Introduction
The mortality rate associated with cardiovascular
disease (CVD) is increasing year by year. CVD is the most prevalent
cause of mortality, according to World Health Organization's
statistics. Myocardial dysfunction, the major cause of mortality in
intensive care units (1), is a major
clinical manifestation of CVD and is easily induced by
endotoxin.
Lipopolysaccharides (LPS), also known as endotoxin,
are a prevalent component of the cell walls of most Gram-negative
bacteria (2). LPS is a pathogen that
may induce endotoxemia, septic shock and multiple organ failure
(3,4). The heart is the most common organ to be
adversely affected by endotoxemia (5,6). The
prevalence of myocardial dysfunction in endotoxemia patients is
>60%. However, the detailed molecular mechanisms remain
undefined and require further elucidation (7).
The fragile X mental retardation protein (FMRP)
gene, which encodes the protein fragile X mental retardation 1
(fmr1), is a determinant of normal cognitive development and female
reproductive function, and is highly prevalent in the brain
(8). Mutations of fmr1 may lead to
fragile X syndrome (9). Studies have
demonstrated that fmr1 is essential for a normal heart rate in
Drosophila (10) and cardiac
function during development (11).
The specific function of fmr1 in the cardiovascular system has
remained largely elusive. It is therefore worthwhile to investigate
whether and how fmr1 is able to inhibit LPS-induced cardiac
injury.
The massive release of inflammatory cytokines,
oxidative stress and mitochondrial dysfunction are the major
characteristics of endotoxemia (12). LPS combines to serum
endotoxin-binding protein to produce massive inflammatory
cytokines, which induce oxidative stress and overproduction of
reactive oxygen species (ROS), causing damage of the mitochondrial
structure and cardiomyocyte dysfunction (13–15),
Oxidative stress and excessive ROS accumulation result in
cytotoxicity and cell apoptosis. Previous studies have indicated
that moderate oxidative stress is one of the major causes of
cardiomyocyte apoptosis (16), which
is an important mechanism of cardiomyocyte death (17). Therefore, inhibition of oxidative
stress-induced cardiomyocyte apoptosis is an important strategy to
prevent cardiomyocyte damage and protect subjects at risk from
cardiac disease.
The phosphatidylinositide-3kinase (PI3K)/Akt pathway
is involved in the regulation of myocardial contraction,
revascularization, physiological or pathological myocardial
hypertrophy and congestive heart failure, having critical roles in
cell proliferation, differentiation and apoptosis (18). In the present study, it was
speculated that the PI3K/Akt pathway also participates in the
processes by which fmr1 inhibits LPS-induced cardiomyocyte
injury.
In the present study, an in vitro model of
LPS-induced cardiomyocyte damage was constructed using the H9c2
cell line, which was subjected to vector-mediated ectopic
overexpression of fmr1, in order to study how fmr1 inhibits
oxidative stress, myocardial injury and cell apoptosis, as well as
the implication of the PI3K/Akt pathway. It is of significant value
to unveil a novel potential biomarker of fmr1 in cardiovascular
systems, which may provide a novel method for the diagnosis and
prognosis of patients with CVD.
Materials and methods
Cell culture
H9c2 rat embryo cardiomyocytes purchased from the
American Type Culture Collection (Manassas, VA, USA) were cultured
in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin (Invitrogen; Thermo Fisher Scientific,
Inc.) at 37°C with 5% CO2. Cells in the logarithmic
growth phase were used in all experiments. The morphology of H9c2
cells were observed under an optical microscope at 48 h after
inoculation.
Cell viability assay
H9c2 cells were divided into 5 groups: LPS groups
treated with LPS (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at
different concentrations (1, 3, 6 and 9 µg/ml), and a control group
without any treatment (n=5 per group). The cell viability was
measured with a Cell Counting Kit-8 (CCK8; Beyotime Institute of
Biotechnology, Haimen, China) after LPS treatment for the set
durations (4, 12 and 24 h). In brief, cells were seeded in 96-well
plates at an initial density of 5×103 cells/well and
incubated with different concentrations of LPS (1, 3, 6 and 9
µg/ml) for the indicated times. CCK-8 stain (20 µl) was then added
to each well of the plate, followed by further incubation for 1 h.
The optical density values at 450 nm (OD450) were read
with a microplate reader (BioTek Instruments, Inc., Winooski, VT,
USA). Values were expressed as the percentage of viable cells as
follows: Relative viability
(%)=[OD450(treated)-OD450
(blank)]/[OD450(control)-OD450(blank)]
×100%.
Cell transfection
Cell transfection experiments were performed after
the construction of an fmr1 overexpression plasmid using the
pGEM-T/pFLAG Vector (Promega Corp., Madison, WI, USA) and
transfection reagent Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.). The empty vector was transfected
into another batch of cells in parallel. In brief, cells were
inoculated in DMEM without antibiotics. When the cell density
reached 90–95%, the plasmid expressing fmr1 and
Lipofectamine® 2000 were added into DMEM without FBS,
and mixed gently at the ratio of DNA toLipofectamine®
2000 of 1:2. The culture media was changed to DMEM with FBS after
incubation for 6 h at 37°C with 5% CO2. The cell
transfection rates were detected by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blot analysis after further culture for 48 h.
RT-qPCR
The mRNA expression levels were determined by
RT-qPCR. Total RNA was extracted from cells with an RNeasy kit
(Qiagen, Valencia, CA, USA), and complementary DNA was reversely
transcribed with 1 µg RNA using the Quantiscript Reverse
Transcriptase kit (Qiagen) according to the manufacturer's
protocol. PCR amplification was performed in an ABI 7300
Thermocycler (Applied Biosystems; Thermo Fisher Scientific, Inc.)
using Fast SYBR Green Master Mix (Applied Biosystems; Thermo Fisher
Scientific, Inc.) with the following reaction conditions: 15 sec at
95°C, followed by 40 cycles of denaturation at 95°C for 15 sec and
annealing/extension at 60°C for 15 sec. The oligonucleotide primer
sequences are displayed in Table
I.
 | Table I.Primers used for polymerase chain
reaction. |
Table I.
Primers used for polymerase chain
reaction.
| Name | Type | Sequence (5′-3′) |
|---|
| Fmr1 | Forward |
GAGGGTGAGGATCGAAGCTG |
|
| Reverse |
GTACCATCCCCCTCTGGACT |
| Bcl-2 | Forward |
CCCCTGGCATCTTCTCCTTCC |
|
| Reverse |
GGGTGACATCTCCCTGTGACG |
| Bax | Forward |
GGATGCGTCCACCAAGAA |
|
| Reverse |
ACGGAGGAAGTCCAGTGT |
| Caspase3 | Forward |
GCCTCTGCCCGGTTAAGAAA |
|
| Reverse |
CATCTGTACCAGACCGAGCG |
| XIAP1 | Forward |
TGGATCTGAATGCCCGATCT |
|
| Reverse |
TCCAACCAGTGTGGAACAGT |
| XIAP2 | Forward |
TGTTGTACCTGCAGACACCA |
|
| Reverse |
AGCTGAGTCTCCATACTGCC |
| GAPDH | Forward |
GGTCATGAGTCCTTCCACGATA |
|
| Reverse |
ATGCTGGCGCTGAGTACGTC |
Western blot analysis
Cells were lysed using lysis buffer (50 mM Tris-Cl,
150 mM NaCl, 0.02% NaN2, 100 µg/ml phenylmethanesulfonyl
fluoride, 1 µg/ml aprotinin, and 1% Triton X-100), and the lysate
was centrifuged at a speed of 12,000 × g for 30 min at 4°C, and the
supernate containing proteins was collected. The protein
concentrations were determined with the bicinchoninic acid assay
(Beyotime Institute of Biotechnology). Subsequently, the total
protein (20 µg) was subjected to each lane of15% SDS-PAGE gel
electrophoresis (SDS-PAGE) and then electroblotted onto a
polyvinylidene fluoride (PVDF) membrane (GE Healthcare, Little
Chalfont, UK). Following blocking with 5% non-fat dry milk in PBS
for 1 h at room temperature, the blotting membranes were probed
overnight at 4°C with the following primary antibodies obtained
from Abcam (Cambridge, UK): Rabbit anti-Fmr1 (cat. no. ab17722;
1:1,000), anti-Bax (cat. no. ab53154; 1:1,000), anti-Bcl-2 (cat.
no. ab196495; 1:1,000), anti-cleaved caspase-3 (cat. no. ab2305;
1:200), anti-XIAP (cat. no. ab2541; 1:200), anti-p-PI3K (cat. no.
ab182651; 1:1,000), anti-PI3K (cat. no. ab227204; 1:2,000),
anti-p-AKT (cat. no. ab38449; 1:1,000), anti-AKT (cat. no. ab64148;
1:200), anti-p-FoxO3a (cat. no. ab47285; 1:1,000), anti-FoxO3a
(cat. no. ab23683; 1:1,000) and anti-GAPDH (cat. no. ab9485;
1:2,500). Samples were then probed with horseradish peroxidase
conjugated Goat anti-rabbit immunoglobulin G secondary antibodies
(Abcam; cat. no. ab6721, 1:5,000) for 2 h at room temperature.
GAPDH was used as loading control. The PVDF membrane was exposed to
X-ray film (Kodak, Rochester, NY, USA) and immunoreactive bands
were detected using enhanced chemiluminescence detection system of
GE ECL Start (GE Healthcare, USA). Western blot bands were
quantified using the Bio-Rad ChemiDoc MP system with Image Lab™
Software version 4.1 (each, Bio-Rad Laboratories, Inc., Hercules,
CA, USA).
Treatment groups
In the present study, 1×105 cells were
inoculated onto each well of a 6-well plate and divided into 5
experimental groups as follows: H9c2 cells treated with 9 µg/ml LPS
for 24 h after being transfected with fmr1 overexpression plasmid
for 12 h (Fmr1+LPS group), H9c2 cells treated with 9 µg/ml LPS for
24 h after being transfected with empty plasmid vector for 12 h
(Vect+LPS group), H9c2 cells only transfected with empty plasmid
vector for 12 h (Vect group), H9c2 cells only treated with 9 µg/ml
LPS for 24 h (LPS group) and H9c2 cells without any treatment
(Control group).
ROS detection
ROS were detected with the oxygen-sensitive
fluorescence probe dichloro-dihydro-fluorescein diacetate (DCTH-DA)
assay. Following culture and pre-treatment as aforementioned, 10
µmol/lDCFH-DA was added to the wells of a 6-well plate and
incubated at 37°C for 20 min. Then, the cells were washed with PBS3
times. Analysis was immediately performed with a flow cytometer (BD
Biosciences, San Diego, CA, USA). BD CellQuest™ Pro
software version 5.1 (BD Biosciences) was used to analyze ROS
levels.
Mitochondrial membrane potential
detection
The mitochondrial membrane potential was determined
with the JC-1 probe assay. In brief, the floating and trypsinized
adherent cells (5×105) of these 5 cell groups (Fmr1+LPS,
Vect+LPS, Vect, LPS and Control group) were collected and
resuspended in 500 µl PBSJC-1 dye (10 µmol/l) was added, followed
by incubation for another 20 min at 37°C. Cells were resuspended in
phosphate buffer and the fluorescence was detected with a flow
cytometer (BD Biosciences). BD CellQuest™ Pro software version 5.1
(BD Biosciences) was used to analyse the mitochondrial membrane
potential.
Apoptosis detection
The apoptotic rate was determined with the
Annexin-V/propidium iodied (PI) double-staining assay (Biovision,
Mountain View, CA, USA) according to the manufacturer's protocol.
In brief, floating and trypsinized adherent cells
(5×105) of the 5 cell groups (Fmr1+LPS, Vect+LPS, Vect,
LPS and Control group) were collected and resuspended in 500 µl
phosphate buffer containing 5 µl Annexin-V fluorescein
isothiocyanate and 5 µl PI, followed by incubation for 5 min in the
dark at the room temperature. Analysis was immediately performed
with a flow cytometer (BD Biosciences). BDCellQuest™ Pro
software version 5.1 (BD Biosciences) was used to analyze the
apoptotic rate.
Oxidative stress factor detection
The levels of antioxidant enzymes, including
superoxide dismutase (SOD) and reduced glutathione/oxidized
glutathione (GSH/GSSG), and the lipid peroxidation product
malondialdehyde (MDA) were determined with specific kits.
Activities of SOD were determined using the Total Superoxide
Dismutase Assay kit with WST-8 (Beyotime Institute of
Biotechnology). The GSH/GSSG ratio was measured with a GSH and GSSG
Assay kit (Beyotime Institute of Biotechnology) with
2,2-dithio-bis-nitrobenzoic acid. The levels of MDA were detected
with a Lipid Peroxidation MDA Assay kit (Beyotime Institute of
Biotechnology); in this assay, MDA reacts with thiobarbituric acid.
All measurements were performed according to the manufacturers'
protocols.
Statistical analysis
All results were expressed as mean ± standard
deviations of five independent experiments. Statistical analysis
was performed using the SPSS 18.0 statistical package (SPSS, Inc.,
Chicago, IL, USA) and data were subjected to one-way analysis of
variance, followed by Dunnett's test. P<0.05 was considered to
indicate a statistically significant difference.
Results
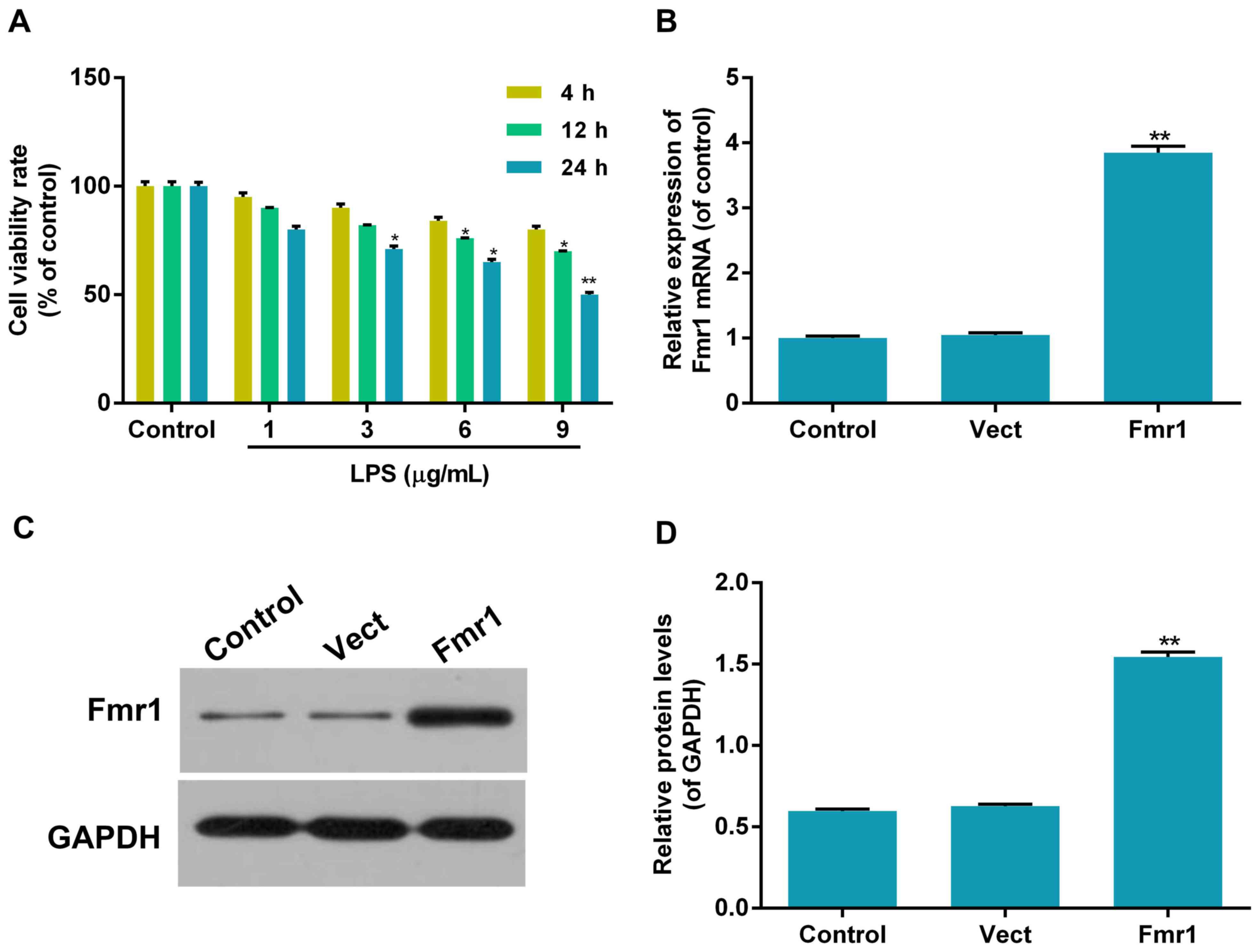
Inhibitory effect of LPS on H9c2 cell
viability
The viability of H9c2 cells treated with LPS at
different concentrations (0, 1, 3, 6 and 9 µg/ml) for determined
durations (4, 12 and 24 h) was evaluated with a CCK8 assay in order
to indicate the damage effect of LPS on H9c2 cells. The results
indicated that the viability of H9c2 cells was decreased by LPS in
a dose- and time-dependent manner. Compared with the control,
treatment with 6 or 9 µg/ml LPS for 24 h significantly decreased
the cell viability by 35 and 50%, respectively (P<0.05; Fig. 1A). Treatment with 9 µg/ml LPS for 24
h was selected as the conditions for the subsequent experiments
(LPS group) due to the severe damage effect.
Transfection rates of overexpression
of fmr1
The ectopic overexpression rates of fmr1 were
determined by RT-qPCR and western blot analysis. The mRNA and
protein levels of fmr1 were significantly increased by ~3-fold in
the fmr1 overexpression group compared with those in the control
group (P<0.01). As expected, mRNA and protein levels in the Vect
group transfected with empty vector were similar to those in the
control group (Fig. 1B and D).
Fmr1 overexpression inhibits oxidative
stress induced by LPS in H9c2 cells
To determine the effect of fmr1 overexpression on
ROS levels when H9c2 cells were injured by LPS, an assay utilizing
the oxygen-sensitive fluorescence probe DCTH-DA was performed to
detect ROS levels in the following 5 cell groups: Fmr1+LPS,
Vect+LPS, Vect, LPS and Control group. The results indicated that
ROS levels in the LPS group increased significantly compared with
those in the control group (P<0.01), and ROS levels in the
Fmr1+LPS group decreased significantly in the Vect+LPS group
(P<0.01), in which the levels were similar to those in the LPS
group. It was demonstrated that fmr1 overexpression inhibited the
promotion effect of LPS on ROS levels in H9c2 cells (Fig. 2A).
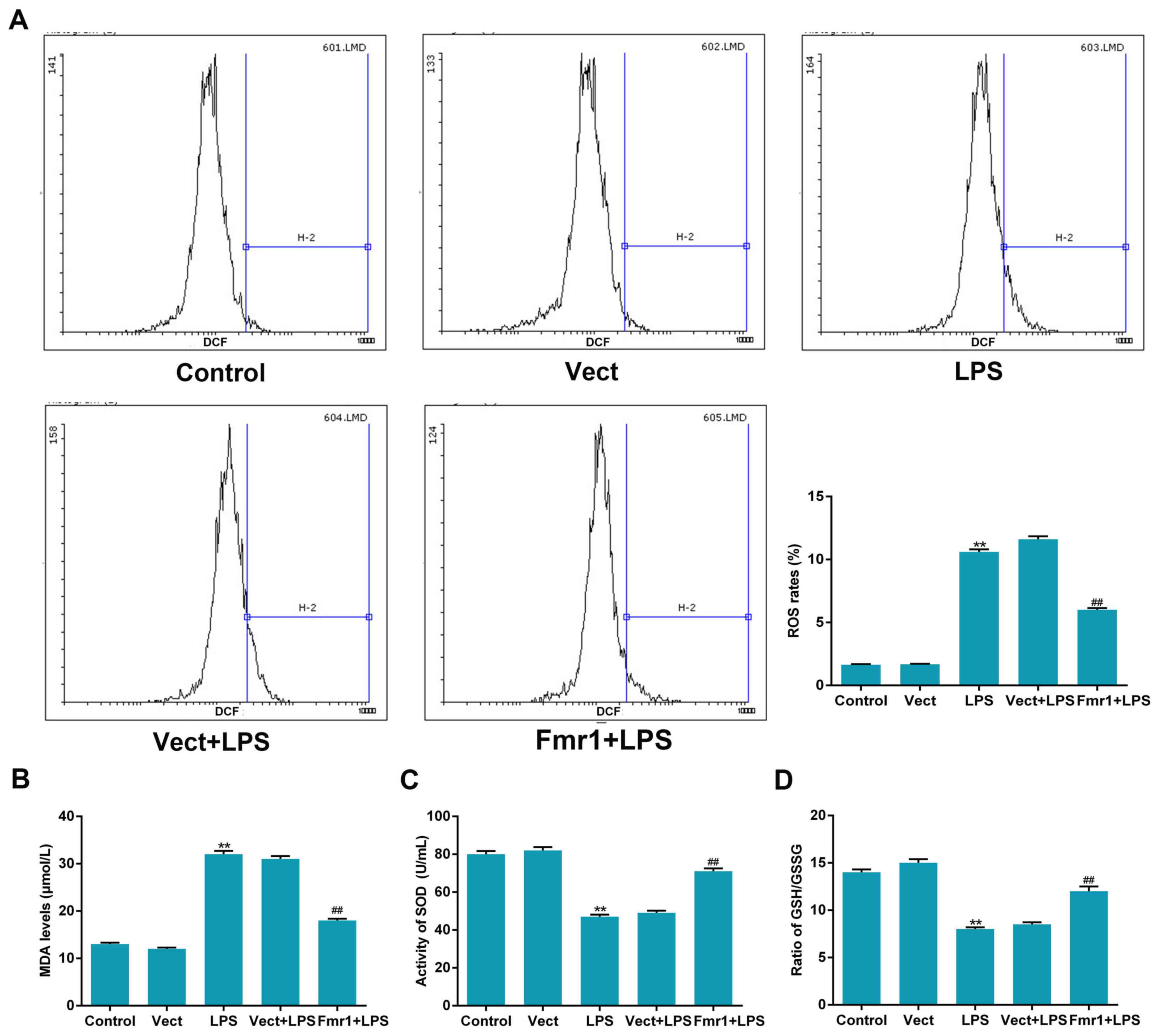 | Figure 2.Fmr1 overexpression alleviates
oxidative stress induced by LPS in H9c2 cells. (A) A
dichloro-dihydro-fluorescein diacetate assay and flow cytometry
were applied to detect ROS levels in the Fmr1+LPS group, Vect+LPS
group, Vect group, LPS group and Control group. (B) The content of
the lipid peroxidation product MDA, (C) the activity of SOD and (D)
the ratio of GSH/GSSG were also detected in these 5 cell groups.
Values are expressed as the mean ± standard deviation (n=5 per
group). **P<0.01 vs. control; ##P<0.01 vs.
Vect+LPS control. LPS, lipopolysaccharide; Vect, empty vector;
fmr1, fragile X mental retardation 1; ROS, reactive oxygen species;
MDA, malondialdehyde; SOD, superoxide dismutase; GSH/GSSG, reduced
vs. oxidized glutathione. |
The levels of oxidative stress-associated factors,
including lipid peroxidation product MDA, the antioxidant enzyme
SOD and the GSH/GSSG ratio were determined by specific kits. The
content of MDA increased by >2-fold in the LPS group compared
with that in the control group (P<0.01), and decreased by 42% in
the Fmr1+LPS group compared with that in the Vect+LPS group
(P<0.01; Fig. 2B). The activity
of SOD decreased by 41% in the LPS group compared with that in the
control group (P<0.01), and increased again in the Fmr1+LPS
group compared with that in the Vect+LPS group (P<0.01; Fig. 2C). Under normal conditions,
mitochondrial thiols, most notably GSH, prevent mitochondrial DNA
from oxidation, but the administration of LPS decreases
mitochondrial GSH. The ratio of GSH/GSSG decreased significantly in
the LPS group compared with that in the control group, and
increased again in the Fmr1+LPS group compared with that in the
Vect+LPS group (P<0.01; Fig.
2D).
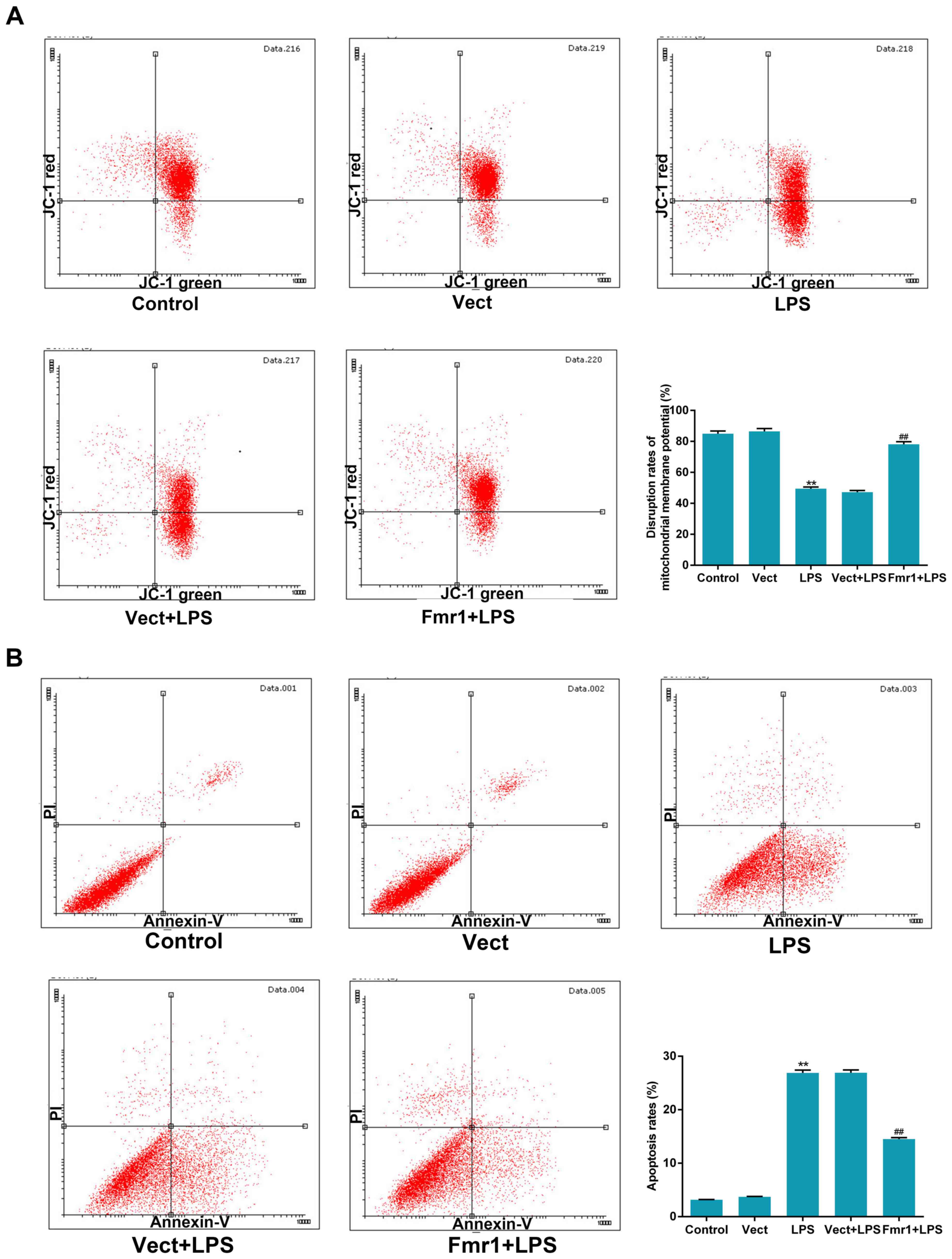
Fmr1 overexpression inhibits cell
apoptosis induced by LPS in H9c2 cells
To assess the function of fmr1 overexpression on the
mitochondrial membrane potential in H9c2 cells after injury by LPS,
the JC-1 probe assay was performed to detect mitochondrial membrane
potential levels in the 5 experimental groups. The results
indicated that the disrupted mitochondrial membrane potential in
the LPS group increased significantly compared with that in the
control group (P<0.01), and decreased significantly in the
Fmr1+LPS group compared with that in the Vect+LPS group
(P<0.01). The values in the Vect+LPS and LPS group were similar.
Overall, the results indicated that fmr1 overexpression reduced the
disruption of the mitochondrial membrane potential following
LPS-induced injury in H9c2 cells (Fig.
3A).
To identify whether LPS induces apoptosis in H9c2
cells and whether fmr1 overexpression inhibits this, an
Annexin-V/PI double-staining assay was performed to detect the
apoptotic rates in the 5 experimental groups. The results indicated
that the apoptotic rate increased significantly in the LPS group
compared with that in the control group (P<0.01), and decreased
to 46% in the Fmr1+LPS group compared with that in the Vect+LPS
group (P<0.01), while the values in the Vect+LPS and LPS group
were similar. It was demonstrated that fmr1 overexpression
inhibited the apoptotic effect of LPS on H9c2 cells (Fig. 3B). These results indicated an
inhibitory effect of fmr1 overexpression on LPS-induced apoptosis
in H9c2 cells.
To detect the mechanism by which fmr1 overexpression
protects H9c2 cells from the LPS-induced reduction of cell
viability and promotion of apoptosis, RT-qPCR and western blot
analysis were performed to detect the mRNA and protein levels of
apoptosis-associated factors, including B-cell lymphoma 2
(Bcl-2)-associated X protein (Bax), Bcl-2, cleaved caspase-3 and
X-linked inhibitor of apoptosis protein (XIAP) in the 5
experimental groups (Fig. 4). The
results indicated that, at the mRNA and protein level, the
apoptosis-activating factors Bax and cleaved caspase-3 increased
significantly in the LPS group compared with those in the control
group (P<0.01), and decreased significantly in the Fmr1+LPS
group compared with those in the Vect+LPS group (P<0.05), while
levels in the Vect+LPS and LPS groups were similar. By contrast,
the mRNA and protein levels of the apoptosis inhibitors Bcl-2 and
XIAP decreased significantly in the LPS group compared with those
in the control group (P<0.01), and increased significantly in
the Fmr1+LPS group compared with those in the Vect+LPS group
(P<0.05), while levels in the Vect+LPS and LPS groups were
similar (Fig. 4).
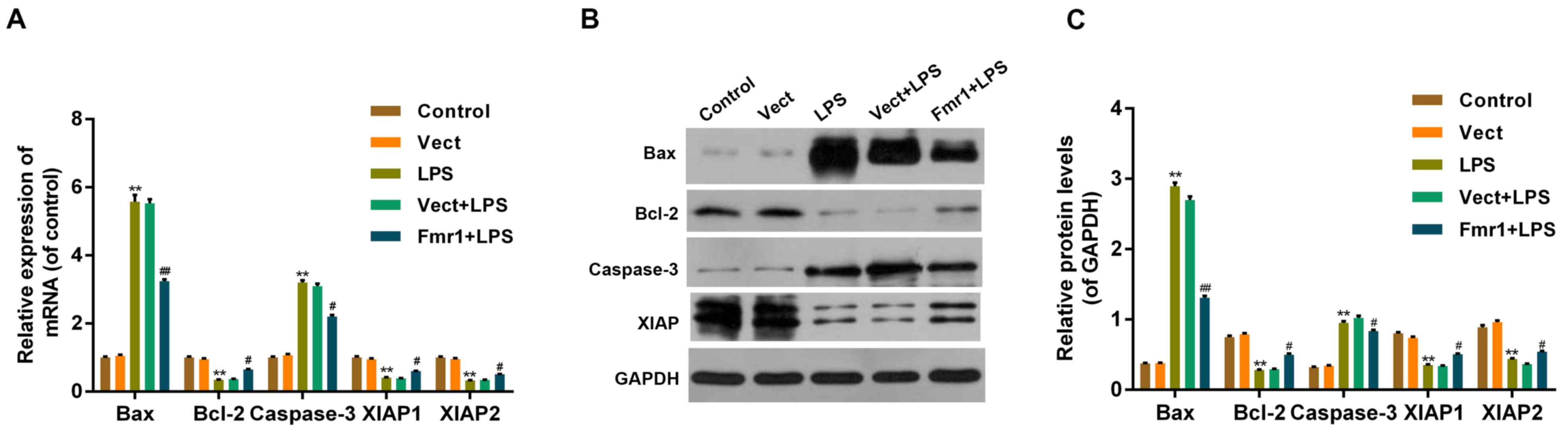 | Figure 4.Effect of fmr1 on apoptosis-associated
factors in H9c2 cells damaged by LPS. (A) Reverse
transcription-quantitative polymerase chain reaction analysis was
performed to detect the mRNA levels of apoptosis-associated
factors, including Bax, Bcl-2, Caspase-3 and XIAP in the following
experimental cell groups: Fmr1+LPS, Vect+LPS, Vect, LPS and Control
group. (B and C) Western blot analysis was performed to detect
protein levels of the abovementioned apoptosis-associated factors
in the 5 cell groups. Values are expressed as the mean ± standard
deviation (n=5 per group). **P<0.01 vs. control;
#P<0.05, ##P<0.01 vs. Vect+LPS control.
LPS, lipopolysaccharide; Vect, empty vector; fmr1, fragile X mental
retardation 1; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X
protein; XIAP, X-linked inhibitor of apoptosis protein. |
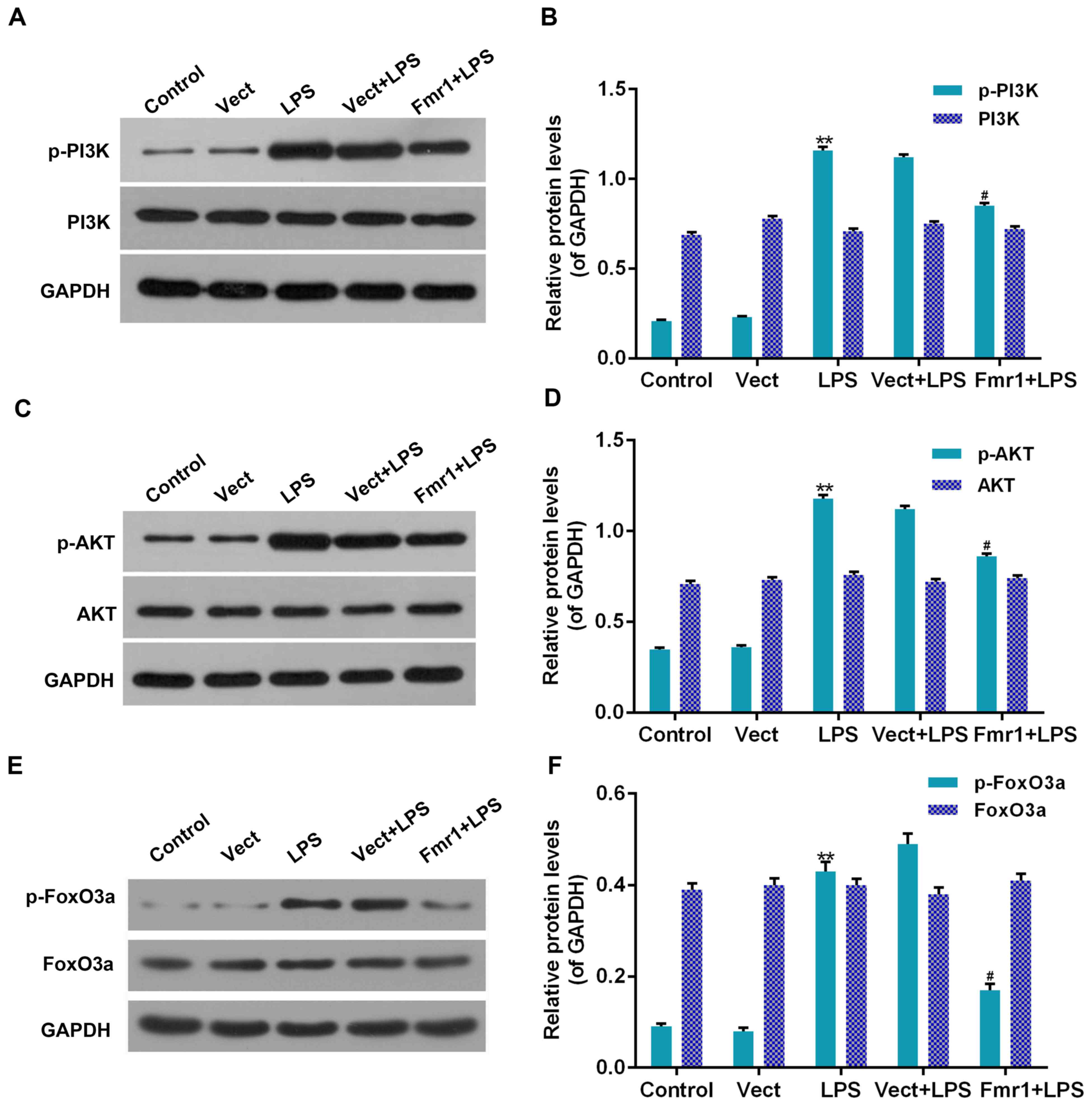
Effect of fmr1 overexpression on the
PI3K/Akt/forkhead box (Fox)O3a pathway in H9c2 cells injured by
LPS
To detect whether the protective function of fmr1
overexpression on LPS-associated inhibition of H9c2 cell viability
and induction of apoptosis were associated with the PI3K/Akt/FoxO3a
pathway, western blot analysis was performed to detect the
phosphorylation levels of PI3K, Akt and FoxO3a in H9c2 cells
treated with LPS (LPS group), which had been optionally transfected
with Fmr1 overexpression vector (Fmr1+LPS group). The
phosphorylation levels were compared with those in the control and
Vect+LPS groups. As displayed in Fig.
5, the phosphorylation levels of PI3K, Akt and FoxO3a in the
LPS group were all significantly increased compared with those in
the control group (P<0.01). In addition, the phosphorylation
levels of PI3K, Akt and FoxO3a in the Fmr1+LPS group were all
significantly decreased by 9, 22 and 37%, respectively, compared
with those in the Vect+LPS group (P<0.05). However, the total
protein levels of PI3K, Akt and FoxO3a were not significantly
affected (P>0.05).
Discussion
LPS, also called endotoxin, induces endotoxemia and
myocardial dysfunction, which is a major clinical manifestation of
CVD (11). Recently, fmr1 was
identified to be associated with a normal heart rate and cardiac
function (11,19). However, the specific mechanism has
remained to be fully elucidated and requires further study.
The present study explored the effect of fmr1
overexpression on LPS-induced cardiomyocyte injury, including
oxidative stress reaction, mitochondrial membrane potential
variation and cell apoptosis. H9c2 cardiomyocytes were used in the
present study. The viability of LPS-injured H9c2 cardiomyocytes was
evaluated with a CCK8 assay and was observed to be decreased by LPS
in a dose- and time-dependent manner. Treatment of H9c2 cells with
9 µg/ml LPS for 24 h was employed in the subsequent experiments due
to the severe damage associated with it.
Mitochondrial dysfunction and oxidative stress are
the major characteristics of endotoxemia (20). Injury of the structural integrity of
mitochondria induces oxidative stress and abundant ROS release,
resulting in mitochondrial dysfunction, cardiomyocyte injury and
cardiac structure damage. The present results indicated that LPS
increases the disruption rate of the mitochondrial membrane
potential, and fmr1 overexpression reduced the increased rates of
mitochondrial membrane potential collapse to repair mitochondrial
dysfunction, which may influence the release of cytochrome c
and mitochondrial ATP production in H9c2 cells treated with
LPS.
ROS are highly reactive oxygenated chemical
substances. Under normal conditions, the antioxidant enzymes,
including SOD and GSH-peroxidase (GSH-Px), eliminate ROS during
cell metabolism, maintaining a balance of ROS generation and
elimination (21). However,
excessive amounts of ROS are generated to resist bacterial
infection-mediated cellular damage. When ROS accumulates
ceaselessly and the endogenic anti-oxygen defence system cannot
eliminate it in time, it induces oxidative stress, cellular
disorders including upregulated lipid peroxidation, and also cell
apoptosis (22). Therefore, LPS
induces cardiomyocyte injury mostly through excessive ROS
generation (23). The present study
proved that the levels of ROS and MDA, the representative product
of the lipid peroxidation process, increased, while activities of
antioxidant enzymes, including SOD and GSH-Px (GSH/GSSG ratio)
declined inH9c2 cells treated with LPS, which was attenuated by
fmr1overexpression. Hence, fmr1 overexpression alleviated oxidative
stress induced by LPS, with decreased peroxide levels and improved
antioxidant enzyme activities.
Partial oxidative stress is one of the major reasons
of cardiomyocyte apoptosis, and apoptosis is an important mechanism
of cardiomyocyte death (24–26). The abnormal expression of bcl-2
family proteins serve important roles in cell apoptosis. Bax
facilitates apoptosis by forming a heterodimer with Bcl-2 and
inhibiting the activities of Bcl-2. Caspase-3 is the downstream
activator of apoptosis, actively causes the dismemberment of the
cell. On the contrary, XIAP has critical roles in inhibiting
apoptosis. The present study provided evidence that the mechanism
underlying the effects of LPS and the inhibitory role of fmr1is
associated with apoptosis and apoptosis-associated factors. The
results indicated that LPS increased the mRNA and protein levels of
apoptosis-activating factors, including Bax and caspase-3, while
overexpression of fmr1 significantly decreased them. Furthermore,
LPS inhibited the mRNA and protein expression of the apoptosis
inhibitors Bcl-2 and XIAP, while overexpression of fmr1 caused a
significant upregulation of these factors. It was indicated that
fmr1 inhibited apoptosis and associated factors to alleviate
LPS-induced oxidative stress in H9c2 cells.
In addition to apoptosis-associated signaling, the
PI3K/Akt/FoxO3a pathway was also indicated to have a role in the
effect of fmr1 to alleviate LPS-induced injury of H9c2 myocardial
cells. The activation of the PI3K/Akt pathway has critical roles in
cell proliferation and differentiation, and alleviates oxidative
stress-induced apoptosis (18).
FoxO3a is a critical transcriptional factor downstream of the
PI3K/Akt pathway, and also has important roles in regulating
oxidative stress-induced apoptosis (27). Consistent with the activating effect
of LPS on Akt by stimulating its phophorylation in macrophages
demonstrated in RAW 264.7 macrophages by Nishina et al
(28), in HT29 cells by Shi et
al (29) and in chondrogenic
cells by Wu et al (30), the
present results indicated that the phosphorylation levels of PI3K,
Akt and FoxO3a increased in the LPS group, which was significantly
reduced when fmr1 was overexpressed. It was confirmed that the
alleviating function of fmr1 on LPS-induced oxidative stress in
H9c2 cells was mediated via the PI3K/Akt/FoxO3a pathway.
In conclusion, in the present study, a model of
LPS-induced H9c2 cardiomyocyte injury was constructed to determine
how the overexpression of fmr1 attenuated LPS-induced oxidative
stress and apoptosis, during which peroxide levels, antioxidant
enzyme activities and levels of apoptosis-associated factors were
regulated via reducing the phosphorylation levels of PI3K, Akt and
FoxO3a. Fmr1may be of significant value as a novel potential
biomarker and an important endogenous protection factor in the
cardiovascular system. The present results provide novel insight
into the cardioprotective effects of fmr1, which may also have
potential use as a diagnostic and prognostic biomarker for CVD.
Acknowledgements
Not applicable.
Funding
This work was supported by the State Administration
of Traditional Chinese Medicine (grant no. 2015ZB003).
Availability of data and materials
The analyzed data sets generated during the study
are included in this published article.
Authors' contributions
JB and ZZho conceived the study; JB performed the
oxidative stress reaction, and assessed mitochondrial membrane
potential variation and cell apoptosis; CY performed RT-qPCR and
western blotting; and ZZhe treated cells.
Ethical approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interest.
References
|
1
|
Fattahi F and Ward PA: Complement and
sepsis-induced heart dysfunction. Mol Immunol. 84:57–64. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Murphy E and Steenbergen C: What makes the
mitochondria a killer? Can we condition them to be less
destructive? Biochim Biophys Acta. 1813:1302–1308. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen JQ, Cammarata PR, Baines CP and Yager
JD: Regulation of mitochondrial respiratory chain biogenesis by
estrogens/estrogen receptors and physiological, pathological and
pharmacological implications. Biochim Biophys Acta. 1793:1540–1570.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lin WJ and Yeh WC: Implication of
Toll-like receptor and tumor necrosis factor alpha signaling in
septic shock. Shock. 24:206–209. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vieillard-Baron A, Caille V, Charron C,
Belliard G, Page B and Jardin F: Actual incidence of global left
ventricular hypokinesia in adult septic shock. Crit Care Med.
36:1701–1706. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Niederbichler AD, Westfall MV, Su GL,
Donnerberg J, Usman A, Vogt PM, Ipaktchi KR, Arbabi S, Wang SC and
Hemmila MR: Cardiomyocyte function after burn injury and
lipopolysaccharide exposure: Single-cell contraction analysis and
cytokine secretion profile. Shock. 25:176–183. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Akira S, Takeda K and Kaisho T: Toll-like
receptors: Critical proteins linking innate and acquired immunity.
Nat Immunol. 2:675–680. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Van't Padje S, Chaudhry B, Severijnen LA,
van der Linde HC, Mientjes EJ, Oostra BA and Willemsen R: Reduction
in fragile X related 1 protein causes cardiomyopathy and muscular
dystrophy in zebrafish. J Exp Biol. 212:2564–2570. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jiraanont P, Hagerman RJ, Neri G, Zollino
M, Murdolo M and Tassone F: Germinal mosaicism for a deletion of
the FMR1 gene leading to fragile X syndrome. Eur J Med Genet.
59:459–462. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Novak SM, Joardar A, Gregorio CC and
Zarnescu DC: Regulation of heart rate in drosophila via fragile X
mental retardation protein. PLoS One. 10:e01428362015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Klusek J, Roberts JE and Losh M: Cardiac
autonomic regulation in autism and Fragile X syndrome: A review.
Psychol Bull. 141:141–175. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schechter MA, Southerland KW, Feger BJ,
Linder D Jr, Ali AA, Njoroge L, Milano CA and Bowles DE: An
isolated working heart system for large animal models. J Vis Exp.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen L, Li W, Qi D and Wang D: Lycium
barbarum polysaccharide protects against LPS-induced ARDS by
inhibiting apoptosis, oxidative stress, and inflammation in
pulmonary endothelial cells. Free Radic Res. 23:1–11. 2018.
|
|
14
|
Dong Z and Yuan Y: Accelerated
inflammation and oxidative stress induced by LPS in acute lung
injury: Inhibition by ST1926. Int J Mol Med. 41:3405–3421.
2018.PubMed/NCBI
|
|
15
|
Zhang H, Zhang W, Jiao F, Li X, Zhang H,
Wang L and Gong Z: The nephroprotective effect of MS-275 on
lipopolysaccharide (LPS)-induced acute kidney injury by inhibiting
reactive oxygen species (ROS)-oxidative stress and endoplasmic
reticulum stress. Med Sci Monit. 24:2620–2630. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tavares AM, da Rosa Araujo AS, Llesuy S,
Khaper N, Rohde LE, Clausell N and Belló-Klein A: Early loss of
cardiac function in acute myocardial infarction is associated with
redox imbalance. Exp Clin Cardiol. 17:263–267. 2012.PubMed/NCBI
|
|
17
|
Selvaraju V, Joshi M, Suresh S, Sanchez
JA, Maulik N and Maulik G: Diabetes, oxidative stress, molecular
mechanism, and cardiovascular disease-an overview. Toxicol Mech
Methods. 22:330–335. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Deng C, Sun Z, Tong G, Yi W, Ma L, Zhao B,
Cheng L, Zhang J, Cao F and Yi D: α-Lipoic acid reduces infarct
size and preserves cardiac function in rat myocardial
ischemia/reperfusion injury through activation of PI3K/Akt/Nrf2
pathway. PLoS One. 8:e583712013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma Y, Tian S, Wang Z, Wang C, Chen X, Li
W, Yang Y and He S: CMP-N-acetylneuraminic acid synthetase
interacts with fragile X related protein 1. Mol Med Rep.
14:1501–1508. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Buja Maximilian L: Mitochondria in
ischemic heart disease. Adv Exp Med Biol. 982:127–140. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang X, Lai R, Su X, Chen G and Liang Z:
Edaravone attenuates lipopolysaccharide-induced acute respiratory
distress syndrome associated early pulmonary fibrosis via
inhibition of oxidative stress and transforming growth
factor-β1/Smad3 signaling. Biochem Biophys Res Commun. 495:706–712.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ding ZM, Jiao XF, Wu D, Zhang JY, Chen F,
Wang YS, Huang CJ, Zhang SX, Li X and Huo LJ: Bisphenol AF
negatively affects oocyte maturation of mouse in vitro
through increasing oxidative stress and DNA damage. Chem Biol
Interact. 278:222–229. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gölz L, Memmert S, Rath-Deschner B, Jäger
A, Appel T, Baumgarten G, Gotz W and Frede S: LPS from P.
gingivalis and hypoxia increases oxidative stress in periodontal
ligament fibroblasts and contributes to periodontitis. Mediators
Inflamm. 2014:9862642014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu XR, Li T, Cao L, Yu YY, Chen LL, Fan
XH, Yang BB and Tan XQ: Dexmedetomidine attenuates H2O2-induced
neonatal rat cardiomyocytes apoptosis through mitochondria- and
ER-medicated oxidative stress pathways. Mol Med Rep. 17:7258–7264.
2018.PubMed/NCBI
|
|
25
|
Wang Y, Lei T, Yuan J, Wu Y, Shen X, Gao
J, Feng W and Lu Z: GCN2 deficiency ameliorates doxorubicin-induced
cardiotoxicity by decreasing cardiomyocyte apoptosis and myocardial
oxidative stress. Redox Biol. 17:25–34. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Z, Wang M, Liu J, Ye J, Jiang H, Xu
Y, Ye D and Wan J: Inhibition of TRPA1 attenuates
doxorubicin-induced acute cardiotoxicity by suppressing oxidative
stress, the inflammatory response, and endoplasmic reticulum
stress. Oxid Med Cell Longev. 2018:51794682018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li D, Qu Y, Mao M, Zhang X, Li J, Ferriero
D and Mu D: Involvement of the PTEN-AKT-FOXO3a pathway in neuronal
apoptosis in developing rat brain after hypoxia-ischemia. J Cereb
Blood Flow Metab. 29:1903–1913. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nishina A, Shimizu K, Koketsu M, Ninomiya
M, Sato D, Suzuki T, Hayakawa S and Kimura H: 5,7-Dihydroxyflavone
analogues may regulate lipopolysaccharide-induced inflammatory
responses by suppressing iκbα-linked AKT and ERK5 phosphorylation
in raw 264.7 macrophages. Evid Based Compl Altern Med.
2017:78989732017.
|
|
29
|
Shi J, Shan S, Li H, Song G and Li Z:
Anti-inflammatory effects of millet bran derived-bound polyphenols
in LPS-induced HT-29 cell via ROS/miR-149/Akt/NF-κB signaling
pathway. Oncotarget. 8:74582–74594. 2017.PubMed/NCBI
|
|
30
|
Wu DP, Zhang JL, Wang JY, Cui MX, Jia JL,
Liu XH and Liang QD: MiR-1246 promotes LPS-induced inflammatory
injury in chondrogenic cells ATDC5 by targeting HNF4γ. Cell Physiol
Biochem. 43:2010–2021. 2017. View Article : Google Scholar : PubMed/NCBI
|