed DEG-lncRNA pairs with an
absolute value of the Pearson correlation coefficient of ≥0.8 were
selected and the co-expression network was established by using
Cytoscape software. The Cytoscape MCODE plug-in (version 3.4.0) was
applied to search for clustered sub-networks of highly connected
nodes from the co-expression network. The resulting network was
subjected to module analyses with the Plugin MCODE with the
following default parameters: Degree cut-off, ≥3; and nodes with
edges, ≥3-core).
Statistical analysis
Numerical data are presented as the mean ± standard
deviation of at least three determinations. Statistical comparisons
between groups of normalized data were performed using the t-test
or Mann-Whitney U-test according to the test conditions. P<0.05
was considered to indicate a statistically significant difference
with a 95% confidence level. R (version 3.2.4; http://www.r-project.org/) was used to perform all
analyses.
Results
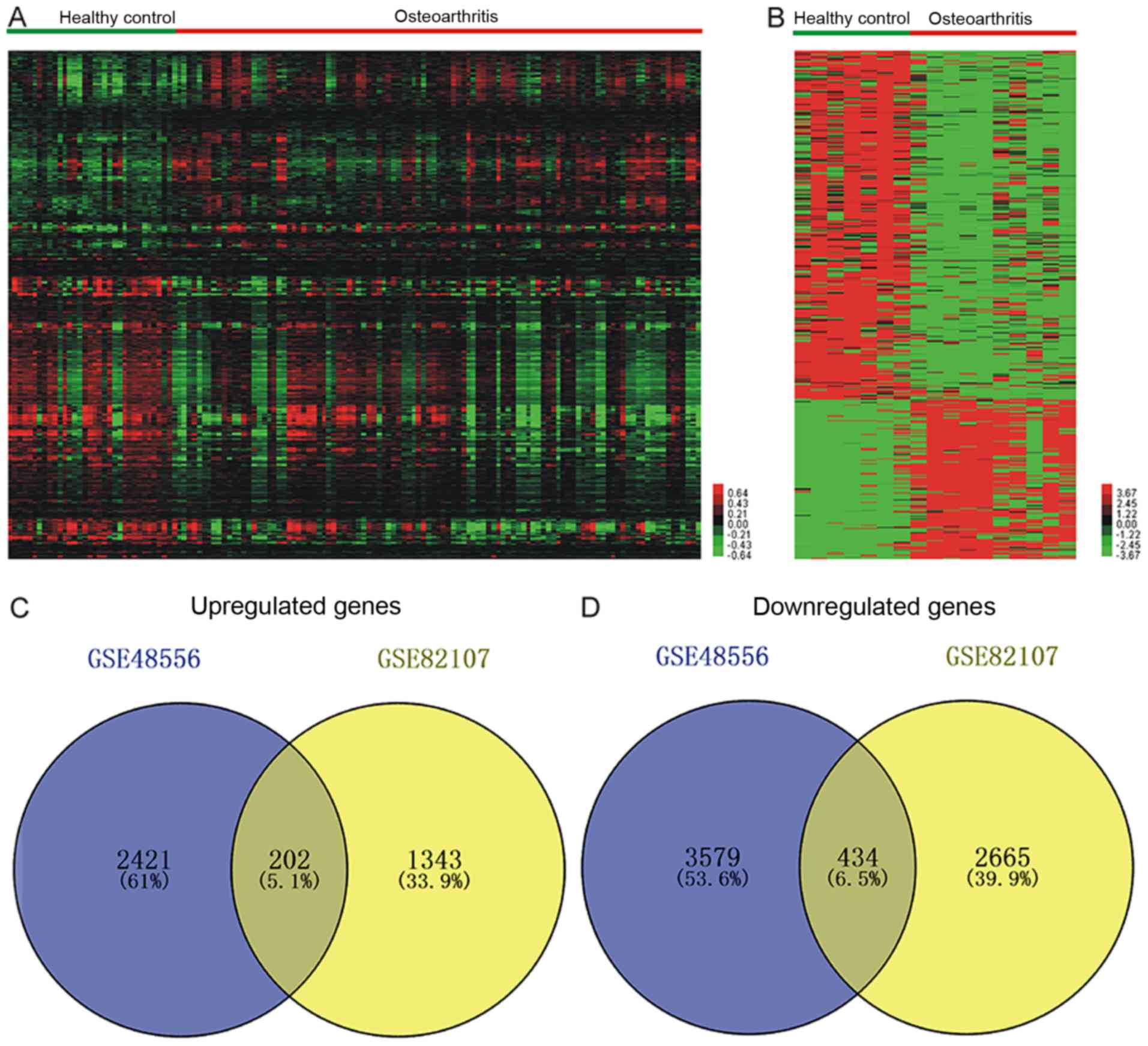
Identification of DEGs in OA
To identify the significantly differentially
expressed mRNAs between OA patients vs. healthy controls, two
publicly available gene expression datasets, GSE48556 and GSE82107,
were analyzed. A total of 2,623 up- and 4,013 downregulated mRNAs
were identified in GSE48556 (Fig.
1A), and from the GSE82107 dataset, 1,545 up- and 3,099
downregulated mRNAs were obtained (Fig.
1B). Integrated analysis of these two datasets revealed that
the two datasets had 202 up- and 434 downregulated mRNAs in common
(Fig 1C and D). The top 10 up- and
downregulated mRNAs are listed in Table
I.
 | Table I.Top 10 up- and downregulated mRNAs in
the GSE82107 dataset. |
Table I.
Top 10 up- and downregulated mRNAs in
the GSE82107 dataset.
| Gene symbol | P-value | Ave (control) | Ave (OA) |
|---|
| Downregulated
genes |
|
|
|
| GPD1 | 0.04825352 | 104.04 | 11.85 |
| MYF6 | 0.046950299 | 529.61 | 60.34 |
|
PCDH15 | 0.018945043 | 7.96 | 0.93 |
|
KIAA1257 | 0.00647357 | 12.73 | 1.49 |
| ARG1 | 0.03656324 | 10.41 | 1.26 |
|
LOC101927734 | 0.035854747 | 26.83 | 3.45 |
|
LOC101929609 | 0.0011542 | 13.47 | 1.88 |
| SHC3 | 0.001053533 | 28.54 | 4.02 |
|
KIAA1661 | 0.035300865 | 11.66 | 1.69 |
|
FBXL21 | 0.011937398 | 9.66 | 1.41 |
| Upregulated
genes |
|
|
|
|
CYR61 | 0.030911933 | 307.24 | 1482.31 |
| DLX1 | 0.031583337 | 1.67 | 8.47 |
|
LRRC15 | 0.049914446 | 145.54 | 747.21 |
|
SOCS3 | 0.014017918 | 159.34 | 850.29 |
| VMO1 | 0.022699188 | 4.83 | 25.96 |
| SELO | 0.001439415 | 2.16 |
11.88 |
|
HORMAD1 | 0.025736549 | 1.53 | 8.46 |
|
RARRES1 | 0.045620106 | 14.71 | 108.20 |
|
STMN2 | 0.020694784 | 5.23 | 39.04 |
| GJB2 | 0.02794137 | 99.03 | 746.29 |
GO and KEGG analysis of differentially
expressed mRNAs
Next, the differentially expressed mRNAs were
subjected to GO and KEGG analyses (Fig.
2). GO analysis indicated that the upregulated genes were
mainly involved in regulating antigen processing and presentation,
interspecies interaction between organisms, immune response,
transcription, protein transport, vesicle-mediated transport,
ubiquitin-dependent protein catabolism, endocytosis, cell adhesion
and apoptosis (Fig. 2A).
Furthermore, the downregulated genes were mainly enriched in
categories associated with transcription, signal transduction,
oxidation/reduction, cell adhesion, ion transport, cell cycle,
protein amino acid phosphorylation, development,
modification-dependent protein catabolism and inflammatory response
(Fig. 2B). The above pathways may
therefore participate in regulating the progression of OA.
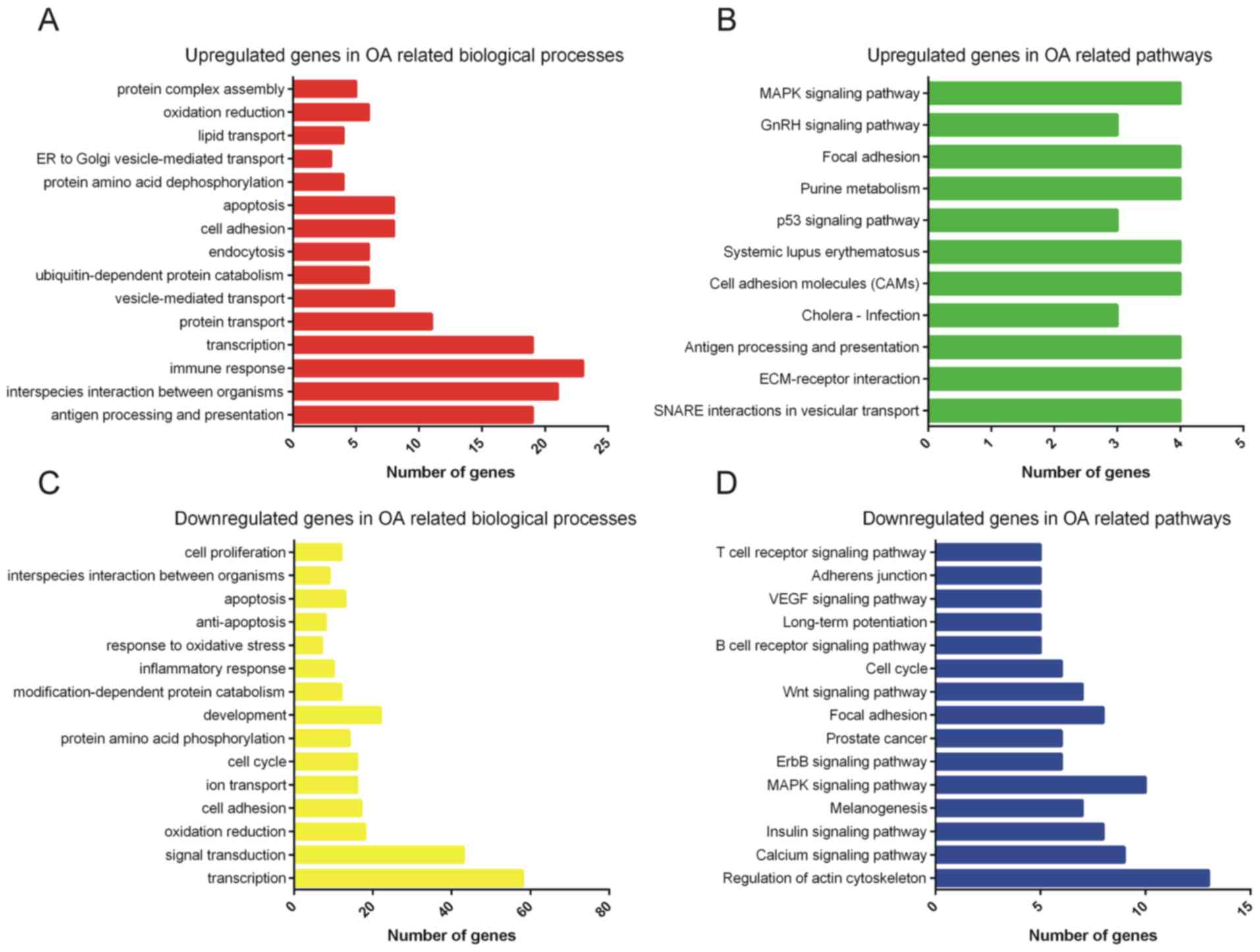 | Figure 2.GO and KEGG pathway analysis of
differentially expressed mRNAs. (A) GO analysis and (B) KEGG
analysis of upregulated mRNAs in OA-associated biological processes
and pathways, respectively. (C) GO analysis and (D) KEGG analysis
of downregulated mRNAs in OA-associated biological processes and
pathways, respectively. GO, gene ontology; KEGG, Kyoto Encyclopedia
of Genes and Genomes; OA, osteoarthritis; ER, endoplasmic
reticulum; ECM, extracellular matrix; VEGF, vascular endothelial
growth factor; MAPK, mitogen-activated protein kinase; GnRH,
gonadotropin-releasing hormone; SNARE, soluble NSF attachment
protein receptor. |
KEGG pathway analysis revealed that upregulated
genes were primarily enriched in pathways associated with the p53,
gonadotropin-releasing hormone and mitogen-associated protein
kinase (MAPK) signaling pathways (Fig.
2C). Downregulated genes were mainly associated with calcium,
insulin, MAPK, ErbB, Wnt, B-cell receptor signaling pathway and
vascular endothelial growth factor (VEGF) signaling pathways
(Fig. 2D).
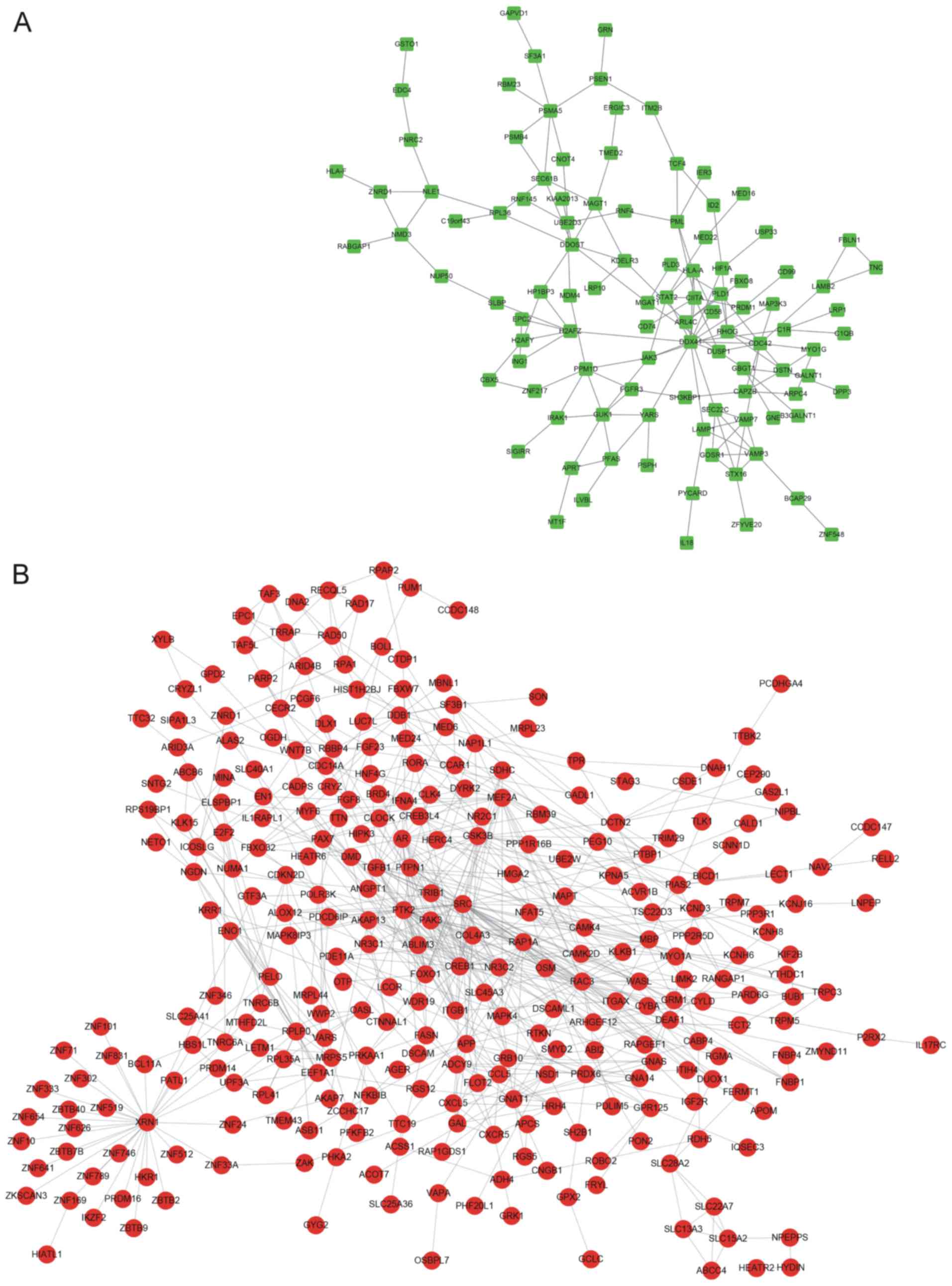
PPI network construction
The PPI networks constructed for the up- and
downregulated genes are presented in Fig. 3. The PPI network for the upregulated
genes contained 78 nodes and 158 edges, and the hub nodes with the
highest connectivity degree were DEAD box helicase 41 (DDX41;
connectivity degree=13) and cell division cycle 41 (CDC42;
connectivity degree=6; Fig. 3A). The
PPI network for the downregulated genes contained 219 nodes and 656
edges, and the hub nodes with the highest connectivity degree were
cyclic AMP responsive element binding protein 1 (CREB; connectivity
degree=28), SH2 domain containing (SRC; connectivity degree=27) and
androgen receptor (AR; d connectivity degree=18; Fig. 3B).
Furthermore, a module analysis of the network was
performed using the MCODE plugin (degree cut-off, ≥3; the nodes
with edges, ≥3-core). In the respective PPI networks, a total of 5
up-regulated proteins, including golgi SNAP receptor complex member
1 (GOSR1), SEC22 homolog C vesicle trafficking protein (SEC22C),
vesicle associated membrane protein 7 (VAMP7), syntaxin 16 (STX16)
and vesicle associated membrane protein 3 (VAMP3; Fig. 4A) and 12 down-regulated proteins
including, C-C motif chemokine ligand 5 (CCL5), galanin and GMAP
prepropeptide (GAL), C-X-C motif chemokine ligand 5 (CXCL5),
amyloid beta precursor protein (APP), adenylate cyclase 9 (ADCY9),
histamine receptor H4 (HRH4), C-X-C motif chemokine receptor 5
(CXCR5), replication protein A1 (RPA1), RAD50 double strand break
repair protein (RAD50), DNA replication helicase/nuclease 2 (DNA2),
RAD17 checkpoint clamp loader component (RAD17) and RecQ like
helicase 5 (RECQL5; Fig. 4B and C)
were identified as key proteins/genes.
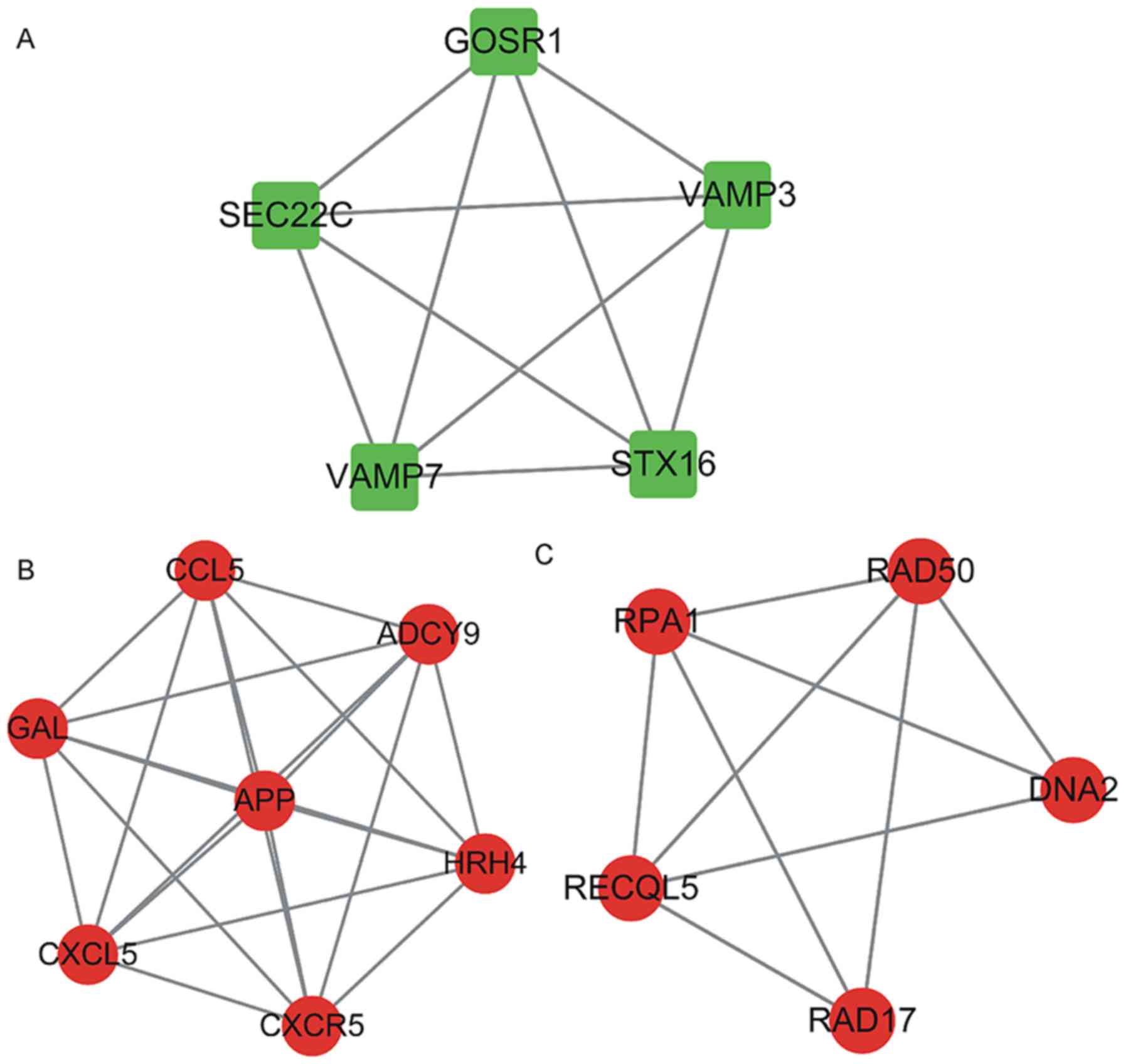 | Figure 4.Hub modules from the PPI network in
osteoarthritis patients were constructed. (A) A total of five
upregulated proteins constituted a hub PPI module: GOSR1, SEC22C,
VAMP7, STX16) and VAMP3. (B and C) Twelve downregulated proteins
constituted 2 hub PPI modules: CCL5, GAL, CXCL5, APP, ADCY9, HRH4,
CXCR5, RPA1, RAD50, DNA2, RAD17 and RECQL5. PPI, protein-protein
interaction. GOSR1, golgi SNAP receptor complex member 1; SEC22C,
SEC22 homolog C vesicle trafficking protein; VAMP7, vesicle
associated membrane protein 7; STX16, syntaxin 16; VAMP3, vesicle
associated membrane protein 3; CCL5, C-C motif chemokine ligand 5;
GAL, galanin and GMAP prepropeptide; CXCL5, C-X-C motif chemokine
ligand 5; APP, amyloid beta precursor protein; ADCY9, adenylate
cyclase 9; HRH4, histamine receptor H4; CXCR5, C-X-C motif
chemokine receptor 5; RPA1, replication protein A1; RAD50, RAD50
double strand break repair protein; DNA2, DNA replication
helicase/nuclease 2; RAD17, RAD17 checkpoint clamp loader
component; RECQL5, RecQ like helicase 5. |
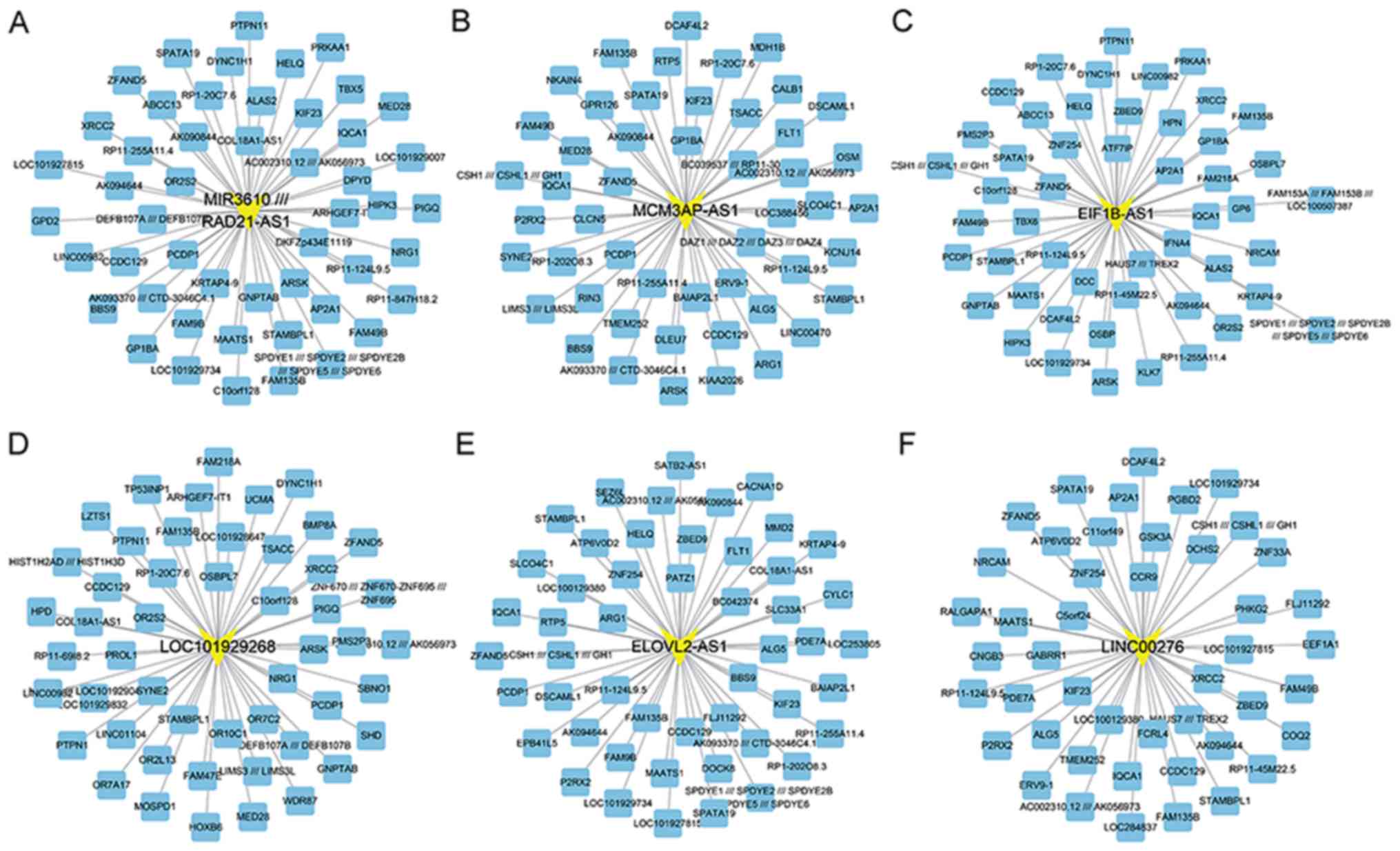
Co-expression network analysis of
differently expressed lncRNAs in OA
Furthermore, the differently expressed lncRNAs were
identified from the GSE82107 dataset. A total of 25 up- and 326
downregulated lncRNAs were identified in OA. The top 10 up- and
down-regulated lncRNAs are presented in Table II. To predict the potential
functional roles of these lncRNAs, the Pearson correlation
coefficient for lncRNA-DEG pairs was first calculated according to
their expression value. The co-expressed DEG-lncRNA pairs with an
absolute value of their Pearson correlation coefficient of ≥0.8
were selected. As presented in Fig.
5A, the network included 348 differentially expressed lncRNAs
and 2,883 DEGs (Fig. 5A).
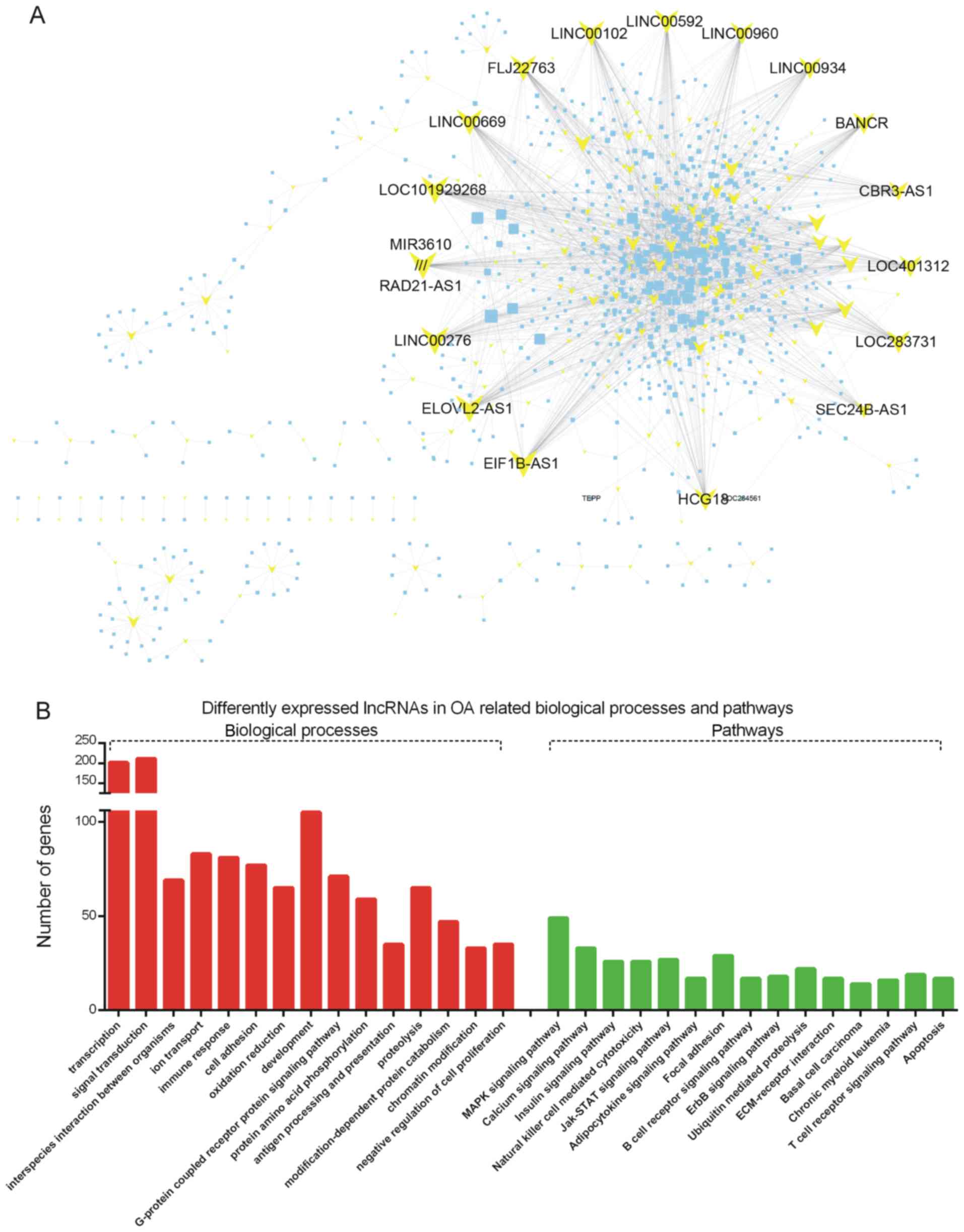 | Figure 5.(A) Differentially expressed
gene-lncRNA co-expression pairs with an absolute value of the
Pearson correlation coefficient of ≥0.9 were selected. (B) Gene
ontology and Kyoto Encyclopedia of Genes and Genomes pathway
analysis of lncRNAs based on the set of co-expressed mRNAs. OA,
osteoarthritis; lncRNA, long non-coding RNA; ER, endoplasmic
reticulum; ECM, extracellular matrix; MAPK, mitogen-activated
protein kinase; Jak, Janus kinase; STAT, signal transducer and
activator of transcription; LINC00592, long intergenic non-protein
coding RNA 592; LINC00960, long intergenic non-protein coding RNA
960; LINC00934, long intergenic non-protein coding RNA 934; BANCR,
BRAF-activated non-protein coding RNA; CBR3-AS1, CBR3 antisense RNA
1; SEC24B-AS1, SEC24B antisense RNA 1; HCG18, HLA complex group 18
(non-protein coding); EIF1B-AS1, EIF1B antisense RNA 1; ELOVL2-AS1,
ELOVL2 antisense RNA 1; LINC00276, long intergenic non-protein
coding RNA 276; RAD21-AS1, RAD21 antisense RNA 1; MIR3610, microRNA
3610; LINC00669, MIR924 host gene; FLJ2276, chromosome 3 open
reading frame 85; LINC00102, long intergenic non-protein coding RNA
102. |
| Table II.Top 10 up- and downregulated lncRNAs
in the GSE82107 dataset. |
Table II.
Top 10 up- and downregulated lncRNAs
in the GSE82107 dataset.
| Gene symbol | P-value | Ave (control) | Ave (OA) |
|---|
| Downregulated
lncRNA |
|
|
|
|
LEMD1-AS1 |
2.68×10−3 | 7.39 | 1.06 |
|
NAV2-AS5 |
1.28×10−2 | 9.59 | 1.4 |
|
LOC101928107 |
4.46×10−2 | 6.70 | 1.07 |
|
LOC286083 |
7.45×10−3 | 10.49 | 1.82 |
|
LOC340107 |
3.90×10−3 | 9.66 | 1.72 |
|
GRIK1-AS2 |
3.08×10−3 | 13.34 | 2.39 |
|
LINC00551 |
1.88×10−2 | 5.94 | 1.07 |
|
LOC401134 |
1.19×10−2 | 9.70 | 1.81 |
|
SNORA74A |
2.98×10−2 | 7.26 | 1.41 |
|
CBR3-AS1 |
5.04×10−3 | 14.81 | 2.89 |
| Upregulated
lncRNA |
|
|
|
|
FLJ32255 |
5.84×10−4 | 21.84 | 46.97 |
|
LINC01094 |
2.11×10−2 | 48.00 | 114.5 |
|
NPHP3-AS1 |
2.75×10−2 | 5.61 | 13.8 |
|
LINC00957 |
9.84×10−3 | 32.21 | 86.68 |
|
LINC00963 |
3.02×10−2 | 3.30 | 9.47 |
|
FLJ27354 |
3.85×10−3 | 4.54 | 14.76 |
|
LOC101927720 |
1.08×10−2 | 4.77 | 15.67 |
|
SIGLEC16 |
1.09×10−2 | 6.30 | 20.8 |
|
LOC100506119 |
4.60×10−2 | 4.43 | 16.33 |
|
LOC101060091 |
1.75×10−2 | 1.56 | 7.68 |
To predict the functions of the differentially
expressed lncRNAs, GO and KEGG pathway analysis was performed for
each given lncRNA by using the respective set of co-expressed
mRNAs. According to the KEGG pathway analysis, the dysregulated
lncRNAs were primarily enriched in pathways associated with the
MAPK, calcium and insulin pathways, natural killer cell-mediated
cytotoxicity, and with the Janus kinase/signal transducer and
activator of transcription, adipocytokine, B-cell receptor and ErbB
signaling pathways (Fig. 5B). GO
analysis revealed that the dysregulated lncRNAs were associated
with transcription, signal transduction, interspecies interaction
between organisms, ion transport, immune response, cell adhesion,
oxidation/reduction, development, G-protein-coupled receptor
protein signaling pathway and protein amino acid phosphorylation
(Fig. 5B).
Identification of key lncRNAs in
OA
In the present study, a total of lncRNAs, including
EIF1B antisense RNA 1 (EIF1B-AS1), ELOVL2 antisense RNA 1
(ELOVL2-AS1), long intergenic non-protein coding RNA 276
(LINC00276), LOC101929268, MCM3AP antisense RNA 1 (MCM3AP-AS1) and
RAD21 antisense RNA 1 (RAD21-AS1) were identified to be
co-expressed with an absolute value of the Pearson correlation
coefficient of ≥0.8. These lncRNAs were identified as key lncRNAs
in OA. The co-expression network of each key lncRNAs is presented
in Fig. 6.
Discussion
OA has become one of the most frequent chronic
diseases, which is considered synonymous with aging (12). Elucidation of the mechanisms
underlying the development and progression of OA is urgently
required in order to identify novel biomarkers for the diagnosis
and prognosis of OA patients, and for the development of drug
targets. In the present study, the GSE48556 and GSE82107 datasets
were analyzed to identify differentially expressed mRNAs and
lncRNAs in OA patients. A total of 202 up- and 434 downregulated
mRNAs were obtained in OA. GO analysis indicated that the
upregulated genes were mainly involved in regulating antigen
processing and presentation, interspecies interaction between
organisms, immune response and transcription, while the
downregulated genes were mainly enriched in transcription, signal
transduction, oxidation reduction and cell adhesion. KEGG pathway
analysis revealed that the upregulated genes were primarily
enriched in pathways associated with the p53, GnRH and MAPK
signaling pathways, while the downregulated genes were mainly
associated with calcium, insulin, MAPK, ErbB, Wnt, B-cell receptor
and VEGF signaling pathways. The GO terms and KEGG pathways that
the dysregulated genes were enriched in provide an insight in the
pathways associated with OA.
PPI network analysis is a useful tool to explore the
pathological mechanisms of human diseases. For instance, Chen et
al (13) identified DEGs in
salivary adenoid cystic carcinoma cells. In the present study, PPI
networks for up- and downregulated genes in OA were constructed.
The PPI network for the upregulated genes contained 78 nodes and
158 edges, and the hub nodes with the highest connectivity degree
were DDX41 (degree=13) and CDC42 (degree=6). The PPI network for
the downregulated genes contained 219 nodes and 656 edges, and the
hub nodes with the highest connectivity degree were CREB1
(degree=28), SRC (degree=27) and AR (degree=18). Of note, it was
observed that the PPI for the down-regulated genes included hub
genes involved in various hormone-associated pathways, suggesting
that hormones are involved in the development and progression of
OA. Furthermore, a module analysis of the network was performed
using the MCODE plugin. A total of upregulated proteins (GOSR1,
SEC22C, VAMP7, STX16 and VAMP3) and 12 downregulated proteins
(CCL5, GAL, CXCL5, APP, ADCY9, HRH4, CXCR5, RPA1, RAD50, DNA2,
RAD17 and RECQL5) were identified as key genes in the PPI network
for the up- and downregulated genes, respectively.
Emerging studies have indicated lncRNAs have
regulatory roles in various physiological processes and are
deregulated in certain human diseases, including prostate cancer
(14), breast cancer (15), colon cancer (16,17) and
OA (5,18). A number of lncRNAs, including
ubiquitin-fold modifier conjugating enzyme 1 (5), HOX transcript antisense RNA (8), maternally expressed 3 (non-protein
coding) (19), growth arrest
specific 5 (non-protein coding) (7)
and imprinted maternally expressed transcript (non-protein coding)
(20), were reported to be
significantly associated with the progression of OA. However, the
functional roles of most lncRNAs in OA remain largely elusive. In
the present study, the lncRNA expression patterns in OA were
identified from the GSE82107 dataset. A total of 25 up- and 326
downregulated lncRNAs were identified in OA. A total of 6 lncRNAs
(EIF1B-AS1, ELOVL2-AS1, LINC00276, LOC101929268, MCM3AP-AS1 and
RAD21-AS1) were identified as key lncRNAs, based on an absolute
value of the Pearson correlation coefficient of ≥0.8. Combined with
co-expression, GO analysis and KEGG analysis, it was identified
that the dysregulated lncRNAs were primarily enriched in pathways
associated with the MAPK, calcium and insulin signaling pathways.
GO analysis revealed that the dysregulated lncRNAs were associated
with transcription, signal transduction, interspecies interaction
between organisms, ion transport, immune response, cell adhesion,
oxidation reduction, development, G-protein-coupled receptor
protein signaling pathway and protein amino acid
phosphorylation.
In conclusion, in the present study, two public
datasets were analyzed to identify differentially expressed lncRNAs
and mRNAs in OA. GO and KEGG pathway analysis indicated that DEGs
were mainly involved in regulating antigen processing and
presentation, interspecies interaction between organisms, immune
response, transcription and signal transduction. A series of
differently expressed lncRNAs in OA was also identified. To provide
novel information on the molecular mechanisms and functional roles
of lncRNAs in OA, a co-expression analysis was performed, which key
provided several key lncRNAs in OA. The present study provided
useful information to explore potential candidate biomarkers for
the diagnosis and prognosis of OA patients, and to identify novel
drug targets.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from Tongji
Hospital of Shanghai (grant nos. KPB1608 and HBRC1607) and the
Sailing Program from the Shanghai Science and Technology Commission
(grant no. 16YF1410300).
Availability of data and materials
The analyzed data sets generated during the study
are available from GEO database. The accession number of dataset
GSE48556 is GDS5363 (https://www.ncbi.nlm.nih.gov/sites/GDSbrowser?acc=GDS5363)
and GSE82107 for GSE82107 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE82107).
Authors' contributions
YQS made substantial contributions to the conception
and design of the study. LC and JCZ made substantial contributions
to the acquisition of data and drafted the manuscript. YQZ was
involved in the analysis and interpretation of data. ZTR made
substantial contributions to the analysis of data and revised the
manuscript critically for important intellectual content. All
authors read and approved the final manuscript.
Ethical approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cho HJ, Morey V, Kang JY, Kim KW and Kim
TK: Prevalence and risk factors of spine, Shoulder, Hand, Hip, and
Knee osteoarthritis in community-dwelling koreans older than age 65
years. Clin Orthop Relat Res. 473:3307–3314. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Greene MA and Loeser RF: Aging-related
inflammation in osteoarthritis. Osteoarthritis Cartilage.
23:1966–1971. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kong B, Yang T, Chen L, Kuang YQ, Gu JW,
Xia X, Cheng L and Zhang JH: Protein-protein interaction network
analysis and gene set enrichment analysis in epilepsy patients with
brain cancer. J Clin Neurosci. 21:316–319. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Imamura K and Akimitsu N: Long non-coding
RNAs involved in immune responses. Front Immunol. 5:5732014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang G, Wu Y, Xu D and Yan X: Long
noncoding RNA UFC1 promotes proliferation of chondrocyte in
osteoarthritis by acting as a sponge for miR-34a. DNA Cell Biol.
35:691–695. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu Q, Hu X, Zhang X, Dai L, Duan X, Zhou
C and Ao Y: The TMSB4 pseudogene lncRNA functions as a competing
endogenous RNA to promote cartilage degradation in human
osteoarthritis. Mol Ther. 24:1726–1733. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Song J, Ahn C, Chun CH and Jin EJ: A long
non-coding RNA, GAS5, plays a critical role in the regulation of
miR-21 during osteoarthritis. J Orthop Res. 32:1628–1635. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang C, Wang P, Jiang P, Lv Y, Dong C,
Dai X, Tan L and Wang Z: Upregulation of lncRNA HOTAIR contributes
to IL-1β-induced MMP overexpression and chondrocytes apoptosis in
temporomandibular joint osteoarthritis. Gene. 586:248–253. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang L, Ma D, Li X, Deng C, Shi Q, You X,
Leng X, Li M, Tang F, Zhang F and Li Y: Gene expression profiles of
peripheral blood mononuclear cells in primary biliary cirrhosis.
Clin Exp Med. 14:409–416. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Broeren MG, de Vries M, Bennink MB, van
Lent PL, van der Kraan PM, Koenders MI, Thurlings RM and van de Loo
FA: Functional tissue analysis reveals successful cryopreservation
of human osteoarthritic synovium. PLoS One. 11:e01670762016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang X, Sun S, Pu JK, Tsang AC, Lee D,
Man VO, Lui WM, Wong ST and Leung GK: Long non-coding RNA
expression profiles predict clinical phenotypes in glioma.
Neurobiol Dis. 48:1–8. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Laulan J, Marteau E and Bacle G: Wrist
osteoarthritis. Orthop Traumatol Surg Res. 101 Suppl 1:S1–S9. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen W, Liu BY, Zhang X, Zhao XG, Cao G,
Dong Z and Zhang SL: Identification of differentially expressed
genes in salivary adenoid cystic carcinoma cells associated with
metastasis. Arch Med Sci. 12:881–888. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wan X, Huang W, Yang S, Zhang Y, Pu H, Fu
F, Huang Y, Wu H, Li T and Li Y: Identification of
androgen-responsive lncRNAs as diagnostic and prognostic markers
for prostate cancer. Oncotarget. 7:60503–60518. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shi SJ, Wang LJ, Yu B, Li YH, Jin Y and
Bai XZ: LncRNA-ATB promotes trastuzumab resistance and
invasion-metastasis cascade in breast cancer. Oncotarget.
6:11652–11663. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yue B, Qiu S, Zhao S, Liu C, Zhang D, Yu
F, Peng Z and Yan D: LncRNA-ATB mediated E-cadherin repression
promotes the progression of colon cancer and predicts poor
prognosis. J Gastroenterol Hepatol. 31:595–603. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
He X, Tan X, Wang X, Jin H, Liu L, Ma L,
Yu H and Fan Z: C-Myc-activated long noncoding RNA CCAT1 promotes
colon cancer cell proliferation and invasion. Tumour Biol.
35:12181–12188. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fu M, Huang G, Zhang Z, Liu J, Zhang Z,
Huang Z, Yu B and Meng F: Expression profile of long noncoding RNAs
in cartilage from knee osteoarthritis patients. Osteoarthritis
Cartilage. 23:423–432. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Su W, Xie W, Shang Q and Su B: The long
noncoding RNA MEG3 is downregulated and inversely associated with
vegf levels in osteoarthritis. Biomed Res Int. 2015:3568932015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Steck E, Boeuf S, Gabler J, Werth N,
Schnatzer P, Diederichs S and Richter W: Regulation of H19 and its
encoded microRNA-675 in osteoarthritis and under anabolic and
catabolic in vitro conditions. J Mol Med (Berl). 90:1185–1195.
2012. View Article : Google Scholar : PubMed/NCBI
|