Introduction
Reactive oxygen species (ROS) are considered to be
normal by-products of cellular respiration and inflammation
(1), nonetheless, extensive evidence
has accumulated over the last four decades that links the excessive
production of ROS with cellular pathology and tissue damage
(1–4). Cells exhibit several mechanisms to
antagonize the cytotoxic effects of ROS, but these mechanisms can
be overwhelmed, producing what is known as ‘oxidative stress’
(5), which is a state that
contributes to the pathogenesis of a number of chronic disease
conditions including Alzheimer's disease (6,7),
traumatic brain injury (8), diabetes
(9,10) and ageing (11).
The well-established role ROS serves in disease
etiology has provided rationale for research aiming to elucidate
the exact mechanism(s) by which ROS can initiate and/or propagate
cellular pathology (12–14). Although this remains to be completely
understood, there is evidence which indicates that ROS can produce
cellular damage by: i) Peroxidation of polyunsaturated fatty acids
(PUFA) of cellular membranes (15),
ii) inducing mutations through the nitration and deamination of DNA
(16,17), and iii) disrupting cellular function
through protein nitration and carbonylation (18). The above effects may be triggered by
ROS directly or through the decomposition of ROS into other highly
reactive and cell damaging radicals (1).
Lipid peroxidation refers to the process by which
the hydrogen atom of the double bonds of PUFAs is attacked by
oxygen radicals (primarily the hydroxyl and hydroperoxyl radicals)
(19,20). The consequence of this attack is the
formation of a lipid radical, which can directly react with oxygen
to form a lipid peroxyl radical (20). The lipid peroxyl radical formed from
the above reaction is highly reactive and can remove another
hydrogen from the double bond of a neighboring PUFA, resulting in a
chain reaction (20). The above
self-propagating chain of events may be terminated by natural
antioxidants like vitamin E that can donate protons to lipid
peroxyl radicals thereby forming lipid hydroperoxides (21). The resulting vitamin E free radical
can react with yet another lipid peroxyl radical to form
non-radical species. Lipid peroxyl radicals formed in this way can
also cyclize into cyclic peroxide species and finally be converted
into secondary aldehyde species like malondialdehyde (MDA) and
4-hydroxynonenal (4-HNE) (22).
The detrimental effects caused by lipid peroxidation
can be attributed to the loss of membrane integrity described above
with the resulting lack of control over ion trafficking and
cellular osmolality (23), or
cytotoxic and mutagenic properties of the secondary aldehyde
species (MDA and 4-HNE) that result from lipid peroxidation chain
reactions (23). In particular, the
role of 4-HNE has received considerable interest in recent years
and has been the target of extensive research (19,24,25). One
mechanism by which 4-HNE produces cell toxicity is believed to
involve the reaction of 4-HNE with the thiol and amino groups of
macromolecules (19). Another
mechanism involves a potential role that 4-HNE serves as a
secondary messenger of lipid peroxidation where it modulates gene
expression through regulating the effect of a number of
transcription factors (26,27). Nonetheless, the exact mechanism by
which 4-HNE produces its effect is not yet completely
understood.
Using blood plasma and isolated platelets as a model
system, the present authors previously performed an in vitro
assay that recapitulated the effects of peroxynitrite, a reactive
nitrogen species, on protein oxidation and lipid peroxidation
(13). Notably, it was demonstrated
that using drugs that catalytically decompose peroxynitrite-derived
free radicals (nitrogen dioxide, carbonate and superoxide)
mitigates the oxidative damage mediated by peroxynitrite treatment
(13). In the present study, using
the in vitro system described above, it was hypothesized
that by using different antioxidant agents that target different
steps of the lipid peroxidation reactions triggered by 4-HNE, the
mechanism by which 4-HNE produces oxidative damage may be
elucidated. The present study reports the protective effects of
U-83836E (a scavenger of lipid peroxyl radicals) and phenelzine (a
scavenger of reactive aldehyde species) against the 4-HNE-mediated
oxidative damage of blood proteins and lipids. The mechanism by
which 4-HNE induces oxidative damage is discussed in the context of
the above observations.
Materials and methods
Subject characteristics and blood
sample collection
The present prospective study was approved by the
Institutional Review Board affiliated with the Jordan University of
Science and Technology (JUST; Irbid, Jordan).
Recruitment of study participants took place at the
Family Medicine Clinics of King Abdullah University Hospital
(KAUH). KAUH is a tertiary hospital affiliated with JUST. Study
participants were initially interviewed with a clinical research
coordinator who explained the research and its objectives. Relevant
medical history was collected into a structured data collection
sheet by the research coordinator. To be eligible to participate in
the study, the study participants needed to be of Jordanian descent
and 22–25 years of age. Individuals who were; i) habitual cigarette
or water pipe smokers, ii) suffered from hypertension, type 1 or
type 2 diabetes mellitus, atherosclerosis or dyslipidemia as
indicated by their medical history; or iii) received
anti-histamines or nonsteroidal anti-inflammatory drugs up ≤2 weeks
prior to the interview were excluded from participating in the
study. Following the application of the eligibility criteria
explained above, 2 male and 1 female subjects consented to
participate and were thus recruited to the study.
Following providing informed written consent, 500 ml
blood was collected from the subject by a certified phlebotomist
into a 250 ml Citrate-Phosphate-Dextrose with Adenine (CPDA1) bag.
The blood was then divided into ten individual CPDA1 tubes for the
purpose of separating the blood plasma and platelets, and to
receive different treatments.
Isolation of plasma and platelets from
whole blood
Plasma and platelets were isolated as previously
described (13). Briefly, whole
blood was centrifuged at 450 × g for 6.20 min at room temperature
to recover platelet-rich plasma. The platelet-rich plasma recovered
from the previous step was then subjected to centrifugation at
2,500 × g for 5.40 min at room temperature; leading to
sedimentation of a platelet-rich pellet. The pellet recovered from
the previous step was then washed twice with Tyrode's buffer
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and re-suspended in
the same buffer. The final platelet number was measured with a
hemocytometer and accordingly adjusted to 109
platelets/ml suspensions.
Sample treatment
The experimental design and sample treatments were
previously described (13). Briefly,
the plasma and isolated platelets were randomly divided into 6-well
plates and treated with either vehicle (DMSO) or an increasing dose
of 4-HNE (0.1, 1 or 10 µM). In another 6-well plate, samples were
treated with a combination of 4-HNE (10 µM) with the following
doses of either phenelzine (25, 50 or 100 µM; MP Biochemicals, LLC,
Santa Ana, CA, USA) or U-83836E (25, 50 or 100 µM; Biomol
International; Enzo Life Sciences, Inc., Farmingdale, NY, USA).
Estimation of total plasma
proteins
The total protein content of plasma samples was
estimated using a Pierce™ BCA Protein Assay kit (cat.
no. 23225; Thermo Fisher scientific, Inc., Waltham, MA, USA)
according to the manufacturer's protocol. Absorbance was determined
at 562 nm on an ELx800 ELISA reader (BioTek Instruments, Inc.,
Winooski, VT, USA).
Detection of carbonyl group and
thiobarbituric acid reactive substances (TBARS)
The methods used for the detection of carbonyl
groups and TBARS were previously detailed (13). Final measurement of the content of
carbonyl groups in each sample was performed using a Protein
Carbonyl Content Assay kit (cat. no. ab126287; Abcam, Cambridge,
UK). Absorbance was measured at 375 nm on an ELx800 ELISA reader
(BioTek Instruments, Inc., Winooski, VT, USA). Final measurement of
TBARS concentration was performed spectrophotometrically using a
Lipid Peroxidation Assay kit (cat. no. MAK085; Sigma-Aldrich; Merck
KGaA). Absorbance was measured at 530 nm on an ELx800 ELISA
reader.
Statistical analysis
Statistical analysis was performed using SPSS
version 17 (SPSS, Inc., Chicago, IL, USA). Data were expressed as
the mean ± standard deviation. A one-way analysis of variance was
used to compare data in the different experimental groups followed
by Fisher's post hoc test. P<0.05 was considered to indicate
statistically significant differences.
Results
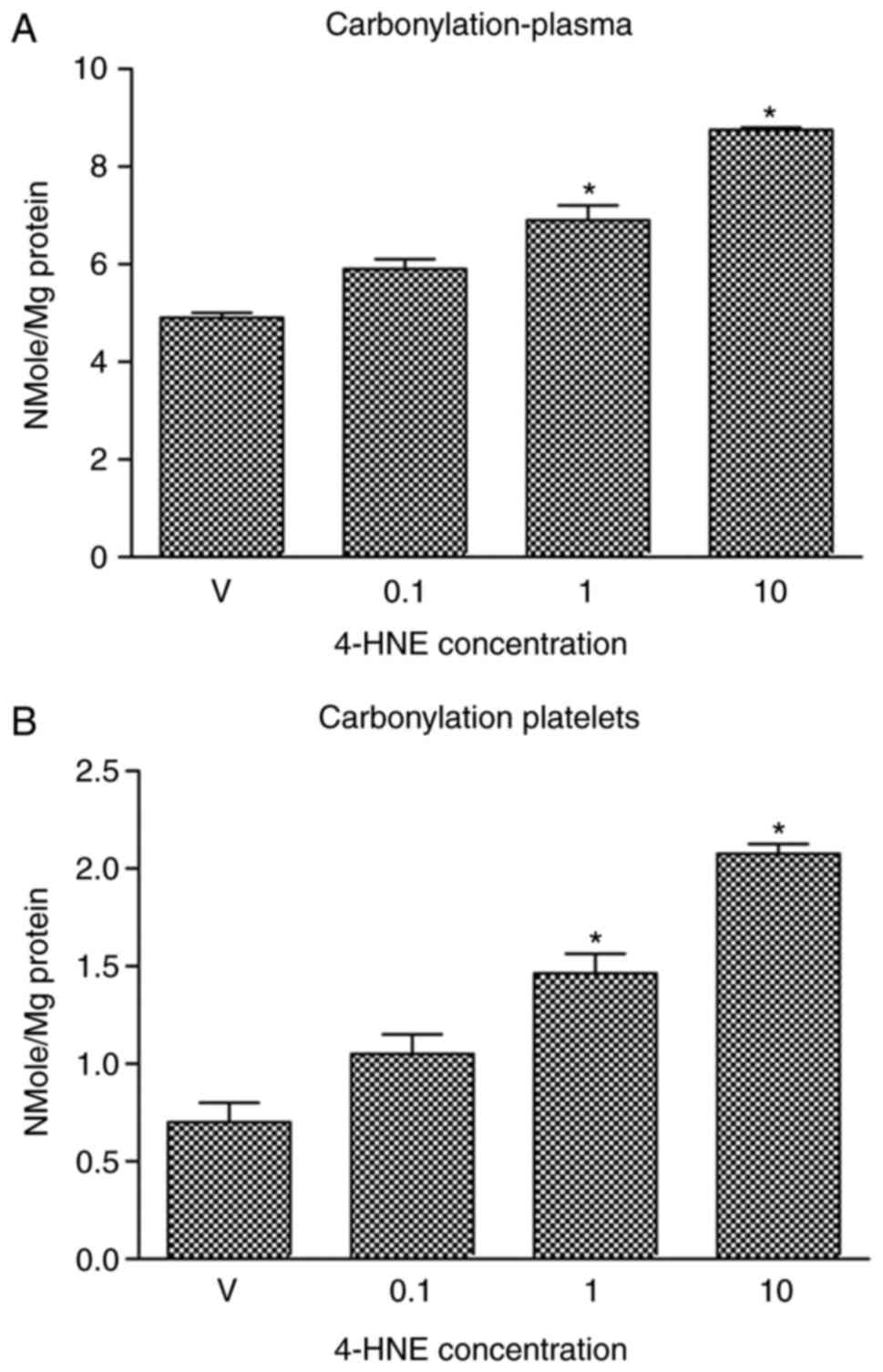
The concentration of protein carbonyl
groups and TBARS in blood plasma and platelets is increased by
4-HNE
In a previous study, the present authors described
an in vitro assay that allowed investigation of the
oxidative damage induced by the reactive nitrogen species
peroxynitrite in blood lipids and proteins (12). Prior to attempting to understand the
mechanism of 4-HNE-induced oxidative damage in the present study,
the aim was to quantitatively evaluate the oxidative damage induced
by 4-HNE using well-established assays. To achieve this goal, blood
plasma and platelet samples were treated with 4-HNE (0.1, 1 or 10
µM) and the concentration of carbonyl group formation (a biomarker
of protein oxidative damage) and TBARS (a biomarker of lipid
peroxidative damage) induced by 4-HNE treatment was then measured.
It was indicated that treating blood plasma (Fig. 1A) or platelets (Fig. 1B) with 1 or 10 µM 4-HNE significantly
increased carbonyl group concentration. Likewise, treatment of
blood plasma (Fig. 2A) or platelets
(Fig. 2B) with 1 or 10 µM 4-HNE
significantly increased TBARS concentration. Treatment with 0.1 µM
4-HNE induced a significant increase in TBARS concentration in
platelets, but not in blood plasma samples.
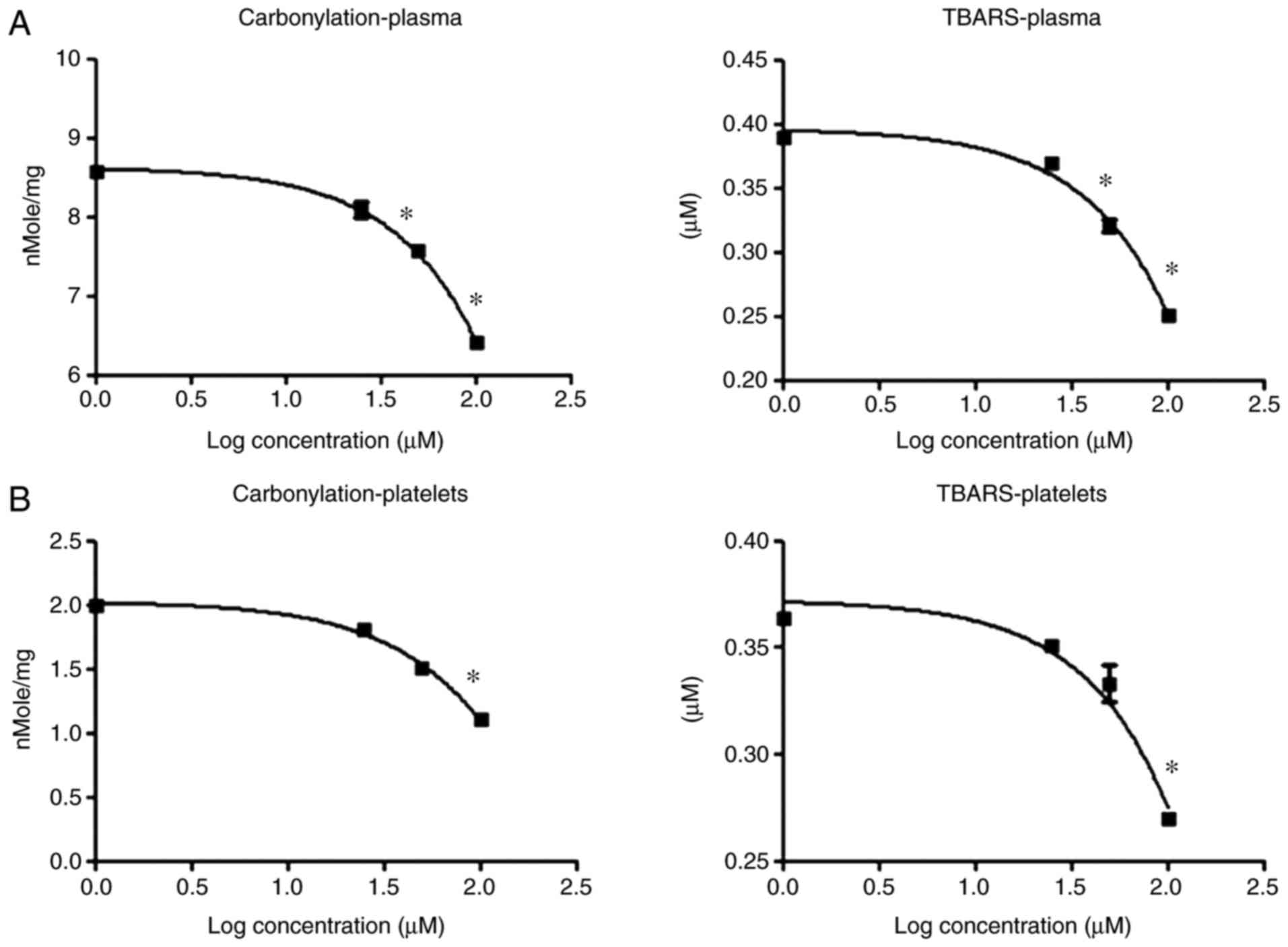
Phenelzine antagonizes the effect of
4-HNE on blood plasma and platelet proteins and lipids
Following quantitatively measuring the oxidative
damage induced by 4-HNE at two end points (protein carbonyl and
TBARS formation), the aim was to elucidate the mechanism by which
4-HNE produces these effects. Phenelzine is a drug that contains a
hydrazine moiety that allows it to function as an aldehyde
scavenger (28), and 4-HNE is an
aldehyde free radical species that results from lipid peroxidation
(29). It was therefore hypothesized
that phenelzine could reverse the oxidative damage produced by
4-HNE if these effects were associated with its chemical character
as a free aldehyde radical. As such, plasma and platelet samples
were treated with 4-HNE accompanied with an increasing dose of
phenelzine, and protein carbonyl and TBARS formation was measured
in the treated samples. Notably, phenelzine partially reversed the
protein carbonyl (Fig. 3A) and TBARS
(Fig. 3B) formation produced by
4-HNE treatment in blood plasma and platelet samples. These
findings suggest that the oxidative damage caused by 4-HNE is at
least partially due to its aldehyde free radical character.
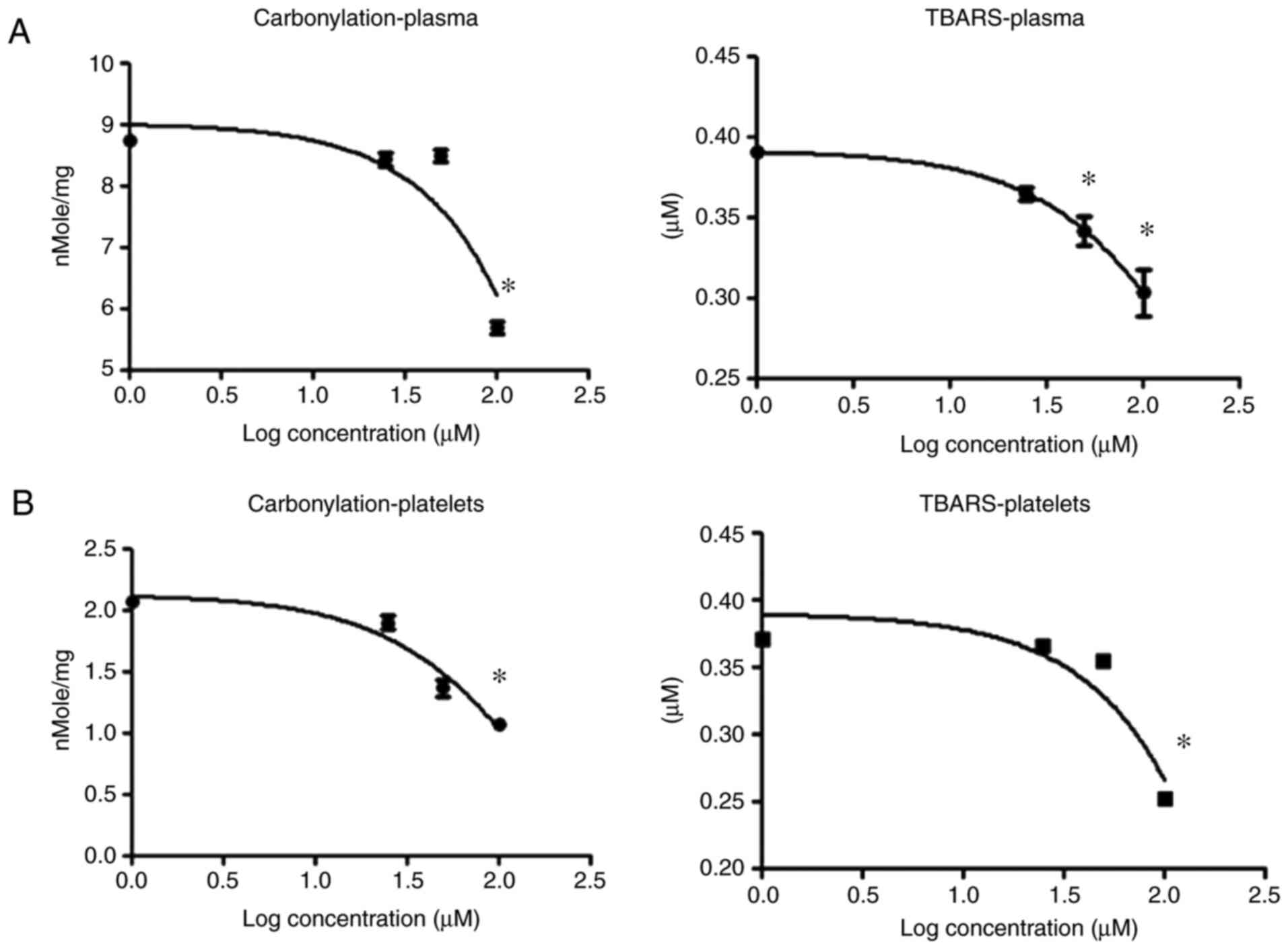
U-83836E antagonizes the effect of
4-HNE on blood plasma and platelet proteins and lipids
Lipid peroxidation proceeds through the formation of
a lipid radical followed by the generation of a lipid peroxyl
radical. If the oxidative damage caused by 4-HNE treatment depends
on initiating secondary lipid peroxidative damage, it may be
concluded that limiting the concentration of lipid peroxyl radicals
would reduce the oxidative damage triggered by 4-HNE. To explore
this, plasma and platelet samples were treated with 4-HNE alone or
in combination with the lipid peroxyl radical scavenger, U-83836E
(30), and protein carbonylation and
TBARS formation were estimated. Treatment with U-83836E antagonized
the effects of 4-HNE on protein carbonylation and TBARS formation
in blood plasma (Fig. 4A) and
platelet (Fig. 4B) samples. These
results suggest that the oxidative damage induced by 4-HNE
treatment is associated with the formation of lipid peroxyl
radicals; radicals known to be produced in lipid peroxidation
reactions.
Discussion
The findings of the present study provide a
mechanistic link between a reactive aldehyde species (4-HNE)
produced from lipid peroxidation of biological membranes and
increased oxidative stress in the intravascular compartment. The
results also highlight several pathways that may be
pharmacologically targeted to antagonize the impact of 4-HNE
production. Furthermore, the correct interpretation of these
results aids in the understanding of the exact mechanism(s) by
which 4-HNE produces its pathophysiological effects whether in the
intravascular compartment or in other biological niches.
Pharmacological mitigation of the effects of 4-HNE may be useful
for the therapeutic intervention of several chronic diseases such
as traumatic brain injury, Alzheimer's disease and diabetes.
Notable in the present study was the observation
that 4-HNE, considered an end product of lipid peroxidation, leads
to secondary lipid peroxidative damage as evidenced by an increase
in TBARS formation (a biomarker of lipid peroxidation) in blood
plasma and isolated platelets treated directly with 4-HNE. A direct
causal relationship between increased 4-HNE concentration and the
peroxidative damage of membrane lipids is supported by the ability
to reverse TBARS formation induced by 4-HNE treatment via the use
of a scavenger of reactive aldehyde species, phenelzine. As 4-HNE
is a reactive aldehyde species, phenelzine will presumably reduce
the concentration of 4-HNE available to induce the peroxidation of
biological membranes. In another experiment, TBARS formation
induced by 4-HNE treatment was reduced via the use of the lipid
peroxyl radical chelator, U-83836E. Lipid peroxyl radicals are
produced during lipid peroxidation following the reaction of oxygen
with lipid radicals, which are themselves produced as a result of
the abstraction of a proton from the double bonds of PUFAs of
biological membranes by oxygen radicals (hydroxyl and hydroperoxyl
radicals). As 4-HNE is not an oxygen radical itself, it is unlikely
to directly cause the formation of lipid radicals. However, it
seems that an increase in the concentration of 4-HNE can indirectly
cause an increase in the concentration of oxygen radicals, which
may initiate a new cycle of lipid peroxidation. In accordance with
the above tentative series of events is the finding that 4-HNE can
deplete the sulfhydryl groups of glutathione (31), which would presumably increase the
concentration of hydrogen peroxide, which can in turn decompose to
hydroxyl radicals.
In addition to demonstrating that 4-HNE direct
treatment causes secondary peroxidative damage of blood and
platelet lipids, it was also demonstrated that 4-HNE induced
oxidative damage of blood and platelet proteins in the form of
increased protein carbonylation. This is not surprising as it is
well documented that electrophiles such as 4-HNE can react in
Michael addition with side chains of cysteine and histidine and
cause increased protein carbonylation (32). The direct role of 4-HNE in increasing
protein carbonylation can be further validated through the present
finding that reducing the effective concentration of 4-HNE in via
chelating it with phenelzine decreased protein carbonylation. It
was also demonstrated that interrupting the cycle of lipid
peroxidation by scavenging lipid peroxyl radicals with U-83836E
reduced the effect of 4-HNE on protein carbonylation. These
findings suggest that 4-HNE treatment can induce secondary lipid
peroxidation reactions, which can lead to the formation of
increasing amounts of 4-HNE (and other reactive aldehyde species),
which may cause yet more protein carbonylation.
In addition to the role of 4-HNE in inducing its
oxidative damage on lipids and proteins in the context of secondary
lipid peroxidation reactions; several observations indicate that
4-HNE can also act as a cell-signaling molecule (33). Alteration of cell signaling pathways,
including the mitogen-activated protein kinase (34) and nuclear factor (erythroid-derived
2)-like 2 (35) pathways, may
contribute to the mechanisms which allow HNE to mediate oxidative
damage, or they may have their own independent effect. The role of
these pathways in 4-HNE mediated oxidative damage is an active area
of research for the present authors.
In conclusion, the present findings demonstrated
that 4-HNE treatment increased the rate of peroxidation of lipids,
and the oxidative damage of proteins in the blood and in isolated
platelets. The data indicates that this may be mitigated by
chelating 4-HNE, or any other reactive aldehyde species, with
phenelzine or by scavenging lipid peroxyl radicals with U-83836E.
The data presented is in accordance with a model in which 4-HNE is
associated a chain reaction that results in secondary lipid
peroxidation and progressively increasing amounts of 4-HNE. Based
on these results, it appears that the therapeutic options to
mitigate the cytotoxic effects of 4-HNE in the intravascular
compartment should involve approaches that reduce 4-HNE itself,
interrupt the lipid peroxidation process, or involve both methods
in combination.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Deanship of
Research at Jordan University of Science and Technology (grant no.
301/2014).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
All the authors participated in the design of the
study, analysis of the data and final review of the manuscript. AGM
and OA-S conceived the study and performed all the biochemical
measurements. AGM, MAA and OA-S collected the data. MAA performed
the statistical analysis. MAA and AGM drafted the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board affiliated with the Jordan University of Science and
Technology (Irbid, Jordan). All participants provided written
informed consent.
Patient consent for publication
All participants provided written informed
consent.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ray PD, Huang BW and Tsuji Y: Reactive
oxygen species (ROS) homeostasis and redox regulation in cellular
signaling. Cell Signal. 24:981–990. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bergamini CM, Gambetti S, Dondi A and
Cervellati C: Oxygen, reactive oxygen species and tissue damage.
Curr Pharm Des. 10:1611–1626. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bandyopadhyay U, Das D and Banerjee RK:
Reactive oxygen species: Oxidative damage and pathogenesis. Current
science. 658–666. 1999.
|
|
4
|
Halliwell B: Reactive oxygen species in
living systems: Source, biochemistry, and role in human disease. Am
J Med. 91:S14–S22. 1991. View Article : Google Scholar
|
|
5
|
Betteridge DJ: What is oxidative stress?
Metabolism. 49 2 Suppl 1:3–8. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shukla V, Mishra SK and Pant HC: Oxidative
stress in neurodegeneration. Adv Pharmacol Sci.
2011:5726342011.PubMed/NCBI
|
|
7
|
Andersen JK: Oxidative stress in
neurodegeneration: Cause or consequence? Nat Med. 10 Suppl:S18–S25.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rodriguez-Rodriguez A, Egea-Guerrero JJ,
Murillo-Cabezas F and Carrillo-Vico A: Oxidative stress in
traumatic brain injury. Current Med Chem. 21:1201–1211. 2014.
View Article : Google Scholar
|
|
9
|
Giacco F and Brownlee M: Oxidative stress
and diabetic complications. Circ Res. 107:1058–1070. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Asmat U, Abad K and Ismail K: Diabetes
mellitus and oxidative stress-A concise review. Saudi Pharm J.
24:547–553. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liochev SI: Reactive oxygen species and
the free radical theory of aging. Free Radic Biol Med. 60:1–4.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mustafa AG, Al-Shboul O, Alfaqih MA,
Al-Qudah MA and Al-Dwairi AN: Phenelzine reduces the oxidative
damage induced by peroxynitrite in plasma lipids and proteins. Arch
Physiol Biochem. 1–6. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mustafa AG, Bani-Ahmad MA, Jaradat AQ and
Allouh MZ: Tempol protects blood proteins and lipids against
peroxynitrite-mediated oxidative damage. Exp Biol Med (Maywood).
240:109–112. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mustafa AG, Singh IN, Wang J, Carrico KM
and Hall ED: Mitochondrial protection after traumatic brain injury
by scavenging lipid peroxyl radicals. J Neurochem. 114:271–280.
2010.PubMed/NCBI
|
|
15
|
Mylonas C and Kouretas D: Lipid
peroxidation and tissue damage. In vivo. 13:295–309.
1999.PubMed/NCBI
|
|
16
|
Jena NR: DNA damage by reactive species:
Mechanisms, mutation and repair. J Biosci. 37:503–517. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hemnani T and Parihar MS: Reactive oxygen
species and oxidative DNA damage. Indian J Physiol Pharmacol.
42:440–452. 1998.PubMed/NCBI
|
|
18
|
Suzuki YJ, Carini M and Butterfield DA:
Protein Carbonylation. Antioxid Redox Signal. 12:323–325. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ayala A, Muñoz MF and Argüelles S: Lipid
peroxidation: Production, metabolism, and signaling mechanisms of
malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid Med Cell Longev.
2014:3604382014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yin H, Xu L and Porter NA: Free radical
lipid peroxidation: Mechanisms and analysis. Chem Rev.
111:5944–5972. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Girotti AW: Lipid hydroperoxide
generation, turnover, and effector action in biological systems. J
Lipid Res. 39:1529–1542. 1998.PubMed/NCBI
|
|
22
|
Reis A and Spickett CM: Chemistry of
phospholipid oxidation. Biochim Biophys Acta. 1818:2374–2387. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dix TA and Aikens J: Mechanisms and
biological relevance of lipid peroxidation initiation. Chem Res
Toxicol. 6:2–18. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhong H and Yin H: Role of lipid
peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing
on mitochondria. Redox Biol. 4:193–199. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schaur RJ, Siems W, Bresgen N and Eckl PM:
4-Hydroxy-nonenal-A bioactive lipid peroxidation product.
Biomolecules. 5:2247–2337. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang Y, Sharma R, Sharma A, Awasthi S and
Awasthi YC: Lipid peroxidation and cell cycle signaling:
4-hydroxynonenal, a key molecule in stress mediated signaling. Acta
Biochim Pol. 50:319–336. 2003.PubMed/NCBI
|
|
27
|
Jang EJ, Jeong HO, Park D, Kim DH, Choi
YJ, Chung KW, Park MH, Yu BP and Chung HY: Src tyrosine kinase
activation by 4-hydroxynonenal upregulates p38, ERK/AP-1 signaling
and COX-2 expression in YPEN-1 Cells. PLoS One. 10:e01292442015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cebak JE, Singh IN, Hill RL, Wang JA and
Hall ED: Phenelzine protects brain mitochondrial function in vitro
and in vivo following traumatic brain injury by scavenging the
reactive carbonyls 4-hydroxynonenal and acrolein leading to
cortical histological neuroprotection. J Neurotrauma. 34:1302–1317.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jaganjac M, Tirosh O, Cohen G, Sasson S
and Zarkovic N: Reactive aldehydes-second messengers of free
radicals in diabetes mellitus. Free Radic Res. 47 Suppl 1:S39–S48.
2013. View Article : Google Scholar
|
|
30
|
Hall ED, Braughler JM, Yonkers PA, Smith
SL, Linseman KL, Means ED, Scherch HM, Von Voigtlander PF, Lahti RA
and Jacobsen EJ: U-78517F: A potent inhibitor of lipid peroxidation
with activity in experimental brain injury and ischemia. J
Pharmacol Exp Ther. 258:688–694. 1991.PubMed/NCBI
|
|
31
|
Raza H and John A: 4-hydroxynonenal
induces mitochondrial oxidative stress, apoptosis and expression of
glutathione S-transferase A4-4 and cytochrome P450 2E1 in PC12
cells. Toxicol Appl Pharmacol. 216:309–318. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Grimsrud PA, Xie H, Griffin TJ and
Bernlohr DA: Oxidative stress and covalent modification of protein
with bioactive aldehydes. J Biol Chem. 283:21837–21841. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schieber M and Chandel NS: ROS function in
redox signaling and oxidative stress. Curr Biol. 24:R453–R462.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Usatyuk PV and Natarajan V: Role of
mitogen-activated protein kinases in 4-hydroxy-2-nonenal-induced
actin remodeling and barrier function in endothelial cells. J Biol
Chem. 279:11789–11797. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang Y, Li W and Kong AN: Anti-oxidative
stress regulator NF-E2-related factor 2 mediates the adaptive
induction of antioxidant and detoxifying enzymes by lipid
peroxidation metabolite 4-hydroxynonenal. Cell Biosci. 2:402012.
View Article : Google Scholar : PubMed/NCBI
|