Introduction
Rheumatoid arthritis (RA), a chronic and systemic
autoimmune disease, primarily affects the peripheral synovial
joints and causes synovitis, which thereby damages the articular
cartilage as well as bone tissues (1). Currently, on account of the
irreversible joint damage and systemic osteoporosis, RA represents
one of the most disabling diseases. It has been published that
0.5–1% of adults are affected by RA in the developed world
(2). It was reported that the
incidence of RA in 2010 was 28.5 per 100,000 person-years in Korea
(3). Additionally, RA is considered
a chronic inflammatory disease and an autoimmune disease, due to
the hyperplasia of synovial tissues in an inflammatory environment
(4). Persistent and progressive
development of inflammation in RA may develop into articular
spasticity, deformity and dysfunction, which seriously affect the
health and quality of life of patients with RA (5). The pathological mechanism of RA is
complex, and basic pathological features are involved in systemic
synovitis and formation of pannus. The immunopathology of pannus is
divided into two parts: Immune and invasion (6–8).
Osteoclasts and synovial fibroblasts (SFBs) are involved in the
invasion. SFBs primarily consist of two types: Type A
(macrophage-like) and type B (fibroblast-like). The number of
fibroblast-like synoviocytes (FLS) is large, and their rich nuclear
material suggests that these cells are in an active state of RNA
metabolism (9). In addition, RAFLS
represent a type of primary cell that can be cultured,
proliferated, and passaged multiple times in vitro to
maintain defined growth and invasion capacities, which is similar
to tumor cells (10). Delays in the
diagnosis and early treatment of RA lead to poor RA prognoses.
Therefore, in order to improve the prognosis of RA patients,
in-depth investigations into the underlying genetic or
pathophysiological factors are necessary for developing effective
treatments for RA.
Mitogen-activated protein kinases (MAPKs), a class
of intracellular serine/threonine protein kinases located in the
majority of cells, can transmit extracellular stimuli into
intracellular signaling pathways leading to a series of cellular
biological reactions, including cell differentiation, proliferation
and apoptosis (11). In mammals,
MAPKs are associated with four major pathways, including p38, c-Jun
N-terminal kinase (JNK), extracellular signal-regulated kinases
(ERKs), which contribute to the cellular responses to various
stimuli, such as inflammatory cytokines, physical and chemical
stress and bacterial products (12).
It has been suggested that both p38 and JNK serve crucial roles in
the biological activity of FLS in RA (13,14). In
addition, previous studies have demonstrated that the p38/JNK
signaling pathway contributes to the modulation of migration in
various cells, including smooth muscle cell subtypes (15), tumor cells (16–18) and
stem cells (19). However, little is
known about the roles and mechanisms of the p38/JNK signaling
pathway in the migration of FLS in RA.
Homeobox D10 (HOXD10), as a member of human homeobox
(HOX) gene family, is associated with cell differentiation and
morphogenesis during embryonic development (20,21).
Previous studies have demonstrated close associations between HOX
gene families and the occurrence, development and prognosis of
multiple malignant tumors (22,23). In
addition, HOX gene families have been demonstrated to participate
in the regulation of tumor cell proliferation, differentiation,
apoptosis via the adenomatous polyposis coli protein/β-catenin
signaling pathways (24,25). Recently, it has been demonstrated
that HOXD10 serves as a modulator in the migration of several
cancer cell types (26–28). Nevertheless, the function of HOXD10
in the migration of RAFLS remains unclear. Therefore, HOXD10 was
selected as the focus of this study to further explore its role and
mechanisms in RA.
In the current study, the correlation between HOXD10
and the migration of RAFLS was analyzed. Furthermore, the exact
role and mechanisms of HOXD10 within thep38/JNK signaling pathway
were investigated with respect to the migration of RAFLS.
Materials and methods
Tissue samples
A total of 30 patients (age range, 37–60;
male:female, 9:21) diagnosed with RA in the Jining No. 1 People's
Hospital (Jining, China) from December 2014 to January 2016 were
included in this study. Immediately following resection, all tissue
samples were snap-frozen in liquid nitrogen and stored at −80°C.
Matched adjacent normal synovium tissue was collected and used as a
negative control. All patients included in the study provided
written informed consent for the utilization of the tissue samples
for clinical research. The project protocol was approved by the
Institutional Review Board of Jining No. 1 People's Hospital.
Cell culture, gene, plasmid and
grouping
Human RAFLS were separated from the RA knee-joint
tissue samples of an RA patient selected at random. RAFLS were
maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.)
in a 5% CO2 atmosphere at 37°C. The HOXD10 small
interfering RNA (siRNA) was cloned into the empty pcDNA3.1 (+)
vector (Invitrogen; Thermo Fisher Scientific, Inc.), further
referred to as si-HOXD10 [this was a gift from Corey Largman
(Addgene plasmid cat. no. 21007)]. Cells were divided into the
following three treatment groups: Control group, untreated RAFLS;
NC group, RAFLS transfected with empty vector; and siHOXD10 group,
RAFLS transfected with si-HOXD10.
Cell viability analysis
A cell counting kit-8 (CCK-8; Beyotime Institute of
Biotechnology, Shanghai, China) was used to study the viability of
RAFLS. RAFLS in the logarithmic phase (6×104 cells/ml)
were seeded into the wells of 96-well plates, and then incubated in
5% CO2 atmosphere at 37°C for 12 h. RAFLS were then
divided into the three aforementioned treatment groups. The cells
were maintained for 24, 48 and 72 h. A total of 10 µl of CCK-8
reagent was then added to the wells. Cells were maintained for a
further 3 h at 37°C. A microplate reader was used to record the
absorbance at 450 nm. Cell viability was quantified as the
percentage of surviving cells relative to the control.
Wound healing assay
Cultured RAFLS were seeded in 24-well plates
(1×105 cells/ml) and divided into the three
aforementioned treatment groups. RAFLS were then maintained for 15
days until the cell confluence reached 100%. Horizontal ‘wounds’
were generated using a 10 µl pipette tip scraping along the surface
of each well. Cells were then washed with Hanks liquor (Beijing
Solarbio Science & Technology Co., Ltd., Beijing, China) three
times to remove the loose cells and serum-free medium (Gibco;
Thermo Fisher Scientific, Inc.) was then added. Pictures were
obtained using an inverted microscope at 0 and 24 h time
points.
Migration assay
Cells were transfected with an empty vector or
siHOXD10. After 36 h, RAFLS were digested using 0.25% trypsin
(Gibco; Thermo Fisher Scientific, Inc.). Cells (1×106
cells/ml; 100 µl) were plated in DMEM without serum in the top
chamber of the Transwell insert (Corning Incorporated, Corning, NY,
USA); while DMEM with 20% FBS was added in the lower well. After 24
h of incubation at 37°C, cells in the lower well were treated with
1 ml paraformaldehyde (4%) for 30 min at room temperature and
subsequently stained with 0.1% crystal violet (Beijing Solarbio
Science & Technology Co., Ltd.) for 20 min at room temperature.
Cells were washed three times with PBS and the optical density was
recorded at 570 nm using a microplate reader.
Western blot analysis
The cells were seeded at a density of
1×103 cells/well into the 24-well plates. Following the
cell confluence reaching 65%, the cells were transfected with empty
vector or siHOXD10. After 36 h, the proteins were isolated with
NP40 lysis buffer (Beyotime Institute of Biotechnology). The
concentration was estimated by a BCA kit provided by Bio-Rad
Laboratories, Inc. (Hercules, CA, USA). The proteins (25 µg/lane)
were separated by 12% SDS-PAGE gels. The separated products were
transferred to a PVDF membrane. The membranes were blocked with 5%
non-skimmed milk at temperature for 2 h at room temperature.
Blotting was performed with specific antibodies at 4°C overnight:
Rabbit anti-human anti-HOXD10 (dilution, 1:1,000; cat. no.
ab138508); anti-cadherin-11 (dilution, 1:500; cat. no. ab151302);
anti-N-cadherin (dilution, 1:1,000; cat. no. ab76057);
anti-E-cadherin (dilution, 1:500; cat. no. ab15148); anti-vimentin
(dilution, 1:500; cat. no. ab137321); anti-zonula occludens-1
(Zo-1; dilution, 1:1,000; cat. no. ab216880); anti-integrin β1
(dilution, 1:2,000; cat. no. ab179471); anti-paxillin (dilution,
1:500; cat. no. ab2264); anti-phosphorylated (p)-p38 (dilution,
1:1,000; cat. no. ab4822); anti-p38 (dilution, 1:2,000; cat. no.
ab170099); anti-p-JNK (dilution, 1:2,000; cat. no. ab124956);
anti-JNK (dilution, 1:1,000; cat. no. ab179461); anti-GAPDH
(dilution, 1:2,500; cat. no. ab9485); anti-β-actin (dilution,
1:1,000; cat. no. ab8227; all Abcam, Cambridge, UK). The membranes
were then incubated with horseradish peroxidase-conjugated
secondary antibodies (goat anti-rabbit; dilution, 1:5,000; cat. no.
ab205718; Abcam) at room temperature for 1 h. Enhanced
chemiluminescent stain (EMD Millipore, Billerica, MA, USA) was used
to evaluate the results. The density of the bands were quantified
with ImageQuant software (version 5.2; Molecular Dynamics; GE
Healthcare, Chicago, IL, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
The cells were seeded at a density of
1×103 cells/well into the 24-well plates. Following the
cell confluence reaching 65%, the cells were transfected with empty
vector or siHOXD10. After 36 h, total RNA was extracted from
cultured RAFLS using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). RNA was reverse transcribed to cDNA using a
Reverse Transcription kit (Beyotime Institute of Biotechnology)
according to the manufacturer's instructions. RT-qPCR analysis was
performed with an ABI 7500 Thermocycler (Applied Biosystems; Thermo
Fisher Scientific, Inc.) PCR cycles were as followed: 4 min
pretreatment at 95°C; followed by 35 cycles of 95°C for 25 sec and
60°C for 30 sec; then extension at 72°C for 10 min. The samples
were then maintained at 4°C. The primers were designed by
Invitrogen (Thermo Fisher Scientific, Inc.): HOXD10, forward,
5′-AAGATGAACGAGCCCGTGAG-3′, and reverse,
5′-TCCAGCGTTTGGTGCTTAGT-3′, (product, 242 bp); cadherin-11,
forward, 5′-GCCGAAGCCTACATCCTCAA-3′, and reverse,
5′-TCAACGCTATTGGGAGCTGG-3′, (product, 332 bp); N-cadherin, forward,
5′-CCAAGTGAACGATAAGGGCG-3′, and reverse,
5′-CCTTACTCTTGCCACCCTGA-3′, (product, 209 bp); E-cadherin, forward,
5′-GTGAACACCTACAATGCCGC-3′, and reverse,
5′-CAAAATCCAAGCCCGTGGTGG-3′, (product, 274 bp); vimentin, forward,
5′-AATAAGATCCTGCTGGCCGA-3′, and reverse,
5′-GGTGTTTTCGGCTTCCTCTC-3′, (product, 225 bp); Zo-1, forward,
5′-GGAGGTAGAACGAGGCATCA-3′, and reverse,
5′-AGGCCTCAGAAATCCAGCTT-3′, (product, 238 bp); integrinβ1, forward,
5′-TCCAACCTGATCCTGTGTCC-3′, and reverse,
5′-CAATTCCAGCAACCACACCA-3′, (product, 174 bp); paxillin, forward,
5′-ATCCTGAGTGCTTTGTGTGC-3′, and reverse,
5′-CCTTGTTGAGCTGCTTGAGG-3′, (product, 225 bp); GAPDH, forward,
5′-GAGTCCACTGGCGTCTTCA-3′, and reverse, 5′-GGTCATGAGTCCTTCCACGA-3′,
(product, 240 bp); β-actin, forward, 5′-GTTACAGGAAGTCCCTCACCC-3′,
and reverse, 5′-CAGACCTGGGCCATTCAGAAA-3′, (product, 194 bp). GAPDH
and β-actin were used as the endogenous controls. The
2−ΔΔCq method was used for the calculation of relative
expression (29).
Statistical analysis
The results are reported as the mean ± standard
error of the mean of at least three independent experiments. All
the experimental data was analyzed by paired Student's t-test, or
one-way analysis of variance followed by a Tukey's test. The
statistical significance was defined as P<0.05.
Results
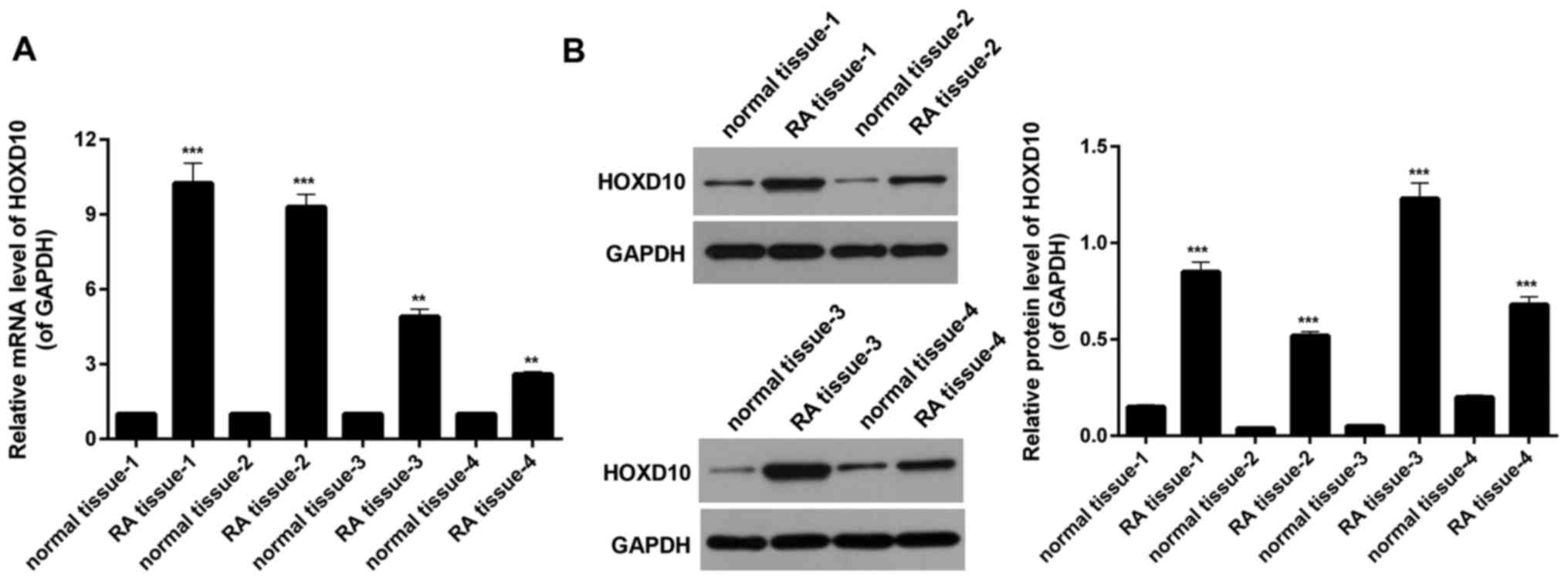
HOXD10 expression is upregulated in
tissues from patients with RA
RT-qPCR analysis was performed to examine the
expression of HOXD10 in human RA tissue samples and matched
adjacent normal synovium tissues of the knee-joint from four
patients with RA, selected at random. It was revealed that the
expression of HOXD10 in RA tissues was significantly higher
compared with that observed in the matched adjacent normal tissues
(P<0.01; Fig. 1A). RT-qPCR and
western blot analyses were performed to evaluate the HOXD10 protein
expression in four random paired human RA and normal adjacent
synovium knee-joint tissues. The results indicated that the HOXD10
expression was significantly enhanced in RA tissue compared with
the control sample (P<0.001; Fig.
1B). This suggests that HOXD10 expression may be upregulated in
RA tissue.
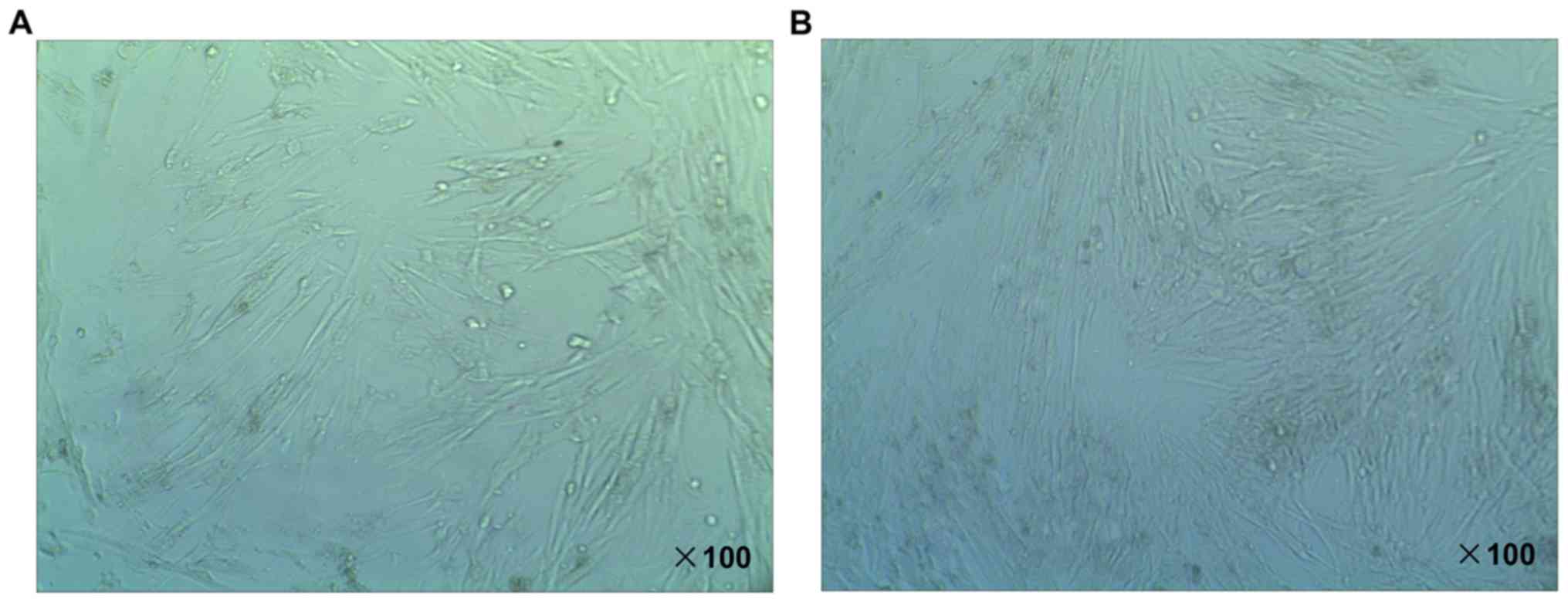
Identification of RAFLS
The SFBs were isolated from RA knee-joint tissue
samples of a single patient with RA selected at random. During the
course of the experiments, SFBs were cultured in DMEM medium
containing 10% FBS. Following 24 h in cell culture, the SFBs were
studied using an inverted microscope. The adherent cells were SFBs.
As shown in Fig. 2A, the morphology
of SFBs was spindle-shaped, stellate and polygonal. The boundaries
of nuclei were clear and oval-shaped. The nucleus was located in
the middle of cells, and the nucleolus was clear. At passage four,
FLS accounted for more than 98% of the total SFBs (Fig. 2B), which were harvested and used in
subsequent studies.
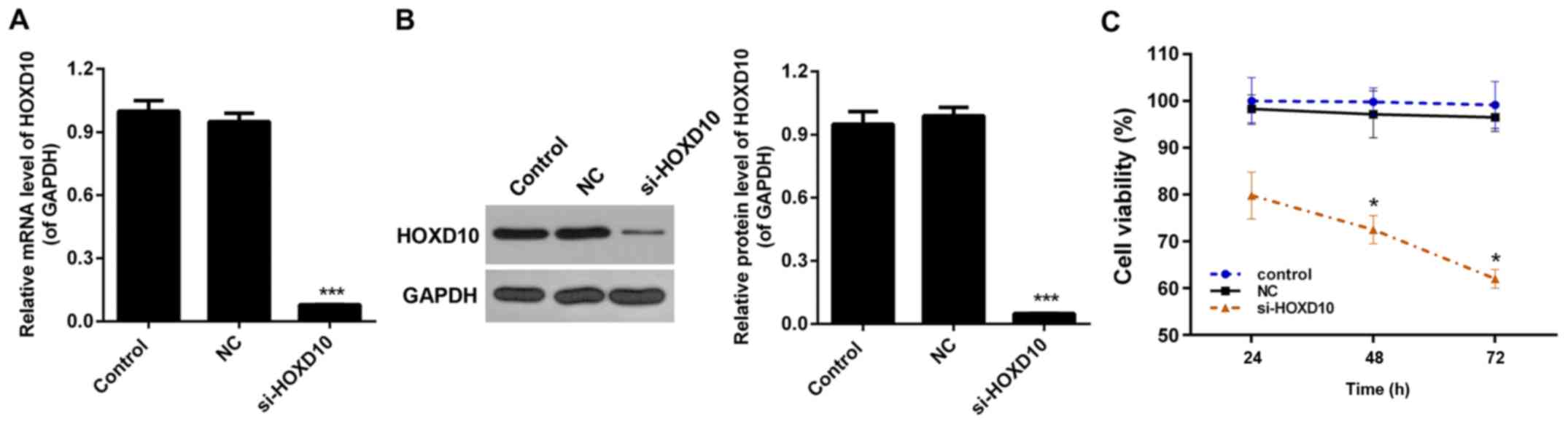
Interference of HOXD10 in RAFLS
An siRNA vector targeting HOXD10, si-HOXD10, was
constructed in the present study. RAFLS separated from RA patients
were transfected with the pcDNA3.1 (+) empty vector or si-HOXD10.
The knockdown efficiency was ~90% in RAFLS following stable
transfection with si-HOXD10 (P<0.001; Fig. 3A). Western blot analysis demonstrated
that following transfection with si-HOXD10, the expression level of
HOXD10 was significantly reduced (P<0.001; Fig. 3B).
HOXD10 silencing inhibits the
viability of RAFLS
A CCK-8 assay was performed to measure the viability
of RAFLS divided into the three aforementioned treatment groups.
The results demonstrated that, compared with the control groups,
HOXD10 silencing inhibited the viability of RAFLS, particularly at
the 48 and 72 h time points (P<0.05; Fig. 3C).
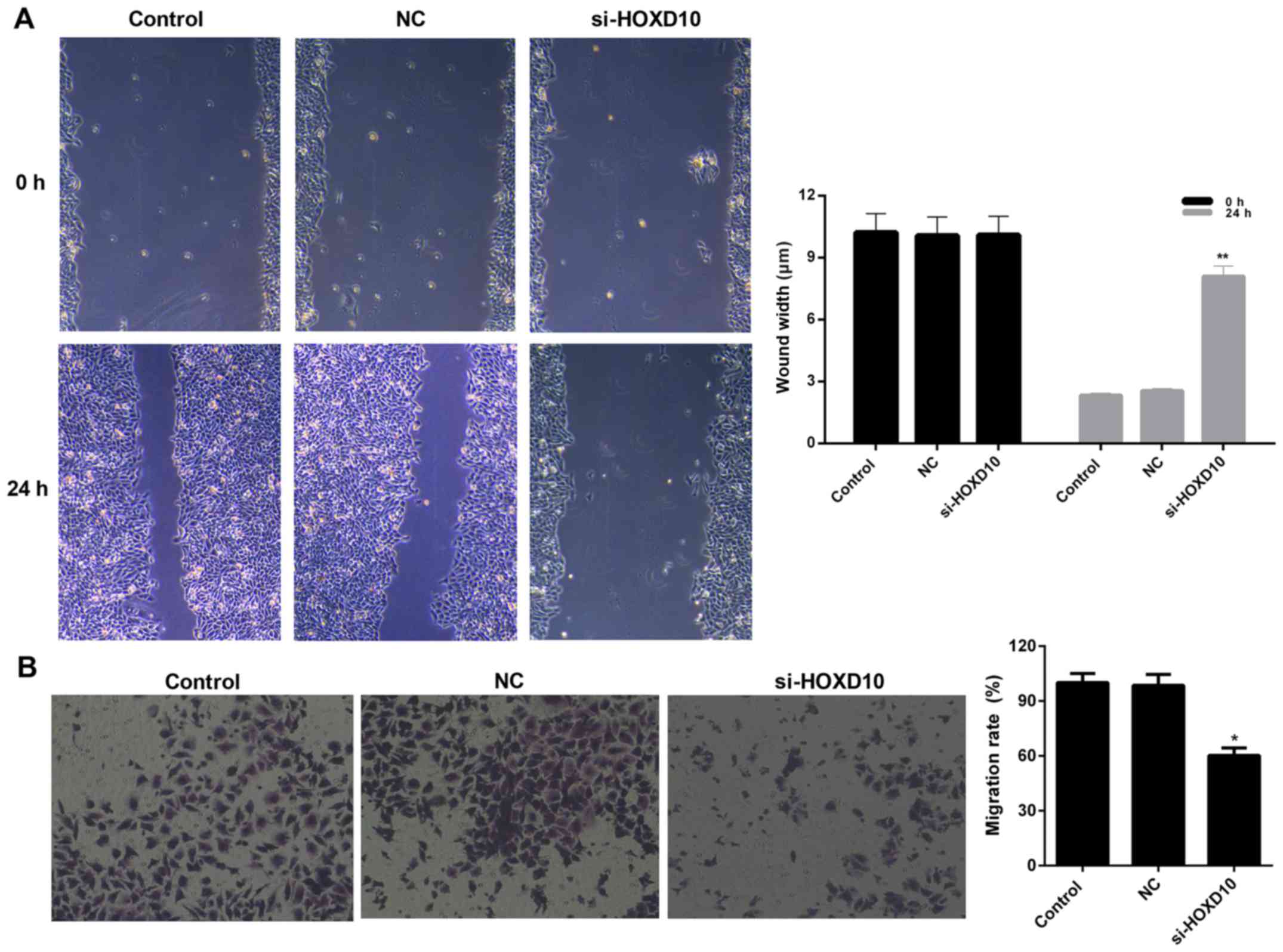
HOXD10 silencing suppresses the
migration of RAFLS
The migration capacity of RAFLS was evaluated using
wound healing and migration assays. The wound healing assay results
revealed that the wound width of RAFLS transfected with si-HOXD10
was significantly increased compared with the NC group (P<0.01;
Fig. 4A). A similar trend was
observed from the migration assay where the number of migrated
RAFLS transfected with si-HOXD10 decreased from 100 to 60.3%
(P<0.05; Fig. 4B). These results
indicate, that HOXD10 silencing may decrease the migration ability
of RAFLS.
HOXD10 silencing modulates the
expression of migration- associated proteins in RAFLS
Considering the results of the migration assay,
migration-associated mechanisms in RAFLS and the expression levels
of migration-associated proteins, includingcadherin-11, N-cadherin,
E-cadherin, vimentin, Zo-1, integrinβ1 and paxillin were assessed.
On the basis of the RT-qPCR results, it was revealed that HOXD10
silencing significantly inhibited the expression levels of
cadherin-11, E-cadherin, Zo-1, integrinβ1 and paxillinin RAFLS
(P<0.01; Fig. 5A). In addition,
it was observed that N-cadherin and vimentin expression in RAFLS
was significantly upregulated following transfection with si-HOXD10
(P<0.001; Fig. 5A). Western blot
analysis revealed similar trends for migration-associated protein
expression in RAFLS (Fig. 5B and C).
Based on these observations, it was identified that HOXD10
silencing may suppress the migration of RAFLS through modulating
the expression levels of cadherin-11, N-cadherin, E-cadherin,
vimentin, Zo-1, integrin β1 and paxillin.
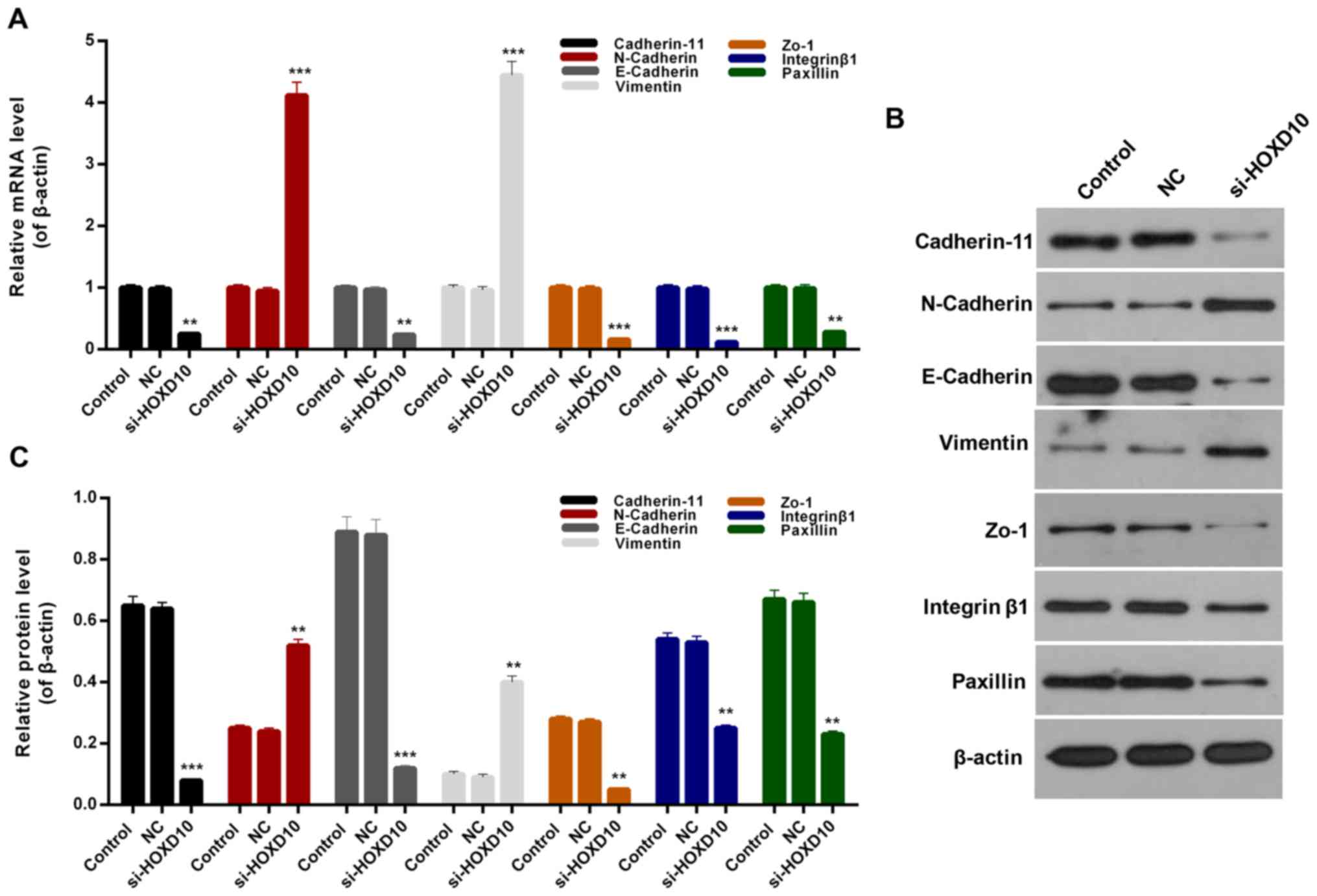 | Figure 5.HOXD10 silencing affects the
expression of migration-associated proteins. (A) Reverse
transcription-quantitative polymerase chain reaction and (B)
western blot assays were performed to determine the expression
levels of cadherin-11, N-cadherin, E-cadherin, vimentin, Zo-1,
integrin β1 and paxillin in RAFLS, RAFLS transfected with empty
vector or si-HOXD10. (C) Quantified western blotting results.
**P<0.01 and ***P<0.001 vs. NC. HOXD10, homeobox D10; Zo-1,
zonula occludens-1; RAFLS, fibroblast-like synoviocytes in
rheumatoid arthritis; si-HOXD10, small interfering RNA targeting
HOXD10; NC, negative control. |
HOXD10 silencing downregulates the
p38/JNK signaling pathway
In order to further investigate the association
between HOXD10 and the p38/JNK signaling pathway in RA, the
expression levels of p-p38, p38, p-cJNK and JNK in RAFLS were
evaluated. Western blot analysis indicated that the expression
levels of p-p38 and p-JNK in RAFLS were significantly downregulated
following transfection with si-HOXD10 (P<0.05; Fig. 6). Therefore, HOXD10 silencing was
demonstrated to suppress the phosphorylation of p38 and JNK in
RAFLS. By contrast, there was no significant difference in p38 and
JNK expression in RAFLS among each experimental group (Fig. 6). Therefore, it was concluded that
HOXD10 silencing may affect the p38/JNK signaling pathway in
RAFLS.
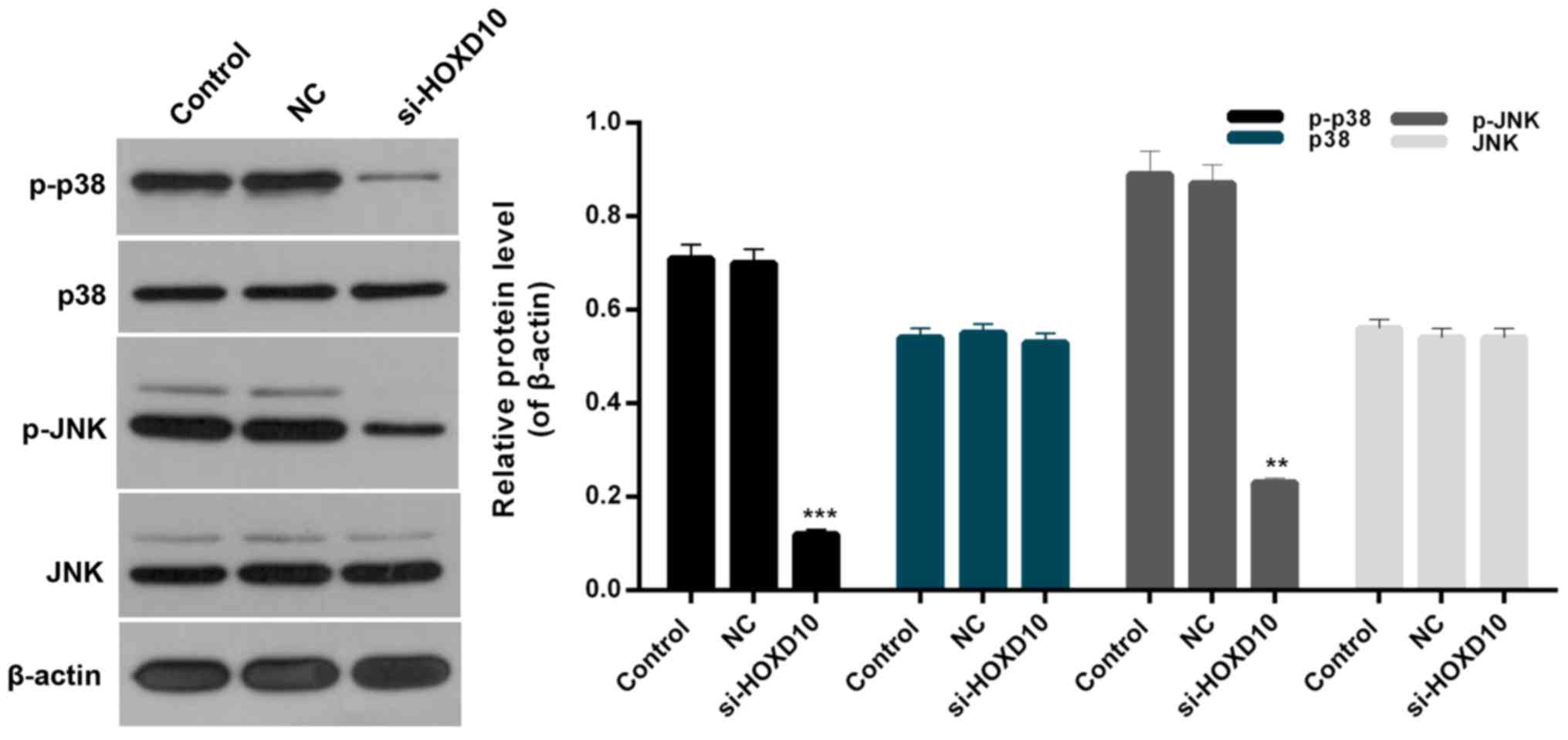 | Figure 6.HOXD10 silencing downregulates the
p38/JNK signaling pathway. Western blot assays were performed to
measure the expression levels of p-p38, p38, p-JNK and JNK in
RAFLS, RAFLS transfected with empty vector or si-HOXD10.
**P<0.01 and ***P<0.001 vs. NC. HOXD10, homeobox D10; JNK,
c-Jun N-terminal kinase; p-p38, phosphorylated p38; p-JNK,
phosphorylated JNK; RAFLS, fibroblast-like synoviocytes in
rheumatoid arthritis; si-HOXD10, small interfering RNA targeting
HOXD10; NC, negative control. |
Discussion
RA is a chronic autoimmune disease with unknown
etiology, which seriously endangers human health and life due to
increased morbidity and the early age of onset (30). A crucial pathological feature of RA
is chronic inflammatory lesions of synovium of joints, which
destroys the articular cartilage and bone tissue. FLS are normally
present in the synovial lining, but abnormal proliferation occurs
in the FLS of patients with RA and gradually erodes the adjacent
cartilage and bone (9). Although
increased RAFLS in the synovial lining has been observed in
situ, the index of enhancement of RA synovial membrane was not
sufficient to account for the observed extent of synovial
hyperplasia (31,32). However, FLS from these tissues have
been activated sustainably in vitro, with increased
proliferative capacity and the ability to express a variety of
growth factors, inflammatory cytokines, oncogenes and cyclins, thus
exhibiting a tumor-like proliferation (33). Recently, studies have demonstrated
that the FLS migration may also represent an important source of
synovial hyperplasia. For example, it has been demonstrated that
CA074Me, an inhibitor of cathepsin B, suppressed the migration and
invasion of FLS in inflamed tissues of patients with RA (34). Therefore, inhibition of RAFLS
migration may serve as a pivotal target for RA therapies.
Previous studies have suggested that HOXD10, a
well-known tumor suppressor gene, serves a crucial role in the
suppression of migration of various tumor cells (26–28),
while the potential functions of HOXD10 in RAFLS migration remain
unclear. Therefore, the aim of the present study was to investigate
the involvement of HOXD10 in the suppression of RAFLS migration,
and the associated mechanisms. Four pairs of human RA tissues and
matched adjacent normal synovium tissues of knee-joints were
randomly selected from RA patients and HOXD10 expression in these
tissue samples was measured. According to the RT-qPCR and western
blot results, it was revealed that the expression levels of HOXD10
in RA tissues were significantly higher compared with those in the
control tissue. Consequently, HOXD10 may serve as an important
target in RA therapies. An siRNA vector targeting HOXD10 was
constructed in this investigation and was used to transfect RAFLS,
with the empty vector as a negative control. The knockdown
efficiency was ~90% in RAFLS following stable transfection with
si-HOXD10. The viability of RAFLS transfected with empty vector and
si-HOXD10 was further assessed. It was revealed that HOXD10
silencing significantly suppressed the viability of RAFLS,
particularly at 72 h following transfection. Moreover, the
migration capacity of RAFLS transfected with empty vector and
si-HOXD10 was evaluated by wound healing and Transwell assays.
Based on the results, it was revealed that HOXD10 silencing
significantly reduced the migration capacity of RAFLS. In order to
investigate the involvement of HOXD10 in the migration of RAFLS
further, the expression of several migration-associated proteins
were measured. N-cadherin, E-cadherin and vimentin are the most
common markers used to evaluate cellular migration abilities
(35,36). In addition, cell adhesion is
associated with cell migration (37,38).
Therefore, specific cell adhesion-associated molecules were
selected to assess cell migration in the present study, including
Zo-1, integrinβ1 and paxillin. It was revealed that HOXD10
silencing significantly downregulated the expression levels of
cadherin-11, E-cadherin, Zo-1, integrinβ1 and paxillin, while it
enhanced the expression of N-cadherin and vimentin in RAFLS.
Collectively, these results suggested that HOXD10 silencing may
suppress the migration of RAFLS through influencing the expression
levels of cadherin-11, N-cadherin, E-cadherin, vimentin, Zo-1,
integrinβ1 and paxillin.
The p38/JNK signaling pathway has been demonstrated
to participate in cell migration processes (39,40).
However, the association between the p38/JNK signaling pathway and
RAFLS migration was not very clear. In the current study, the
expression levels of p-p38, p38, p-JNK and JNK in RAFLS transfected
with empty vector and si-HOXD10 were evaluated. The results
indicated that HOXD10 silencing reduced the phosphorylation of p38
and JNK in RAFLS, whereas no significant difference was detected in
p38 and JNK expression in RAFLS among all groups. These results
indicated that HOXD10 silencing affected the p38/JNK signaling
pathway in RAFLS.
In conclusion, the results of the present study
demonstrated that HOXD10 silencing inhibited the migration of RAFLS
potentially via the p38/JNK signaling pathway. The results provide
valuable insight into the mechanisms of HOXD10 and FLS. The
potential effects of HOXD10 on the migration of RAFLS suggest that
HOXD10 may presentan effective target for RA therapies.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated and/or analyzed during the
present study are included in this published article.
Authors' contributions
LL wrote the main manuscript. LL and YM performed
the experiments. LZ designed the study. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Jining No. 1 People's Hospital. Informed consent was
obtained from the patients.
Patient consent for publication
Written informed consent was obtained from all
participants for the publication of their data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Smolen JS, Aletaha D, Koeller M, Weisman
MH and Emery P: New therapies for treatment of rheumatoid
arthritis. Lancet. 370:1861–1874. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Smolen JS, Aletaha D and McInnes IB:
Rheumatoid arthritis. Lancet. 388:2023–2038. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Won S, Cho SK, Kim D, Han M, Lee J, Jang
EJ, Sung YK and Bae SC: Update on the prevalence and incidence of
rheumatoid arthritis in Korea and an analysis of medical care and
drug utilization. Rheumatol Int. 38:649–656. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Feldmann M, Brennan FM, Foxwell BM and
Maini RN: The role of TNF alpha and IL-1 in rheumatoid arthritis.
Curr Dir Autoimmun. 3:188–199. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pugner KM, Scott DI, Holmes JW and Hieke
K: The costs of rheumatoid arthritis: An international long-term
view. Semin Arthritis Rheum. 29:305–320. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brennan FM, Hayes AL, Ciesielski CJ, Green
P, Foxwell BM and Feldmann M: Evidence that rheumatoid arthritis
synovial T cells are similar to cytokine-activated T cells:
Involvement of phosphatidylinositol 3-kinase and nuclear factor
kappaB pathways in tumor necrosis factor alpha production in
rheumatoid arthritis. Arthritis Rheum. 46:31–41. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lutzky V, Hannawi S and Thomas R: Cells of
the synovium in rheumatoid arthritis. Dendritic cells. Arthritis
Res Ther. 9:2192007. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weyand CM, Kurtin PJ and Goronzy JJ:
Ectopic lymphoid organogenesis: A fast track for autoimmunity. Am J
Pathol. 159:787–793. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bartok B and Firestein GS: Fibroblast-like
synoviocytes: Key effector cells in rheumatoid arthritis. Immunol
Rev. 233:233–255. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huber LC, Distler O, Tarner I, Gay RE, Gay
S and Pap T: Synovial fibroblasts: Key players in rheumatoid
arthritis. Rheumatology (Oxford). 45:669–675. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cuschieri J and Maier RV:
Mitogen-activated protein kinase (MAPK). Crit Care Med. 33 12
Suppl:S417–S419. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Inoue T, Hammaker D, Boyle DL and
Firestein GS: Regulation of p38 MAPK by MAPK kinases 3 and 6 in
fibroblast-like synoviocytes. J Immunol. 174:4301–4306. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Inoue T, Hammaker D, Boyle DL and
Firestein GS: Regulation of JNK by MKK-7 in fibroblast-like
synoviocytes. Arthritis Rheum. 54:2127–2135. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kavurma MM and Khachigian LM: ERK, JNK,
and p38 MAP kinases differentially regulate proliferation and
migration of phenotypically distinct smooth muscle cell subtypes. J
Cell Biochem. 89:289–300. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu QR, Liu JM, Chen Y, Xie XQ, Xiong XX,
Qiu XY, Pan F, Liu D, Yu SB and Chen XQ: Piperlongumine inhibits
migration of glioblastoma cells via activation of ROS-dependent p38
and JNK signaling pathways. Oxid Med Cell Longev. 2014:6537322014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ma CY, Ji WT, Chueh FS, Yang JS, Chen PY,
Yu CC and Chung JG: Butein inhibits the migration and invasion of
SK-HEP-1 human hepatocarcinoma cells through suppressing the ERK,
JNK, p38, and uPA signaling multiple pathways. J Agric Food Chem.
59:9032–9038. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Song H and Moon A: Glial cell-derived
neurotrophic factor (GDNF) promotes low-grade Hs683 glioma cell
migration through JNK, ERK-1/2 and p38 MAPK signaling pathways.
Neurosci Res. 56:29–38. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kuang D, Zhao X, Xiao G, Ni J, Feng Y, Wu
R and Wang G: Stem cell factor/c-kit signaling mediated cardiac
stem cell migration via activation of p38 MAPK. Basic Res Cardiol.
103:265–273. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Botas J: Control of morphogenesis and
differentiation by HOM/Hox genes. Curr Opin Cell Biol. 5:1015–1022.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Samuel S and Naora H: Homeobox gene
expression in cancer: Insights from developmental regulation and
deregulation. Eur J Cancer. 41:2428–2437. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Grier DG, Thompson A, Kwasniewska A,
Mcgonigle GJ, Halliday HL and Lappin TR: The pathophysiology of HOX
genes and their role in cancer. J Pathol. 205:154–171. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cantile M, Pettinato G, Procino A,
Feliciello I, Cindolo L and Cillo C: In vivo expression of the
whole HOX gene network in human breast cancer. Eur J Cancer.
39:257–264. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jung C, Kim RS, Zhang H, Lee SJ, Sheng H,
Loehrer PJ, Gardner TA, Jeng MH and Kao C: HOXB13 is downregulated
in colorectal cancer to confer TCF4-mediated transactivation. Br J
Cancer. 92:2233–2239. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shah N and Sukumar S: The Hox genes and
their roles in oncogenesis. Nat Rev Cancer. 10:361–371. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nakayama I, Shibazaki M, Yashima-Abo A,
Miura F, Sugiyama T, Masuda T and Maesawa C: Loss of HOXD10
expression induced by upregulation of miR-10b accelerates the
migration and invasion activities of ovarian cancer cells. Int J
Oncol. 43:63–71. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang YY, Li L, Ye ZY, Zhao ZS and Yan ZL:
MicroRNA-10b promotes migration and invasion through Hoxd10 in
human gastric cancer. World J Surg Oncol. 13:2592015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xiao H, Li H, Yu G, Xiao W, Hu J, Tang K,
Zeng J, He W, Zeng G, Ye Z and Xu H: MicroRNA-10b promotes
migration and invasion through KLF4 and HOXD10 in human bladder
cancer. Oncol Rep. 31:1832–1838. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gibofsky A: Overview of epidemiology,
pathophysiology, and diagnosis of rheumatoid arthritis. Am J Manag
Care. 18 13 Suppl:S295–S302. 2012.PubMed/NCBI
|
|
31
|
Henderson B and Pettipher ER: The synovial
lining cell: Biology and pathobiology. Semin Arthritis Rheum.
15:1–32. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zvaifler NJ: Relevance of the stroma and
epithelial-mesenchymal transition (EMT) for the rheumatic diseases.
Arthritis Res Ther. 8:2102006. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Firestein GS: Invasive fibroblast-like
synoviocytes in rheumatoid arthritis. Passive responders or
transformed aggressors? Arthritis Rheum. 39:1781–1790. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tong B, Wan B, Wei Z, Wang T, Zhao P, Dou
Y, Lv Z, Xia Y and Dai Y: Role of cathepsin B in regulating
migration and invasion of fibroblast-like synoviocytes into
inflamed tissue from patients with rheumatoid arthritis. Clin Exp
Immunol. 177:586–597. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nijkamp MM, Span PN, Hoogsteen IJ, van der
Kogel AJ, Kaanders JH and Bussink J: Expression of E-cadherin and
vimentin correlates with metastasis formation in head and neck
squamous cell carcinoma patients. Radiother Oncol. 99:344–348.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang X, Liu G, Yi K, Dong Z, Qian Q and
Ma X: N-cadherin expression is associated with acquisition of EMT
phenotype and with enhanced invasion in erlotinib-resistant lung
cancer cell lines. PLoS One. 8:e576922013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Newgreen DF and Gooday D: Control of the
onset of migration of neural crest cells in avian embryos. Role of
Ca++-dependent cell adhesions. Cell Tissue Res. 239:329–336. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Clay MR and Halloran MC: Regulation of
cell adhesions and motility during initiation of neural crest
migration. Curr Opin Neurobiol. 21:17–22. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen HJ, Lin CM, Lee CY, Shih NC, Peng SF,
Tsuzuki M, Amagaya S, Huang WW and Yang JS: Kaempferol suppresses
cell metastasis via inhibition of the ERK-p38-JNK and AP-1
signaling pathways in U-2 OS human osteosarcoma cells. Oncol Rep.
30:925–932. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Desai S, Laskar S and Pandey BN: Autocrine
IL-8 and VEGF mediate epithelial-mesenchymal transition and
invasiveness via p38/JNK-ATF-2 signalling in A549 lung cancer
cells. Cell Signal. 25:1780–1791. 2013. View Article : Google Scholar : PubMed/NCBI
|