Introduction
Cardiovascular disease is one of the most common
non-cancer-related death and disability in the world (1,2). The
mortality of acute myocardial infarction has been decreased in the
past years (3). However, heart
failure-(HF-) induced mortality is still increasing due to the
deteriorating cardiac contractile function and left ventricular
remodeling (4,5). Unfortunately, poor knowledge is known
about the mechanism of metabolic dysfunction in HF.
p38MAPK signaling is suggested to play a key role in
the stressed cardiomyocytes (6–8). It is
reported that oxidative stress or ischemia reperfusion could
activate the p38MAPK, thereby inducing cardiomoycyte apoptosis
(9). Studies have shown that
inhibition of p38MAPK could improve inflammatory reactions and
protect cardiomyocytes from apoptosis (6,7).
The application of herbal medicines is common in
Asian countries due to the lower adverse effects and effectiveness
in various human diseases (10).
Rhein is isolated from the rhizome of rhubarb and is characterized
by broad pharmacological effects, including antidiabetic activity,
anti-inflammation and inhibition of interleukin-1-induced
chondrocyte activation (11,12). However, its application is largely
limited due to the poor water insolubility. And Rhein lysinate
(RHL) is modified with lysine, which is water soluble in drinking
water (11,12). However, whether RHL could improve the
cardiac function after HF has never been explored.
In the current study, we first explored the
protective role of RHL in HF. Our data showed that RHL could
inactivate p38MAPK signaling in cardiomyocytes, thereby protecting
cardiac function from HF.
Materials and methods
Experimental animals
A total of forty 11–14 week-old male healthy
Sprague-Dawley rats were obtained from Experimental Animal Center
of Tengzhou Central People's Hospital affiliated to Jining Medical
University. They were divided into 3 groups randomly: coarctation
of abdominal aorta group (A group, n=20) sham operation group (SH
group, n=10) and control group (C group, n=10). Laparotomy was
performed after anesthesia by intraperitoneal injection of 3%
pentobarbital sodium. Abdominal artery was stripped at
approximately 5 mm from the above left renal artery opening, a 6/0
silk suture was tied around and made up to 65–70% constriction of
abdominal aorta. RHL treatment rats (A+RHL group) were pretreated
with RHL (1.5 g/kg) for 3 days by gavage for 14 additional days.
Sham operated animals underwent the same procedure except the
ligation. Housing and procedures involving experimental animals
were in accordance with the Guide for the Care and Use of Tengzhou
Central People's Hospital affiliated to Jining Medical University.
All animal experiments were approved by the Animal Care and Studies
committee of Tengzhou Central People's Hospital affiliated to
Jining Medical University.
Preparation of RHL
RHL was purchased from the Shi-Feng Biological Co.,
Shanghai, China. The RHL was dissolved in PBS to 10 mg/ml and then
diluted with DMEM culture medium containing 10% FBS at different
concentrations.
Echocardiography
Rats were lightly anesthetized with 1–1.5% isolurane
in oxygen until the heart rate stabilized to 400 to 500 beats per
minute. Echocardiography was carried out with Vevo 770 and Vevo
2100 (VisualSonics) instruments. Fraction shortening (FS), ejection
fraction (EF), let ventricular internal diameter (LVID) during
systole, LVID during diastole, end-systolic volume, and
end-diastolic volume were calculated with Vevo Analysis software
(version 2.2.3). After that, rats were euthanatized by cervical
dislocation, and their hearts were collected for further
analyses.
Histology, immunoluorescence, and
immunohistochemistry
Heart tissues were cut into cryosections and
subsequently analyzed by H&E staining according to the
manufacturer's protocol (Sigma-Aldrich). For the histological
analysis, 8 µm sections were incubated with primary antibodies
overnight at 4°C. Then, the sections were washed with 0.25% Triton
X-100 in PBS and incubated with either fluorescently labeled
(Molecular Probes; Invitrogen) or biotinylated secondary (Vector)
antibodies for 2 h. Then, the sections were observed using
microscopy.
Terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling (TUNEL) staining
Nuclear fragmentation was detected by TUNEL staining
with an apoptosis detection kit (Roche) or by incubating fixed
cells using an apoptosis detection kit (R&D Systems) according
to the manufacturer's protocol. Cells (500–700) in 10 randomly
chosen fields from each dish were counted to determine the
percentage of apoptotic nuclei. Each data point indicates results
from 1,600 to 2,000 cells from 4 independent experiments.
Isolation and culture of rat cardiac
myocytes
Neonatal rat ventricular myocytes (NRVMs) were
isolated from 1–3-day-old Sprague Dawley rats via combined 0.2%
trypsin and 0.1% collagenase type II digestion. The cardiac
myocytes were plated at a density of 6.6×104
cells/cm2 in DMEM supplemented with 10% FBS supplemented
with 0.1 mM 5-bromo-2-deoxyuridine. Fibroblasts were not detected
in these cultures as determined by immunocytochemical staining with
an anti-fibronectin antibody.
Protein extraction and western blot
analysis
Proteins samples were extracted from cardiomyocytes
or myocardial tissue in RIPA buffer (1% TritonX-100, 15 mmol/l
NaCl, 5 mmol/l EDTA, and 10 mmol/l Tris-HCl (pH 7.0) (Solarbio,
China) supplemented with a protease and phosphatase inhibitor
cocktail (Sigma) and then separated by 10% SDS-PAGE, followed by
electrophoretic transfer to a PVDF membrane. After soaking with 8%
milk in PBST (pH 7.5) for 2 h at room temperature, the membranes
were incubated with the following primary antibodies: anti-p-p38,
anti-p38, Bcl-2, Bax and anti-GAPDH (Cell signaling).
Immunodetection was performed by enhanced chemiluminscence
detection system (Millipore) according to the manufacturer's
instructions. The house-keeping gene GAPDH was used as the internal
control.
MTT assay
NRVMs (5,000 cells/well) were plated in 24-well
plates and pretreated with RHL for 1 h and then treated with the
indicated concentrations of H2O2 for 24 h.
All assays were carried out in triplicate. The cells were incubated
with 0.5 mg/ml
3-[4,5-dimethylthiazol-2-yl]-2,5-di-phenyl-tetrazolium bromide for
4 h. And the relative fluorescence was determined at 490 nm as
previously described (13). The MTT
kit was purchased from Roche Applied Science (Indianapolis,
IN).
DHE staining
The living NRVMs were stained with 10 µmol/l DHE
(Sigma) for 30 min in a dark humidified chamber at 37°C. ROS
generation was indicated by red fluorescence and visualized with
fluorescence microscopy. The fluorescence intensity was analyzed as
previously described (13).
Statistical analysis
Data were presented as mean ± SD from 3 independent
experiments or 5 rats. Statistical analysis was carried out with
Student's t test. Multiple comparisons were evaluated by ANOVA
followed by Turkey's multiple-comparison test. P<0.05 was
considered as statistically significant difference.
Results
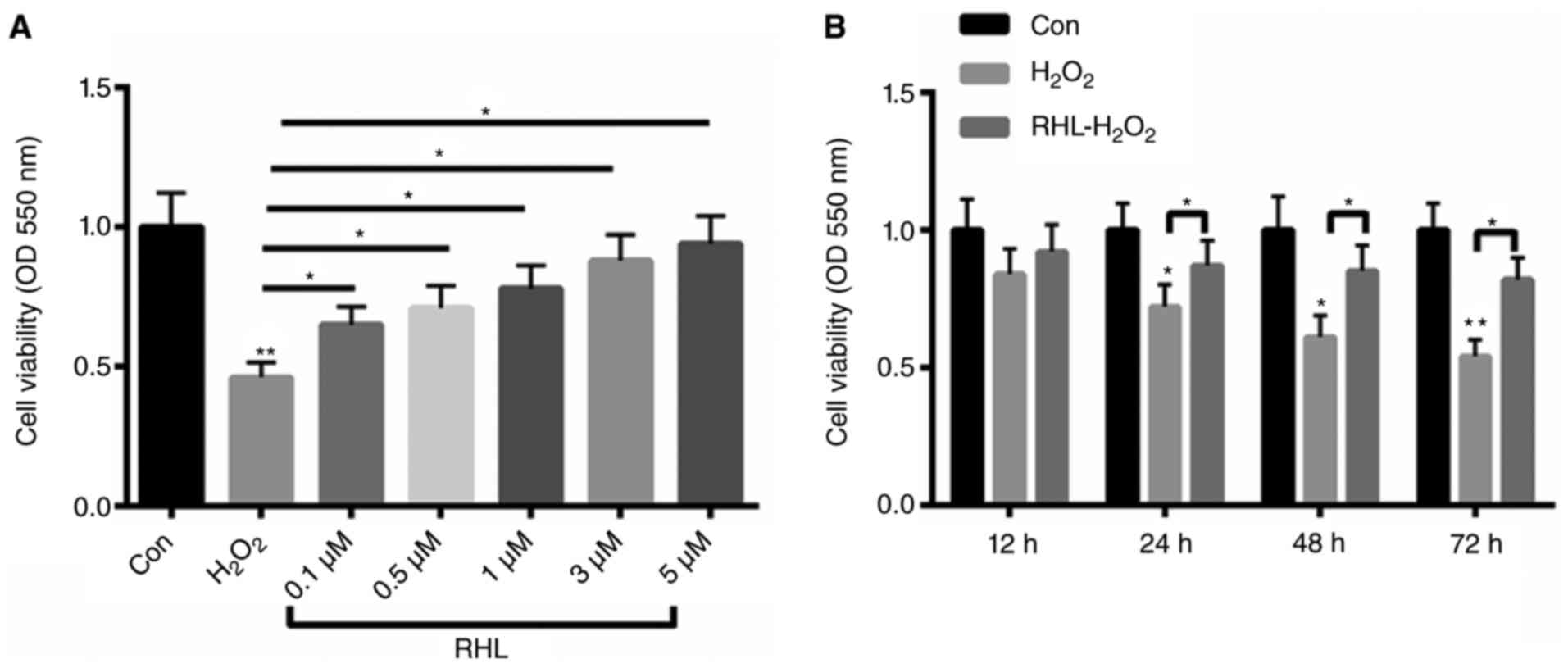
H2O2-induced cell viability could be
reversed by RHL in a dose- and time-dependent manner
Firstly, primary cardiomyocytes were treated with 10
µM H2O2 for 24 h. Then, the cells were
incubated with 0.1, 0.5, 1, 3, 5 µM RHL for 24 h. Treatment with 10
µM H2O2 decreased cell viability by more than
55%. In contrast, primary cardiomyocytes viability was increased by
19, 25, 32, 42, 48% with 0.1, 0.5, 1, 3, 5 µM RHL incubation by in
a dose- dependent manner (Fig. 1A).
Meanwhile, the cells were incubated with 1 µM RHL for 12, 24, 48,
72 h. As shown in Fig. 1B,
H2O2 treatment decreased cardiomyocyte
viability by 16, 28, 39, 46% at 12, 24, 48, 72 h, repectively.
However, RHL could improve H2O2-reduced
cardiomyocyte viability by 8, 15, 24, 28% in a time- dependent
manner (Fig. 1B).
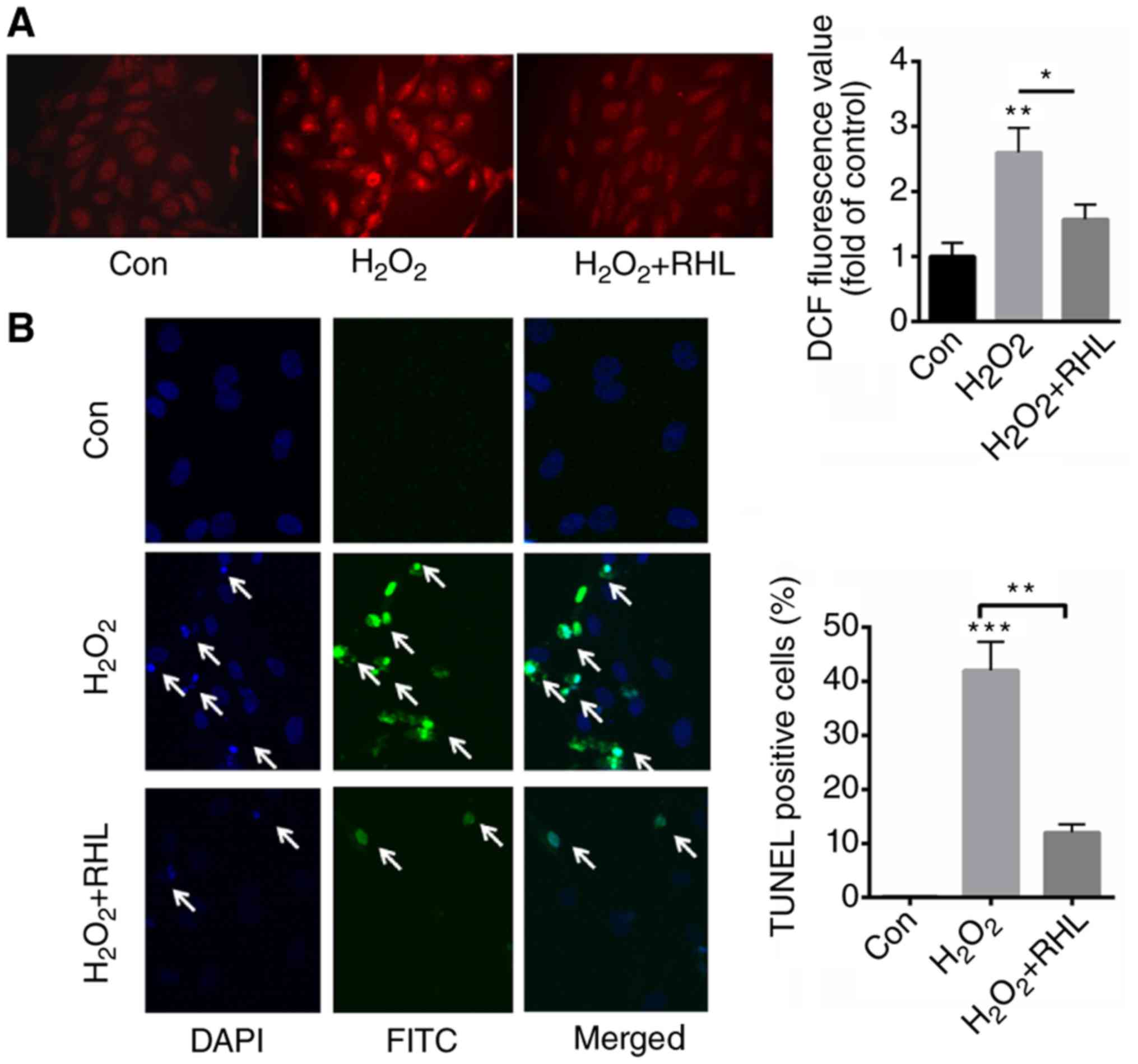
RHL reduced ROS production and cell
apoptosis induced by H2O2 treatment
DHE staining demonstrated that
H2O2 induced the production of ROS by
approximately 3.6 fold. In comparison, incubation with 1 µM RHL
decreased ROS production by about 2.1 fold than that of
H2O2 (Fig.
2A). Next, we further evaluated the role of RHL in
H2O2-induced cardiomyocytes apoptosis. TUNEL
staining indicated that H2O2 treatment
significantly increased apoptotic cells by 42% than that of normal
control (0.1%) (Fig. 2B). In
comparison, RHL incubation significantly decreased
H2O2-induced cardiomyocytes apoptosis by 30%
(Fig. 2B).
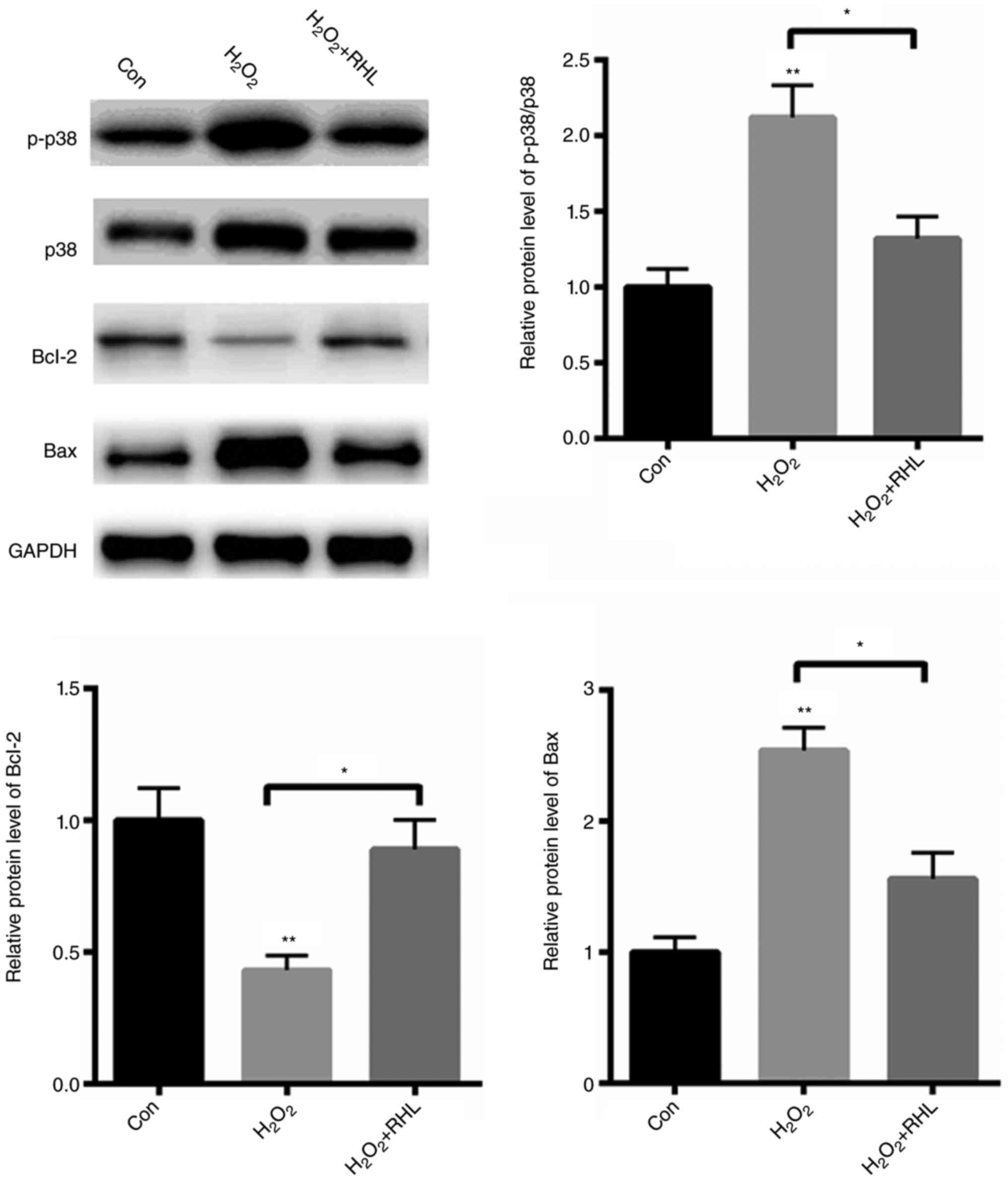
RHL reduced p38MAPK signaling
activation
Next, we explored the potential molecular mechanism
in which RHL protects cardiomyocytes from apoptosis. We found that
H2O2 treatment with markedly activated
p38MAPK signaling by ~1.12 fold (Fig.
3). Furthermore, the pro-apoptotic protein, Bax, was
significantly increased by about ~1.54 fold, while an
anti-apoptotic protein, Bcl-2, was decreased by 57% (Fig. 3). In comparison, RHL markedly reduced
p38MAPK signaling activation by 80%. Furthermore, RHL treatment
reduced the expression of Bax by 98%, but increased the protein
level of Bcl-2 by approximately ~1 fold (Fig. 3). These data indicated that RHL
protected primary cardiomyocytes from
H2O2-induced apoptosis mainly by suppressing
p38MAPK activation.
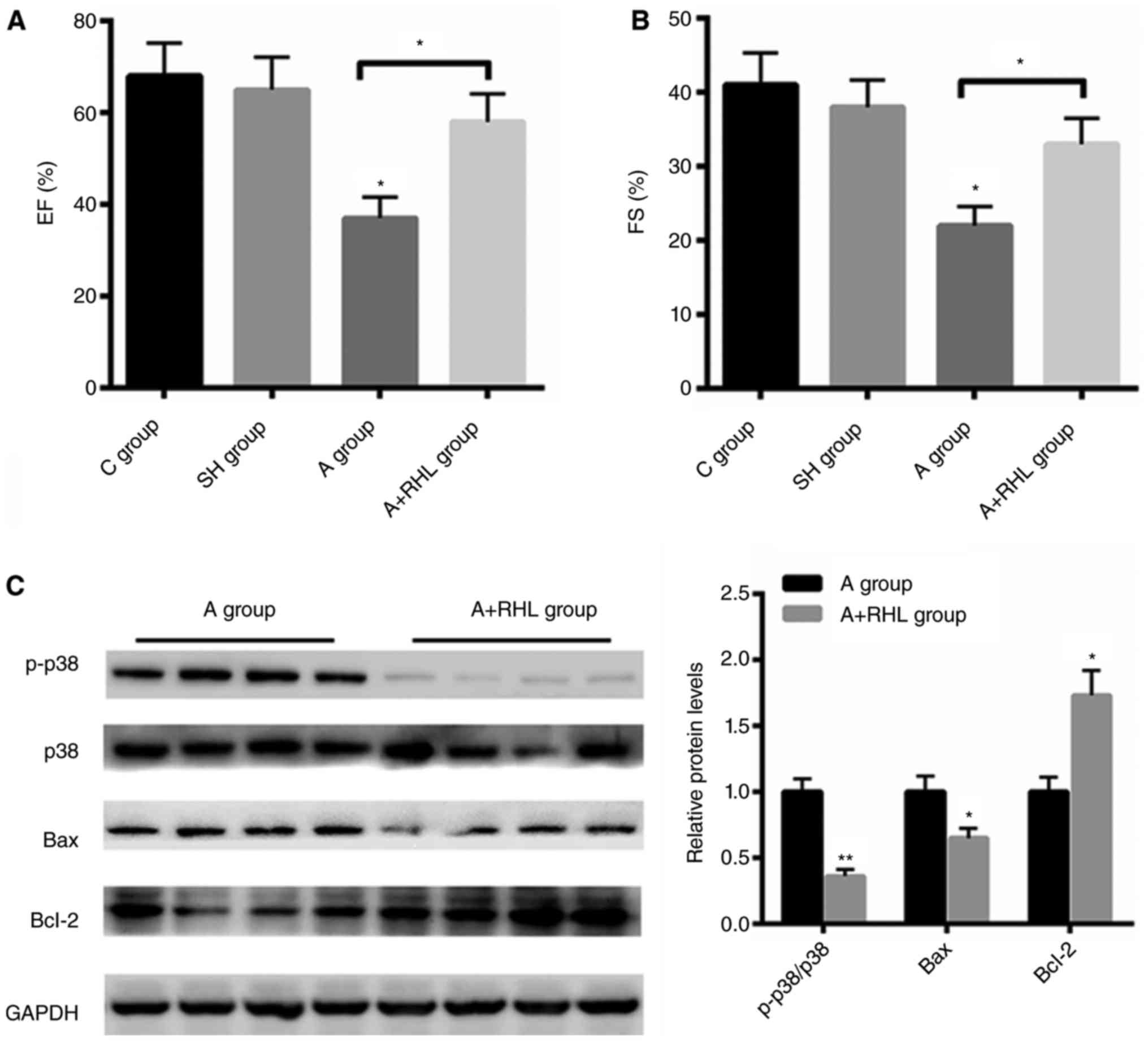
The improved heart function by RHL was
associated with p38MAPK signaling pathway
To further explore the effect of RHL on heart
function, echo analysis was carried out. Compared with control and
sham group, coarctation of abdominal aorta group demonstrated
reduced ejection fraction (EF)% and fraction shortening index (FS)%
by 28 and 16%, respectively (Fig. 4A and
B). But pre-treatment with RHL markedly increased heart
function by 21 and 11% than that of the operation group,
respectively (Fig. 4A and B).
Furthermore, we also found that RHL treatment markedly inactivated
p38MAPK signaling in coarctation of abdominal aorta group.
Moreover, the protein level of Bcl-2 was increased by ~1.73 fold
after RHL treatment, while the expression of Bax was decreased by
35% after RHL incubation in NRVMs (Fig.
4C). These data demonstrated that RHL protects heart failure
mainly by suppressing p38MAPK in vivo.
Discussion
Heart failure refers to a progressive circumstance
when the heart is unable to pump sufficient blood to fulfill the
body's requirements at a normal filling pressure (14). The pathology includes multiple
abnormities in heart muscle (15).
In the past years, enhanced oxidative stress is found to be
involved in the pathophysiology of congestive heart failure (CHF)
(16). Clinical data have shown that
patients with established CHF demonstrat increased oxidative stress
markers (8,17). Furthermore, the level of oxidative
stress is closely related to the severity of heart failure. Thus,
it is important to improve ROS production in failed hearts.
RHL has received attention for its protective role
in vitro. For instance, RHL was shown to reduce inflammation
and adipose infiltration in KK/HlJ diabetic mice with non-alcoholic
fatty liver disease (10). And RHL
was suggested to suppress the progression of breast and ovarian
cancer, hepatocellular carcinoma, cervical cancer and lung
carcinoma mainly by downregulation of Bcl-2 and cyclin D expression
and upregulation of BAX and Bim expression (18). In addition, RHL was demonstrated to
protect the livers by reducing the expression of TNF-α and IL-6 and
the phosphorylation of SREBP-1c and ERK1/2 in diabetic mice
(19). In the present study, we
mainly evaluated the effects of RHL on failed heart. In
vitro study showed that H2O2 treatment
reduced primary cardiomyocytes viability in a time- and
dose-dependent manner, while RHL could abolish the detrimental
effects of H2O2, indicating a protective role
of RHL. Further study found that H2O2-induced
ROS production could largely be reversed by RHL. Oxidative stress
is also suggested to activate cell apoptosis, thereby enhancing CHF
especially in the advanced stages (20,21).
Then, TUNEL staining was carried out and the results showed that
H2O2 markedly primary cardiomyocytes
apoptosis. In contrast, RHL incubation decreased
H2O2-induced cell apoptosis, indicating the
protective role of RHL in oxidative stressed primary
cardiomyocytes.
The mitogen-activated protein kinase p38 is an
important Ser/Thr kinase that is involved in heart failure
(6,8). Multiple studies have been performed to
explore the effects of p38 in heart failure (22,23). In
animal models, abnormal activation of p38 has been identified in
heart failure. Compared with healthy heart, enhanced p38 activity
is identified in the myocardial biopsies from heart failure
patients (24,25). In addition, in vitro studies
have shown that p38 activation enhances cardiomyocyte hypertrophy,
but inhibition of p38 signaling could diminish such effects
(26). In line with previous
studies, we found abnormal p38 activation in failed heart. More
importantly, treatment with RHL could reduce p38 activation.
p38 activation is indicated as a pro-apoptotic
process in cardiomyocytes (9). In
Raf-1-knockout mice which is characterized by left ventricular
systolic dysfunction and heart dilatation, enhanced cardiomyocyte
apoptosis is identified accompanied with an increase in p38 kinase
activity and cell apoptosis (27).
Furthermore, upregulation of p38α in cultured neonatal
cardiomyocytes (28) and expression
of transforming growth factor-β-activated kinase-1 are related to
significant cardiac apoptosis in the mouse heart (29). In the current study, we found that
treatment of RHL significantly enhanced the expression of
anti-apoptotic protein, Bcl-2, but markedly reduced the protein
level of Bax in primary cardiomyocytes, indicating its
anti-apoptotic role in the cardiac setting.
In summary, RHL protects heart failure mainly by
reducing ROS production and cardiomyocyte apoptosis through
reducing p38MAPK activation.
References
|
1
|
Suzuki H, Nodera M, Kamioka M, Kaneshiro
T, Kamiyama Y and Takeishi Y: Intracardiac impedance after cardiac
resynchronization therapy is a novel predictor for worsening of
heart failure. Heart Vessels. 32:926–931. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Senni M, D'Elia E, Emdin M and Vergaro G:
Biomarkers of heart failure with preserved and reduced ejection
fraction. Handb Exp Pharmacol. 243:79–108. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pecoraro A, Crescenzi L, Carucci L,
Genovese A and Spadaro G: Heart failure not responsive to standard
immunosuppressive therapy is successfully treated with high dose
intravenous immunoglobulin therapy in a patient with eosinophilic
granulomatosis with polyangiitis (EGPA). Int Immunopharmacol.
45:13–15. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Marx N: Heart failure and
diabetes-underestimated, underdiagnosed and poorly understood: A
call for action. Diab Vasc Dis Res. 14:67–68. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mathur A, Fernández-Avilés F, Dimmeler S,
Hauskeller C, Janssens S, Menasche P, Wojakowski W, Martin JF and
Zeiher A: BAMI Investigators: The consensus of the Task Force of
the European Society of Cardiology concerning the clinical
investigation of the use of autologous adult stem cells for the
treatment of acute myocardial infarction and heart failure: Update
2016. Eur Heart J. Feb 15–2017.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arabacilar P and Marber M: The case for
inhibiting p38 mitogen-activated protein kinase in heart failure.
Front Pharmacol. 6:1022015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cook SA, Sugden PH and Clerk A: Activation
of c-Jun N-terminal kinases and p38-mitogen-activated protein
kinases in human heart failure secondary to ischaemic heart
disease. J Mol Cell Cardiol. 31:1429–1434. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Di Lisa F, Kaludercic N and Paolocci N:
β2-Adrenoceptors, NADPH oxidase, ROS and p38 MAPK:
Another ‘radical’ road to heart failure? Br J Pharmacol.
162:1009–1011. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marber MS, Rose B and Wang Y: The p38
mitogen-activated protein kinase pathway-a potential target for
intervention in infarction, hypertrophy, and heart failure. J Mol
Cell Cardiol. 51:485–490. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wei J, Zhen YZ, Cui J, He FL, Shen T, Hu
G, Ren XH and Lin YJ: Rhein lysinate decreases inflammation and
adipose infiltration in KK/HlJ diabetic mice with non-alcoholic
fatty liver disease. Arch Pharm Res. 39:960–969. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin YJ, Zhen YZ, Wei J, Liu B, Yu ZY and
Hu G: Effects of Rhein lysinate on H2O2-induced cellular senescence
of human umbilical vascular endothelial cells. Acta Pharmacol Sin.
32:1246–1252. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin YJ, Zhen YZ, Zhao YF, Wei J and Hu G:
Rhein lysinate induced S-phase arrest and increased the anti-tumor
activity of 5-FU in HeLa cells. Am J Chin Med. 39:817–825. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hannan RD, Luyken J and Rothblum LI:
Regulation of ribosomal DNA transcription during
contraction-induced hypertrophy of neonatal cardiomyocytes. J Biol
Chem. 271:3213–3220. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Song B, Li T, Chen S, Yang D, Luo L, Wang
T, Han X, Bai L and Ma A: Correlations between MTP and ros levels
of peripheral blood lymphocytes and readmission in patients with
chronic heart failure. Heart Lung Circ. 25:296–302. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rojas A, Mercadal E, Figueroa H and
Morales MA: Advanced Glycation and ROS: A link between diabetes and
heart failure. Curr Vasc Pharmacol. 6:44–51. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Astashkin EI, Glezer MG, Vinokurov MG,
Egorova ND, Orekhova NS, Novikova AN, Grachev SV, Yurinskaya MM and
Sobolev KE: Actovegin reduces the ROS level in blood samples of
heart failure patients and diminishes necrosis of SK-N-SH human
neuroblastoma cells. Dokl Biol Sci. 448:57–60. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Borchi E, Bargelli V, Stillitano F,
Giordano C, Sebastiani M, Nassi PA, d'Amati G, Cerbai E and Nediani
C: Enhanced ROS production by NADPH oxidase is correlated to
changes in antioxidant enzyme activity in human heart failure.
Biochim Biophys Acta. 1802:331–338. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu J, Zhang K, Zhen YZ, Wei J, Hu G, Gao
JL, Tian YX and Lin YJ: Antitumor activity of rhein lysinate
against human glioma U87 cells in vitro and in vivo. Oncol Rep.
35:1711–1717. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin YJ, Hu G, Li KJ, Zhao YF, Wei J and
Zhen YZ: The protection of Rhein lysinate to liver in diabetic mice
induced by high-fat diet and streptozotocin. Arch Pharm Res.
38:885–892. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hafstad AD, Nabeebaccus AA and Shah AM:
Novel aspects of ROS signalling in heart failure. Basic Res
Cardiol. 108:3592013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zuo L, Chuang CC, Hemmelgarn BT and Best
TM: Heart failure with preserved ejection fraction: Defining the
function of ROS and NO. J Appl Physiol (1985). 119:944–951. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Heusch P, Canton M, Aker S, van de Sand A,
Konietzka I, Rassaf T, Menazza S, Brodde OE, Di Lisa F, Heusch G
and Schulz R: The contribution of reactive oxygen species and p38
mitogen-activated protein kinase to myofilament oxidation and
progression of heart failure in rabbits. Br J Pharmacol.
160:1408–1416. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hoefer J, Azam MA, Kroetsch JT, Leong-Poi
H, Momen MA, Voigtlaender-Bolz J, Scherer EQ, Meissner A, Bolz SS
and Husain M: Sphingosine-1-phosphate-dependent activation of p38
MAPK maintains elevated peripheral resistance in heart failure
through increased myogenic vasoconstriction. Circ Res. 107:923–933.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kerkela R and Force T: p38
mitogen-activated protein kinase: A future target for heart failure
therapy? J Am Coll Cardiol. 48:556–558. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kita T, Ogawa M, Sato H, Kasai K, Tanaka T
and Tanaka N: Role of p38 mitogen-activated protein kinase pathway
on heart failure in the infant rat after burn injury. Int J Exp
Pathol. 89:55–63. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nemoto S, Sheng Z and Lin A: Opposing
effects of Jun kinase and p38 mitogen-activated protein kinases on
cardiomyocyte hypertrophy. Mol Cell Biol. 18:3518–3526. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Irukayama-Tomobe Y, Miyauchi T, Kasuya Y,
Sakai S, Goto K and Yamaguchi I: Activation of peroxisome
proliferator-activated receptor-alpha decreases
endothelin-1-induced p38 mitogen-activated protein kinase
activation in cardiomyocytes. J Cardiovasc Pharmacol. 44 Suppl
1:S358–S361. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Inagaki K, Satoh T, Yagi-Utsumi M, Le
Gulluche AC, Anzai T, Uekusa Y, Kamiya Y and Kato K: Redox-coupled
structural changes of the catalytic a' domain of protein disulfide
isomerase. FEBS Lett. 589:2690–2694. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang D, Gaussin V, Taffet GE, Belaguli
NS, Yamada M, Schwartz RJ, Michael LH, Overbeek PA and Schneider
MD: TAK1 is activated in the myocardium after pressure overload and
is sufficient to provoke heart failure in transgenic mice. Nat Med.
6:556–563. 2000. View
Article : Google Scholar : PubMed/NCBI
|