Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is an
aggressive hematological malignancy, which accounts for 25% of
adult ALL cases (1). Although the
clinical outcome has been dramatically improved by a combination of
chemotherapy and hematopoietic stem cell transplantation, the
prognosis of T-ALL remains poor due to a high frequency of
induction failure and early relapse. Therefore, continued studies
to identify innovative modes of action in T-ALL and the development
of specific targeting therapies are still urgently required
(2).
The effect of prostaglandin E2 (PGE2) on cell growth
has attracted attention in recent years. Previous studies have
demonstrated that the secretion of PGE2 in endometrial cancer,
colon cancer, colorectal cancer, ovarian cancer and other malignant
cells is significantly increased (3,4),
indicating that PGE2 is implicated in the occurrence and
progression of cancer. Microsomal prostaglandin E synthase-1
(mPGES-1) is the terminal synthase responsible for converting
COX-derived PGH2 into PGE2 (5).
Non-steroidal anti-inflammatory drugs (NSAIDs) may reduce the
synthesis of PGE2 by inhibiting COX and affecting various
biological functions of tumors (6–8).
However, due to the gastrointestinal and cardiovascular side
effects of NSAIDs, their clinical application has been limited
(9,10). In recent years, overexpression of
mPGES-1 was observed in a number of solid tumors (11–13). A
preliminary study by the authors confirmed for the first time that
mPGES-1 is highly expressed in human acute myeloid leukemia (AML)
primary cells and AML cell lines such as HL-60. Inhibiting
mPGES-1/PGE2 may induce apoptosis, inhibit proliferation, arrest
the cell cycle and improve chemosensitivity (14–16);
however, the roles of mPGES-1/PGE2 in T-ALL cells are largely
unknown.
Mitogen-activated protein kinases (MAPKs), including
extracellular signal-regulated kinase (ERK1/2), c-Jun N-terminal
kinase (JNK) and P38 subtypes, are a highly-conserved family of
serine/threonine kinases that serve an important role in regulating
cell growth, differentiation, inflammatory reactions and cancer
progression (17,18). Previous studies have revealed that
different MAPKs may be involved in the regulation of mPGES-1
expression induced by inflammatory stimuli (19–21).
Interestingly, certain previous studies have hypothesized that the
MAPK signaling pathway resides upstream of PGE2 and regulates the
synthesis of PGE2 through early growth response protein-1 (EGR-1)
(19), while in other contexts, it
is located downstream of mPGES-1/PGE2 (20,21). The
MAPK signaling pathway, once activated, may be further regulated by
complex feedback loops exerting either positive or negative effects
on cascade components (22). The
present study aimed to investigate the effects of mPGES-1 on T-ALL
jurkat cells in vitro and attempted to determine the
interaction between mPGES-1 and MAPKs in jurkat cells.
Materials and methods
Materials
Human T-ALL jurkat cell line was obtained from the
Hematology Research Institute (Tianjin, China). The EP4 receptor
antagonist L-161982 was purchased from Cayman Chemical Company (Ann
Arbor, MI, USA). The MEK1/2 inhibitor U0126, JNK inhibitor SP600125
and the P38 inhibitor SB203580 were purchased from Selleck
Chemicals (Shanghai, China). The anti-ERK1/2 (cat. no. 9926;
dilution, 1:1,000), anti-p-ERK1/2 (Thr202/Tyr204; cat. no. 9910;
dilution, 1:2,000), anti-P38 (cat. no. 9926; dilution, 1:1,000),
anti-p-P38 (Thr180/Tyr182; cat. no. 9910; dilution, 1:1,000),
anti-JNK (cat. no. 9926; dilution, 1:1,000), anti-p-JNK
(Thr183/Tyr185; cat. no. 9910; dilution, 1:1,000), anti-EGR-1 (cat.
no. 4153; dilution, 1:1,000), anti-GAPDH (cat. no. 2118; dilution,
1:1,000) and anti-rabbit IgG, horseradish peroxidase-conjugated
(cat. no. 7074; dilution, 1:2,000) antibodies were purchased from
Cell Signaling Technology, Inc. (Danvers, MA, USA) and anti-mPGES-1
antibody (cat. no. 10004350; dilution, 1:1,000) was purchased from
the Cayman Chemical Company.
Cell culture
Jurkat cells were cultured in RPMI 1640 medium
containing 10% fetal bovine serum (both Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) at 37°C in 5% CO2.
To test the effect of EP4 receptor, jurkat cells
(8×105/well) were plated in 6-well plate and incubated
with antagonist L-161982 (10 mM stock solution in dimethyl
sulfoxide, further dissolved with RPMI 1640 to give a 33.3 µM
working solution) for 24 h at 37°C in 5% CO2.
Lentivirus infection
Gene knockdown was performed using lentivirus short
hairpin RNA (shRNA), which was synthesized by GeneChem Co., Ltd.,
(Shanghai, China). The shRNA was cloned into pLKO.1 (GV115)
lentiviral vectors (hU6-MCS-CMV-EGFP, GeneChem Co., Ltd., Shanghai,
China) at 40 nmol/l. Four shRNA-mPGES-1 targeting sequences
(27740-1, 5′-GGGCTTCGTCTACTCCTTT-3′; 27741-1,
5′-TGCTGGTCATCAAGATGTA-3′; 27742-1, 5′-GGCTAAGAATGCAGACTTT-3′ and
27743-1, 5′-TTTCTGGTCCCTTCAGTAT-3′) were designed. The
shRNA-negative control (NC) targeting sequence was
5′-TTCTCCGAACGTGTCACGT-3′. The culture containing lentivirus was
added to the jurkat cells in the presence of 5 µg/ml polybrene
(Shanghai GeneChem Co., Ltd.). Positively transfected cells were
selected by 1 µg/ml puromycin after 24 h incubation at 37°C in 5%
CO2. Stable cell lines were verified by western blot
analysis as described below.
Cell proliferation assay
Cell proliferation was measured using a Cell
Counting kit-8 (Dojindo Molecular Technologies, Inc., Kumamoto,
Japan) in vitro. A total of 1×104 cells were
plated per well in 96-well plates and incubated at 37°C with 5%
CO2 for 24, 48 and 72 h. The cells were divided into
three groups: i) KD group, jurkat cells transfected with shRNA
(27743–1) targeting mPGES-1; ii) NC
group, jurkat cells transfected with NC shRNA; and iii) Control
(CON) group, jurkat cells without any treatment. A total of 10 µl
CCK-8 was added to each well and the samples were incubated for a
further 4 h. The optical density (OD) values were measured using a
microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
at 450 nm.
Flow cytometry
Following incubation in a serum-free RPMI 1640
medium overnight, jurkat cells were collected (115 × g, 5 min, room
temperature) and rinsed twice with PBS. For cell cycle analysis, a
total of 5×105 cells were fixed with 70% pre-chilled
ethanol overnight at 4°C and stained with propidium iodide for 10
min at room temperature. The DNA content was analyzed by a BD
FACStar flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). A
total of 1×106 cells were washed and re-suspended in
binding buffer and subsequently incubated with 5 ml Annexin
V-fluorescein isothiocyanate (PE Annexin V Apoptosis Detection kit
1; BD Biosciences) for 15 min at room temperature in the dark. A
total of 2.5 ml allophycocyanin (BD Biosciences) was added and the
cells were analyzed with a FACScan flow cytometer (FACSCalibur; BD
Biosciences). Fluorophores were excited at 640 nm. Data acquisition
and analysis were performed using CellQuest software (v6.1×; BD
Biosciences).
GeneChip assay
GeneChip assays were performed by Shanghai GeneChem
Co., Ltd. (Shanghai, China). General steps were as follows: Total
RNA was extracted using TRIzol reagent (Takara Bio, Inc., Otsu,
Japan) according to the manufacturer protocol. The quantity and
quality of the RNA were determined by spectrophotometer and 1%
formaldehyde denaturing gel electrophoresis. An Affymetrix Gene
Chip® Prime View™ Human Gene Expression array was used
for the microarray analysis. Hybridization, data capture and
analysis were performed by Shanghai GeneChem Co., Ltd. (Shanghai,
China). Briefly, 100 ng total RNA was used for cDNA synthesis and
biotin-tagged cRNA was produced by the Gene Chip IVT Labeling kit
(Affymetrix; Thermo Fisher Scientific, Inc.). A total of 15 µg
fragmented cRNA, with the controls oligo B2 and eukaryotic
hybridization, were hybridized to each GeneChip array at 45°C for
16 h (Affymetrix Gene Chip Hybridization Oven 640) according to the
manufacturer protocol. Following hybridization, the Gene Chip
arrays were washed three times at room temperature and stained with
streptavidin phycoerythrin onan (3×; 35°C; 300 sec) with Affymetrix
Fluidics Station 450 followed by scanning with the Affymetrix Gene
Chip Scanner 30007G. Molecular function and signaling pathways were
analyzed using Gene Ontology (GO; http://www.geneontology.org/) and Kyoto Encyclopedia
of Genes and Genomes (KEGG; http://www.kegg.jp/kegg/kegg4.html), respectively.
Western blot analysis
Cells were lysed with an appropriate volume of the
radioimmunoprecipitation buffer (CWBIO; Biotechnology Co., Ltd.,
Beijing, China) supplemented with protease inhibitor cocktail
(CWBIO; Biotechnology Co., Ltd., Beijing, China) and the protein
concentrations were determined by bicinchoninic acid assays with
bovine serum albumin (BSA; CWBIO, Biotechnology Co., Ltd., Beijing,
China) as the standard. A total of 30 ng/20 µl protein was
separated by 10% SDS-PAGE and transferred to polyvinylidene
difluoride membranes (EMD Millipore, Billerica, MA, USA). Following
blocking with Tris-buffered saline (TBS) containing 5% BSA diluted
in TBS with Tween-20 for 1 h, the membranes were incubated
overnight at 4°C with primary antibodies diluted according to the
instruction followed by incubation with the horseradish
peroxidase-conjugated secondary antibodies for 1 h at room
temperature. The immunoreactive bands were detected using a
chemiluminescent system (Thermo Fisher Scientific, Inc.) and
quantified using ImageJ 1.43 (National Institutes of Health,
Bethesda, MD, USA).
Statistical analysis
All experiments were performed three times. Data
were processed using SPSS 20.0 software (IBM Corp., Armonk, NY,
USA) and presented as the mean ± standard deviation. Statistical
analysis was conducted using one-way analysis of variance followed
by Student-Newman-Keuls post-hoc tests. P<0.05 was considered to
indicate a statistically significant difference.
Results
The results of gene silencing by RNA
interference
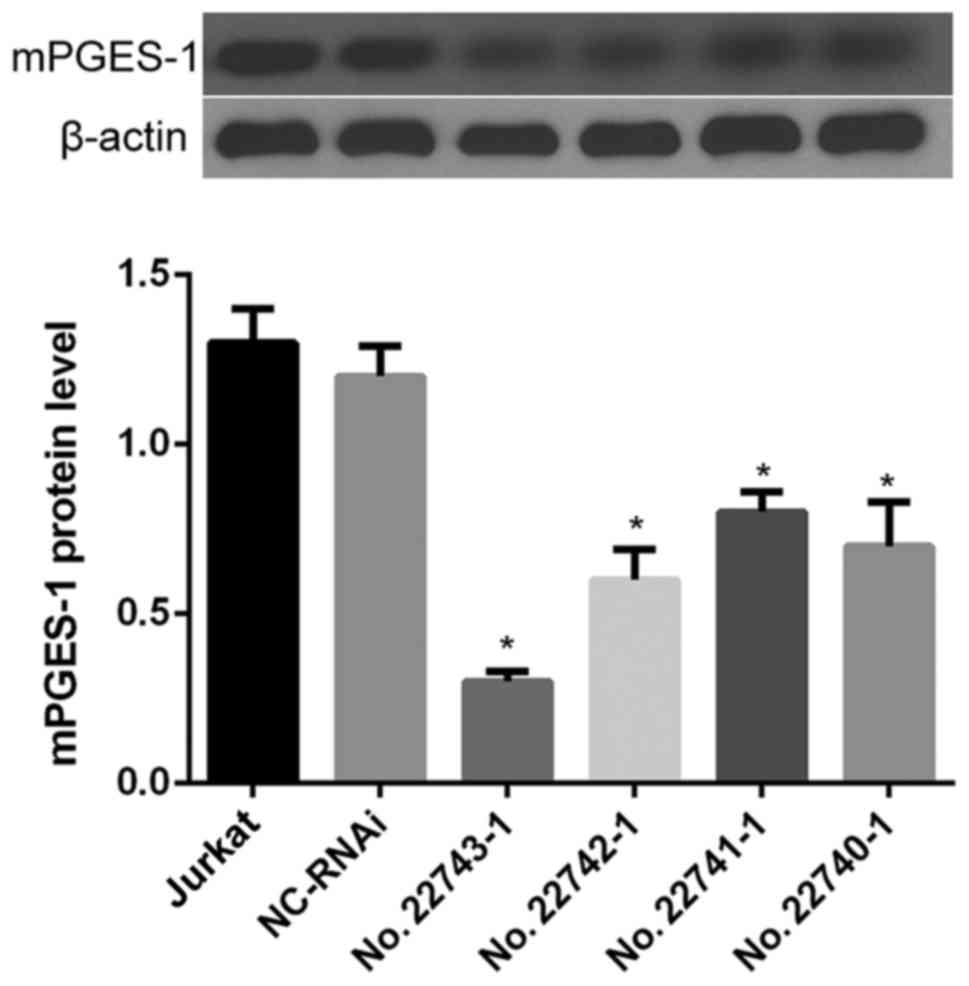
To establish a useful cell line, the expression of
mPGES-1 was decreased via lentivirus shRNA interference. Following
western blot analysis of the results, sequence 27743-1 was selected
for use in the following experiments (as the KD group) as it
displayed the highest interference rate when transfected into
jurkat cells (P<0.05; Fig. 1). An
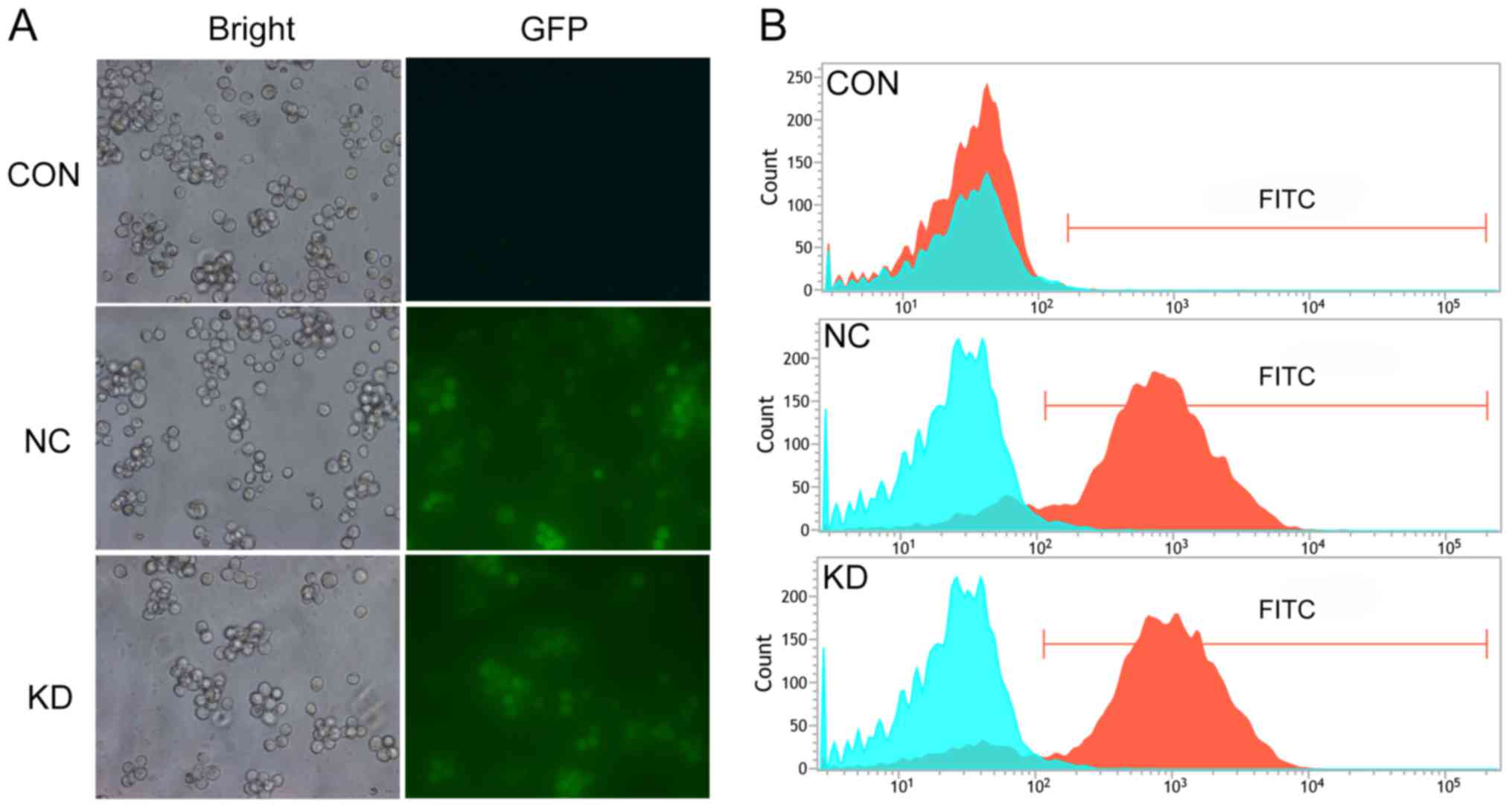
inverted fluorescence microscope was used to observe the cells; it
revealed that the KD and NC groups had a similar morphology to the
CON group (Fig. 2A), indicating that
the lentivirus infection had no notable effects on cell morphology.
Subsequently, flow cytometry was utilized to detect the infection
efficiency. The results revealed that the infection efficiency of
the KD and NC groups were 84.87 and 83.17%, respectively
(P<0.05; Fig. 2B). Infection
efficiency allowed for cells to be used in further experiment.
mPGES-1 silencing inhibits
proliferation, induces apoptosis and arrests the cell cycle in
jurkat cells
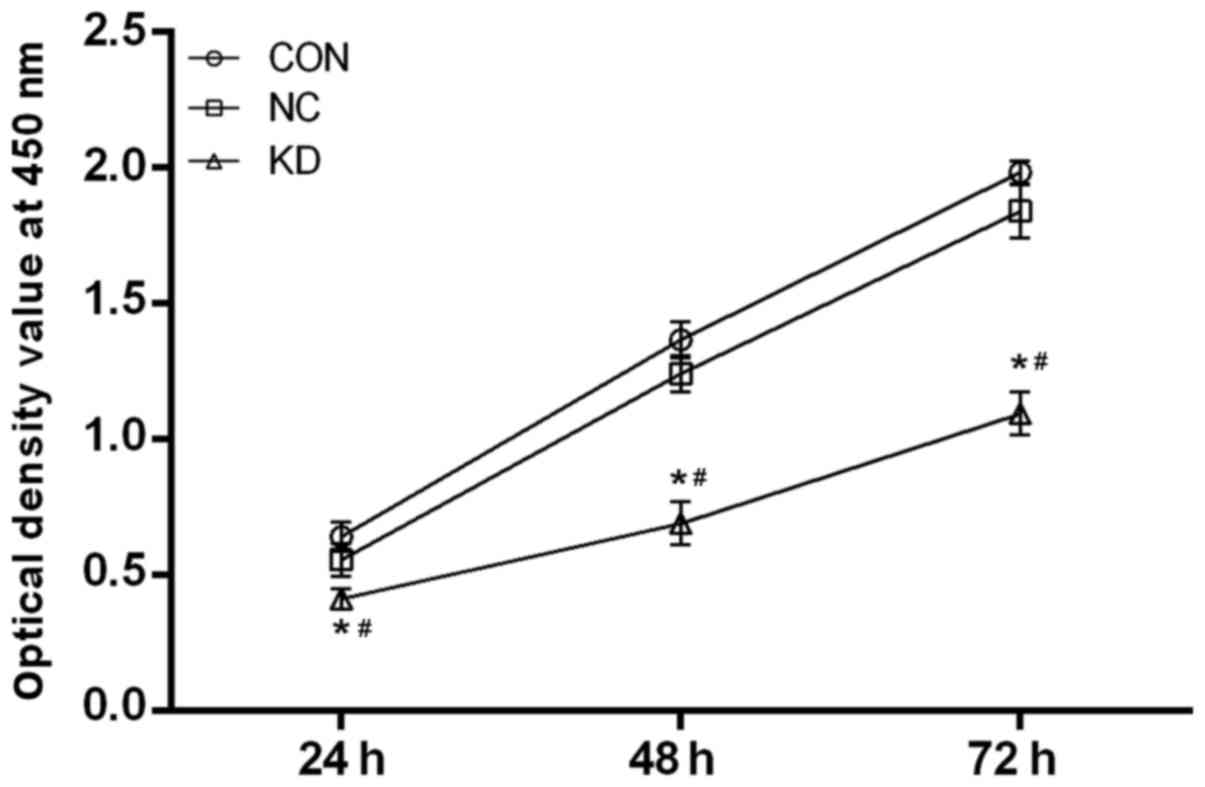
To evaluate the effect of mPGES-1 on the
proliferation of jurkat cells, a CCK-8 experiment was conducted.
The results revealed that the proliferation of the KD group was
significantly slower compared with the NC and CON groups at 24, 48
and 72 h (P<0.05), whereas no significant difference was
observed between the NC group and the CON group at any time point
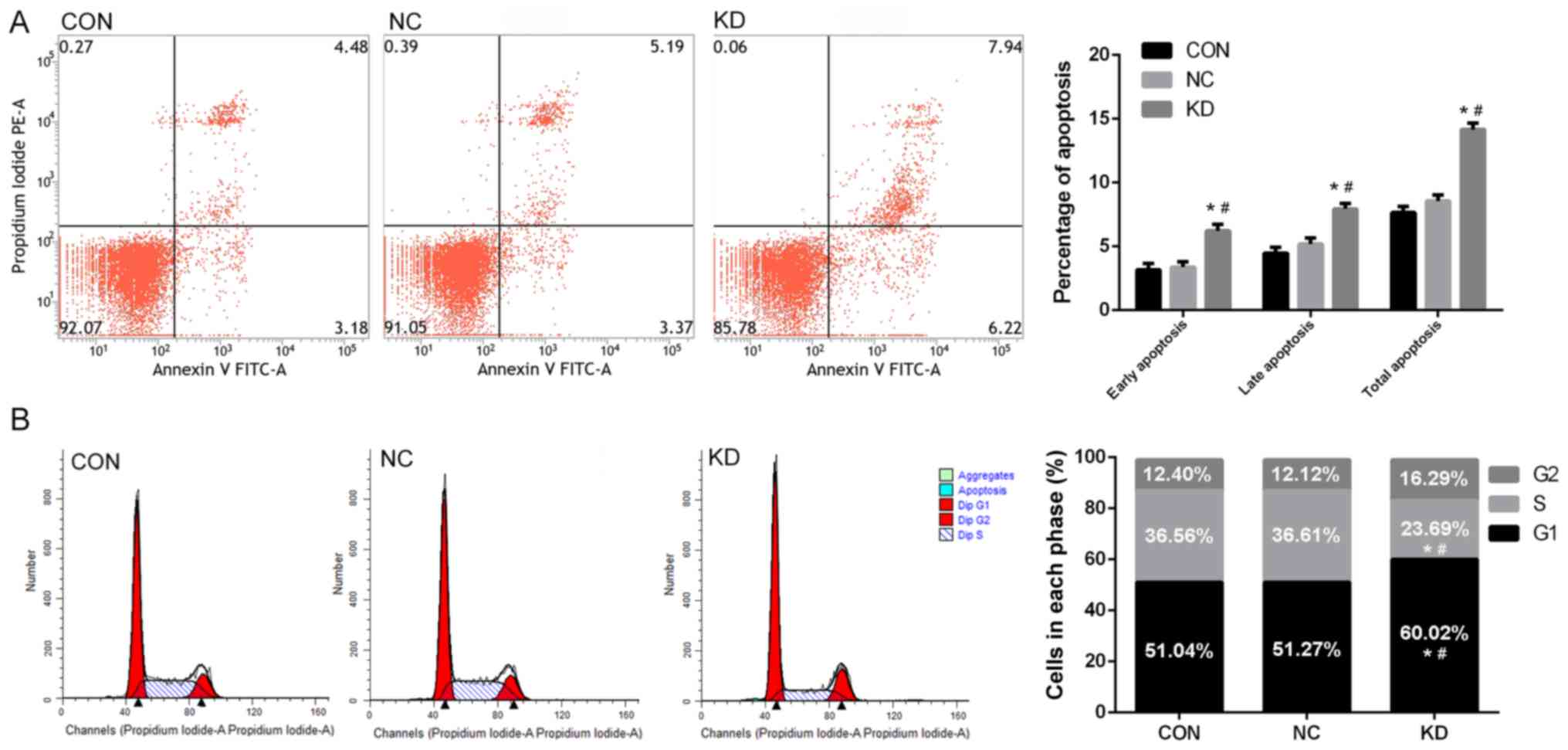
(Fig. 3). In addition, the
percentage of total apoptotic cells (total Annexin-V-FITC+ cells)
was significantly increased in the KD group compared with the CON
and NC groups when assayed by flow cytometry (P<0.05; Fig. 4A). The populations of early
(Annexin-V-FITC+, PI-cells) and late apoptotic cells
(Annexin-V-FITC+, PI+ cells) were also significantly increased in
the KD group compared with the NC and CON groups (P<0.05;
Fig. 4A). Silencing mPGES-1 may also
influence the cell cycle of jurkat cells. The percentage of cells
in the G1 phase was significantly increased, while the percentage
at the S phase was significantly reduced in the KD group compared
with the NC and CON groups (P<0.05; Fig. 4B). These results indicated that
decreasing mPGES-1 inhibited proliferation, induced apoptosis and
arrested the cell cycle the G1 phase in jurkat cells.
Reducing the expression of mPGES-1
inhibits the MAPK signaling pathway
To further understand the mechanism of mPGES-1's
effects on jurkat cells, microarray analysis was used to detect the
changes in gene expression following knockdown of mPGS-1. It was
revealed that 456 genes had changed significantly following the
knockdown of mPGES-1. These genes primarily participated in cell
proliferation, apoptosis, protein metabolism and immune response as
indicated by GO analysis (data not shown). The associated signaling
pathways were analyzed via KEGG software and the results indicated
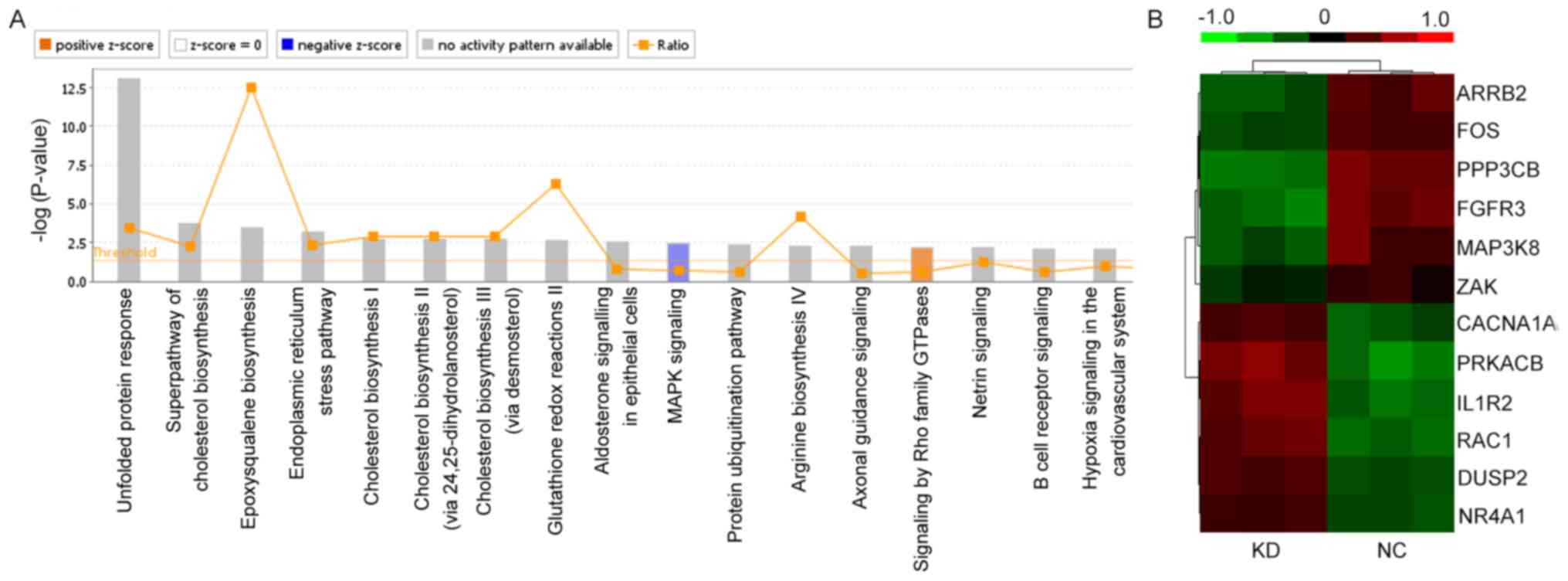
that MAPK had the clearest change in all relevant signaling
pathways. The MAPK signaling pathway was significantly inhibited
(P=6.78×10−4) following knockdown of mPGES-1 (Fig. 5A). A total of 12 genes in the MAPK
signaling pathway, including ARRB2, FOS, PPP3CB, FGFR3, MAP3K8,
ZAK, CACNA1A, PRKACB, IL1R2, RAC1, DUSP2 and NR4A1 were involved
(Fig. 5B and Table I). These findings revealed that the
biological function of jurkat cells may be associated with the MAPK
signaling pathway, and that mPGES-1 may be located upstream of
it.
 | Table I.Gene expression values in the MAPK
signaling pathway following mPGES-1 silencing. |
Table I.
Gene expression values in the MAPK
signaling pathway following mPGES-1 silencing.
|
| Treatment
group |
|
|---|
|
|
|
|
|---|
| Gene | KD | NC | Direction of
regulationa |
|---|
| ARRB2 | −0.329±0.059 |
0.328±0.062 | Down |
| FOS | −0.276±0.039 |
0.280±0.025 | Down |
| PPP3CB | −0.449±0.035 |
0.424±0.050 | Down |
| FGFR3 | −0.447±0.080 |
0.429±0.059 | Down |
| MAP3K8 | −0.320±0.072 |
0.327±0.146 | Down |
| ZAK | −0.156±0.061 |
0.164±0.089 | Down |
| CACNA1A | 0.279±0.032 | −0.324±0.074 | Up |
| PRKACB | 0.476±0.079 | −0.491±0.092 | Up |
| IL1R2 | 0.442±0.089 | −0.404±0.067 | Up |
| RAC1 | 0.381±0.061 | −0.407±0.027 | Up |
| DUSP2 | 0.298±0.017 | −0.300±0.014 | Up |
| NR4A1 | 0.239±0.029 | −0.298±0.028 | Up |
The MAPK signaling pathway is one of the most
important signal transduction systems. It participates in cell
growth, development, differentiation, and other physiological and
pathological processes (23).
Previous studies have demonstrated that the major MAPK signaling
pathway subfamilies associated with COX-2/mPGES-1 or
lipopolysaccharide-activated inflammatory responses may be ERK1/2,
JNK and P38 (23). There is
cross-talk between these three components, which leads to either
coordination or inhibition (24). To
identify which signaling pathway is associated with the function of
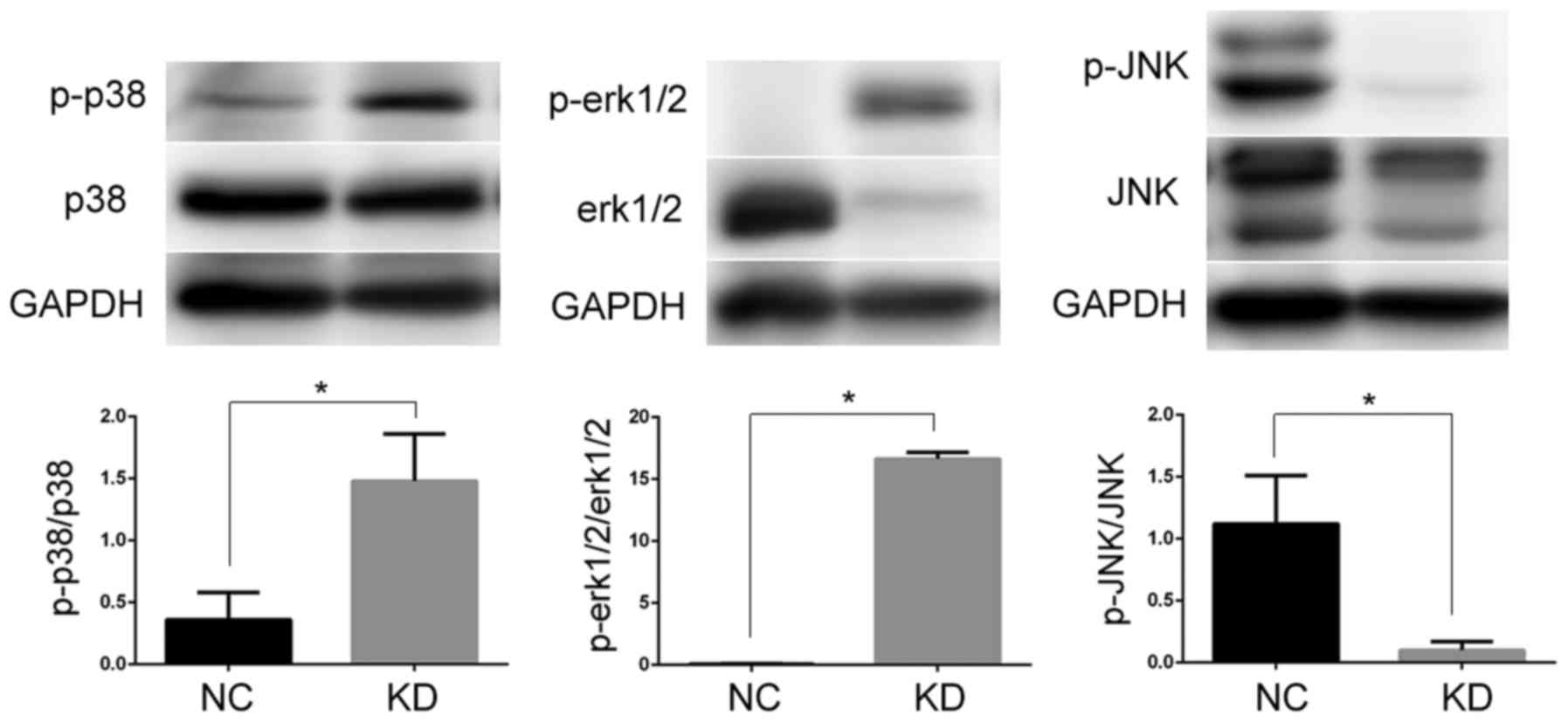
mPGES-1 in jurkat cells, the phosphorylation status of major MAPK
subfamilies was investigated following mPGES-1 silencing. The
expression levels of phosphorylated (p)-P38 and p-ERK1/2 were
significantly increased, while p-JNK was significantly decreased
compared with the NC group (P<0.05; Fig. 6). Based on these results, the authors
speculated that mPGES-1 may affect the growth of jurkat cells
through the JNK/MAPK signaling pathway. Alternatively, decreasing
mPGES-1 may activate the P38 MAPK and ERK1/2/MAPK signaling
pathways. Whether the subfamilies are regulated independently or as
a result of cross-talk is a question, which requires further
investigation.
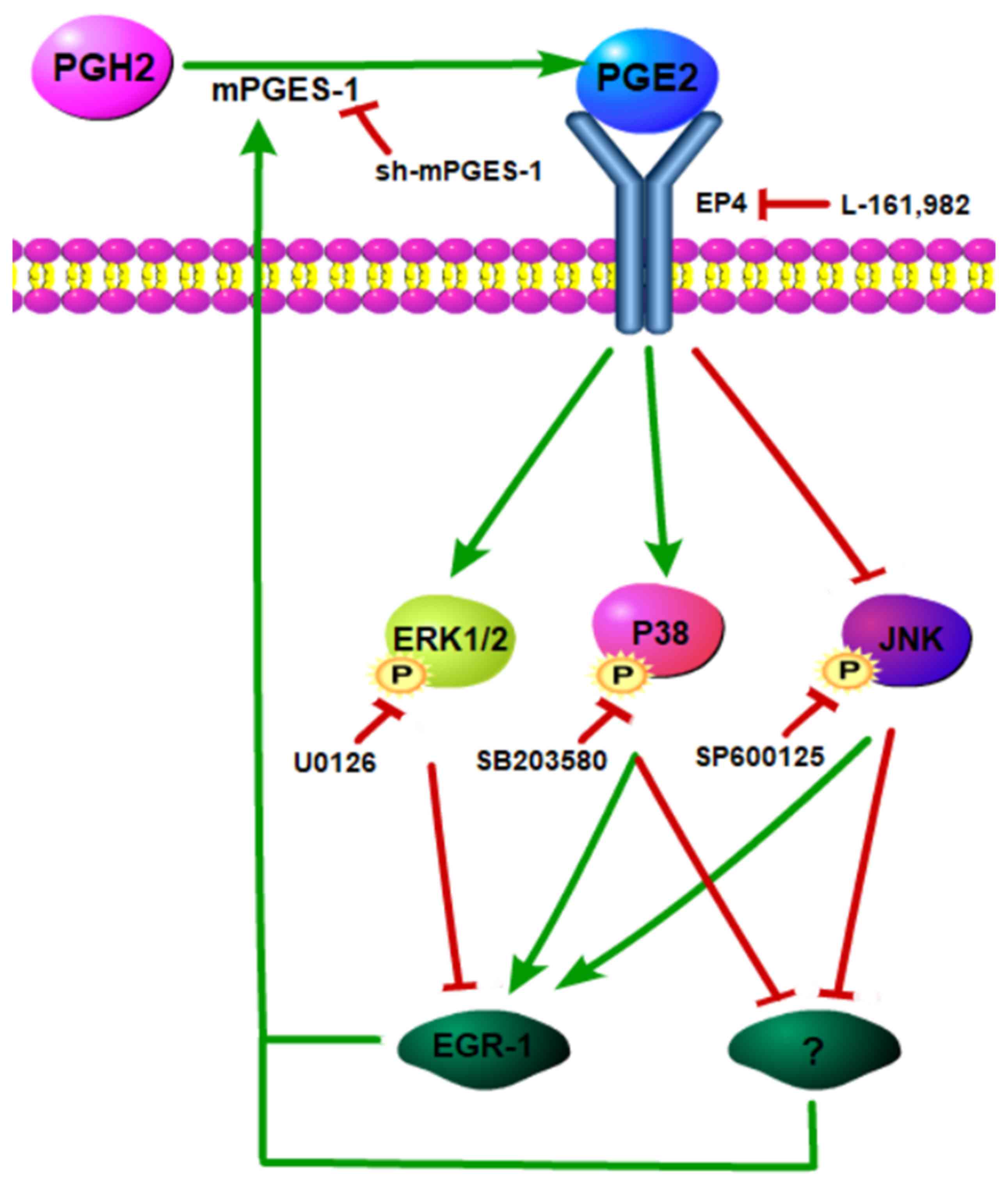
EP4 receptor mediates the regulation
of mPGES-1 on the MAPK signaling pathway
PGE2 has diverse actions and stimulates key
downstream signal transduction pathways by binding to its
prostanoid receptors (EP1, EP2, EP3 and EP4) (25). Binding of PGE2 to different receptors
may lead to the activation of different signaling pathways
(25). A previous study by the
authors reported that mPGES-1/PGE2 was closely associated with
MAPKs, however, which subtype of prostanoid receptors mediated the
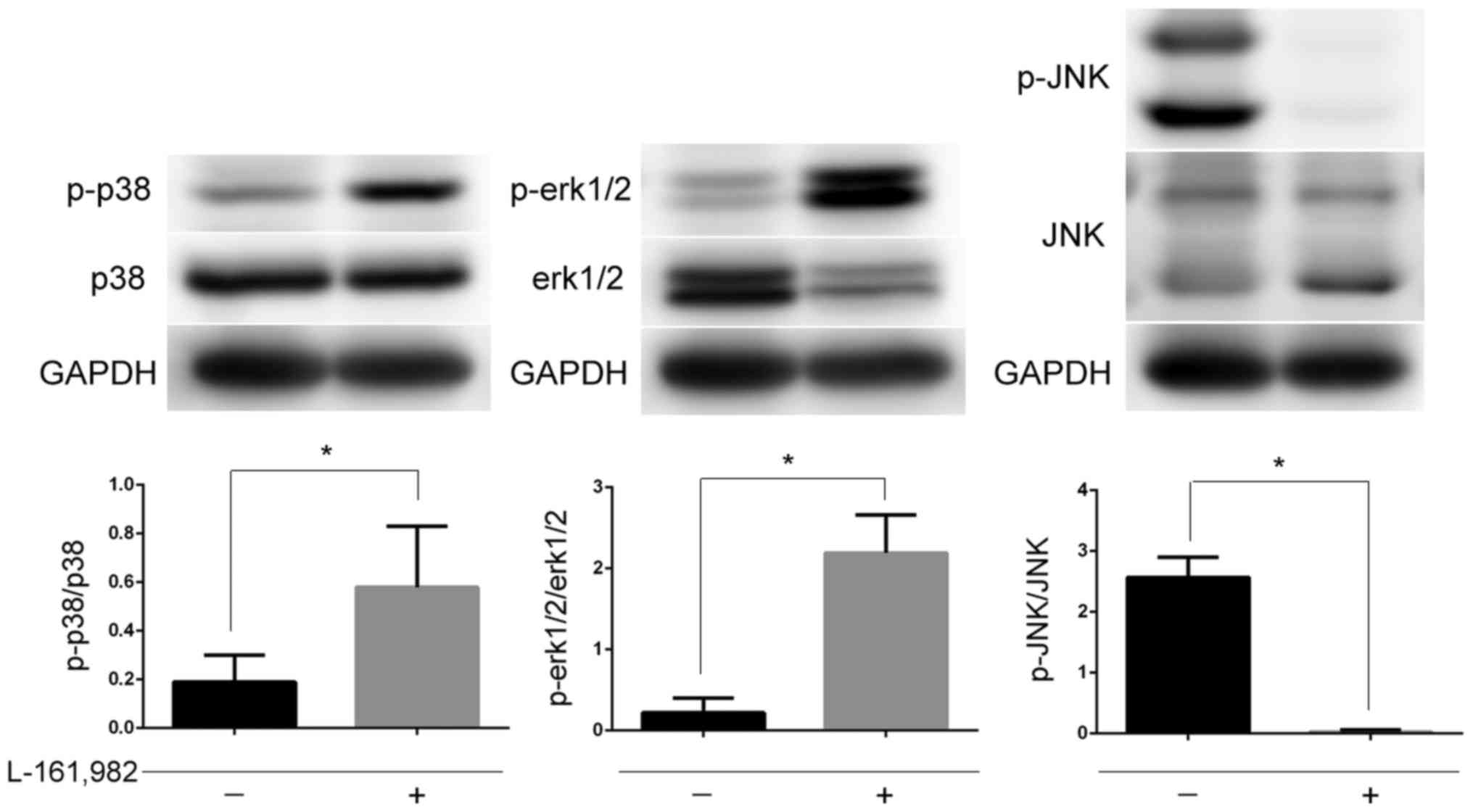
activation of MAPKs remained unknown. In the present study jurkat
cells were pre-incubated with EP4 receptor antagonist L-161982 and
then the phosphorylation of MAPKs was examined. It was observed
that the changes in the MAPKs were consistent with the results
obtained following silencing of mPGES-1 (P<0.05; Fig. 7). This may indicate that mPGES-1/PGE2
regulates MAPKs by combining with the EP4 receptor.
MAPKs feedback on mPGES-1 may be
partly via EGR-1
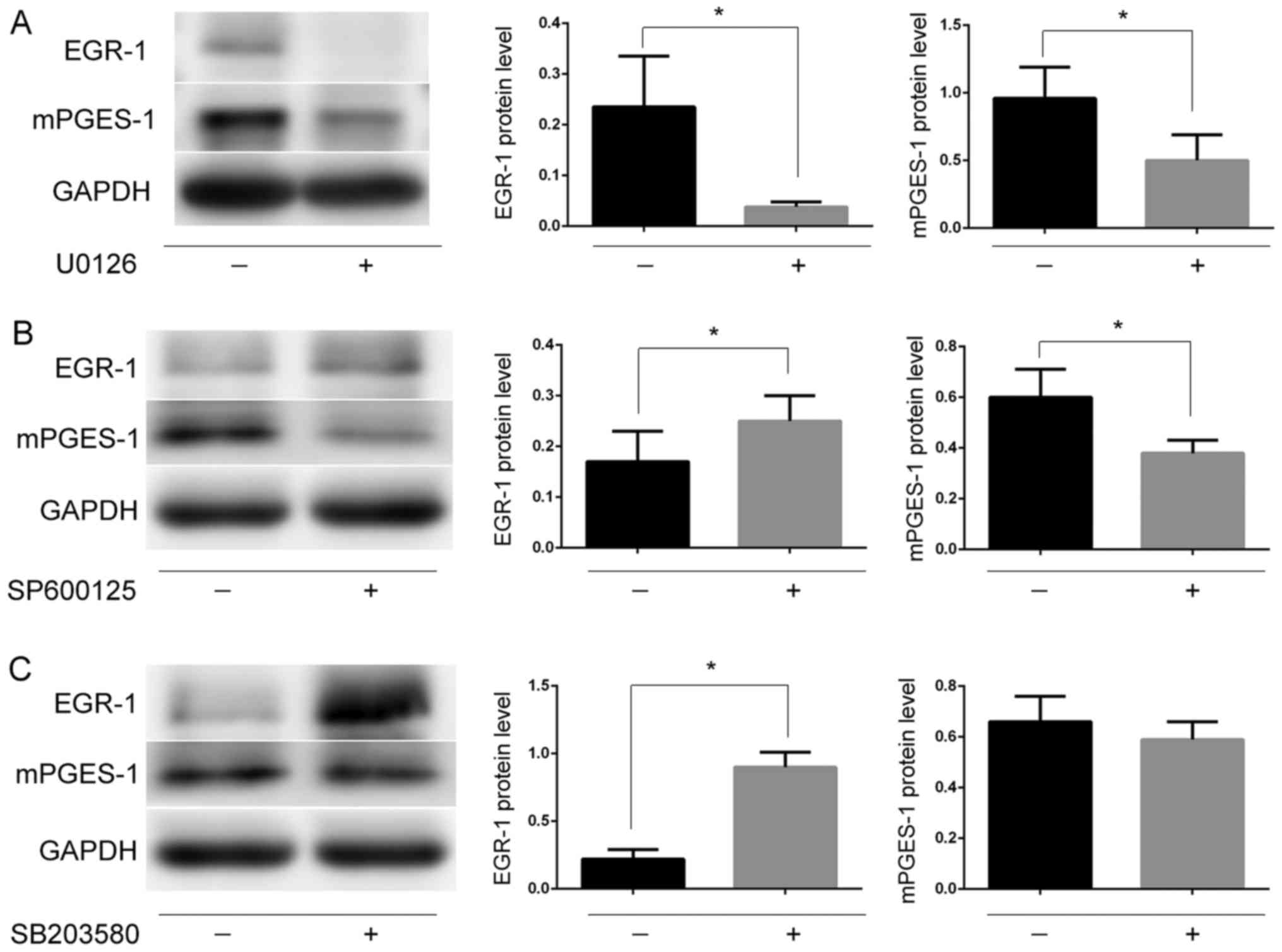
Several previous studies seem to indicate that MAPKs
regulate mPGES-1 via EGR-1, a transcription factor that regulates
the composition of mPGES-1 (26–29). The
authors hypothesized that MAPKs may regulate the expression of
mPGES-1 by regulating EGR-1 in jurkat cells. To confirm this
hypothesis, the jurkat cells were treated with inhibitors U0126,
SB203580 and SP600125 against the phosphorylation of ERK1/2, P38
and JNK, respectively. It was revealed that following the
inhibition of the ERK1/2/MAPK signaling pathway, the expression of
mPGES-1 and EGR-1 was significantly reduced (P<0.05; Fig. 8A), while the JNK and P38 inhibitors
reduced the changes observed in EGR-1 and mPGES-1 (Fig. 8B and C). These results suggest that
the ERK1/2/MAPK signaling pathway may regulate the expression of
mPGES-1 through EGR-1.
Discussion
In the present study, the functional role of mPGES-1
silencing was elucidated in the proliferation, apoptosis and cell
cycle of jurkat cells. Since it was first raised as a promising
therapeutic target in 1999 (30),
studies have revealed that prolonged and excessive production of
mPGES-1 can alter a number of biological processes, leading to
intractable pathologies, including inflammation and cancer
(11–13). Therefore, blocking the expression of
mPGES-1 is often an important strategy in treating these
conditions. Previous studies by the authors have confirmed that
mPGES-1 is expressed highly in a variety of leukemia cells,
including HL-60 (16), K562, jurkat
and Raji (unpublished data). In the present study, it was revealed
that decreasing mPGES-1 affected the growth of T-ALL jurkat cells
and was also associated with the MAPK signaling pathway. As only
one cell line was used in the experiments, it is not clear if such
phenomenon exists in other T-ALL cell lines or primary cells; the
authors believe that this is intriguing and worth exploring in
future studies.
The roles of MAPKs in T-ALL have been previously
described (31–33). The major MAPK signaling pathway
subfamilies, including ERK1/2, JNK and P38 serve key roles in the
regulation of the expression of various inflammatory genes, such as
mPGES-1 (23). Based on these
previous results, it was speculated that MAPKs may be involved in
the regulation of mPGES-1 in T-ALL. Using microarray and western
blot analysis, it was confirmed that mPGES-1 may affect the growth
of jurkat cells via MAPKs. Interestingly, the effects of mPGES-1 on
MAPK subfamilies were completely different. The JNK/MAPK signaling
pathway was activated in the NC group, whereas this activation was
inhibited in the KD group. However, the other two subfamilies,
ERK1/2 and P38, displayed the opposite response following knockdown
mPGES-1. These results indicated that mPGES-1 is located upstream
of MAPKs, that MAPKs may be involved in the impact of mPGES-1 on
jurkat cells and that the three subfamilies of MAPKs had different
responses to mPGES-1 and there may be cross-talk between them;
however, the exact mechanism requires further investigation.
mPGES-1 affects the function of tumor cells by
increasing PGE2 synthesis. PGE2 exerts its biological actions by
binding to four specific receptor subtypes known as EP1, EP2, EP3
and EP4 (34). The EP receptors are
involved in the generation and progression of tumors through the
activation of different signaling pathways (35–37).
Qian et al (38) reported
that PGE2 stimulates human brain natriuretic peptide expression via
the EP4-ERK1/2/MAPK signaling pathway. Mendez and LaPointe
(39) also reported that PGE2
induces protein synthesis in cardiac myocytes, partly via
activation of the EP4 receptor and subsequent activation of the
ERK1/2/MAPK signaling pathway. In the present study the EP4
receptor was blocked by its antagonist, L-161982; this lead to a
similar effect on MAPKs as those caused by mPGES-1 silencing. These
results indicated that mPGES-1/PGE2 affected the growth of jurkat
cells via EP4-dependent activation of the MAPK signaling pathway.
However, the phosphorylated and activated subtype of MAPKs
regulated by mPGES-1/PGE2/EP4 was different from that in Qian and
Mendez's studies. This may be due to the different cell line used
in the present experiment.
Accumulating evidence indicates that the activation
of MAPKs is crucial for the expression of EGR-1 (40,41).
EGR-1 is a zinc finger transcription factor, which binds to GC-rich
sequences, such as mPGES-1, in the regulatory region of its target
genes (42). In the present study,
blocking the ERK1/2/MAPK pathway induced a decrease in EGR-1 and
mPGES-1, so it was speculated that the ERK1/2/MAPK signaling
pathway may regulate mPGES-1 expression through EGR-1 (Fig. 9). Blocking the P38/MAPK or JNK/MAPK
pathways induced an increase in EGR-1, while it decreased mPGES-1.
These results may indicate the possibility of other potential
mechanism underlying the regulation of mPGES-1, in addition to
EGR-1.
Based on the above findings, it is clear that
mPGES-1 serves an important role in T-ALL jurkat cells by
activating the JNK/MAPK signaling pathway, which in turn is
required to achieve high levels of mPGES-1. This suggests that a
positive feedback loop mediated by the JNK/MAPK signaling pathway
promotes mPGES-1 induction. The results of the present study
contradict the findings of a previous study, which mentioned a
positive feedback loop between mPGES-1 and the ERK1/2/MAPK
signaling pathway in macrophages (43). One potential explanation is that the
present study performed experiments with all three of the classical
subfamilies (ERK1/2/MAPK, JNK/MAPK and P38/MAPK) in jurkat cells,
instead of macrophages. By exploring the growth of jurkat cells, it
was observed that the effect of the positive feedback loop induced
by the JNK/MAPK signaling pathway may be greater than that of the
negative feedback induced by the ERK1/2/MAPK and P38/MAPK signaling
pathways, subsequently leading to an inhibition of proliferation,
induction of apoptosis and arrest of the cell cycle. However, the
involvement of negative feedback loops may induce drug resistance
or even treatment failure (44,45). The
findings of the present study raised the possibility that combined
treatments of mPGES-1 with ERK1/2 and P38 inhibitors may be a novel
therapeutic strategy for T-ALL.
Acknowledgments
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81200342),
the Guangdong Science and Technology Department (grant nos.
2014A020212085 and 2016A020215062), the Natural Science Foundation
of Guangdong Province (grant no. 2016A030313360), the State
Scholarship Fund of China (grant no. CSC 201606380189), the Key
Laboratory of Malignant Tumor Molecular Mechanism and the
Translational Medicine of Guangzhou Bureau of Science and
Information Technology [(grant no. 163 (2013)] and the Key
Laboratory of Malignant Tumor Gene Regulation and the Target
Therapy of Guangdong Higher Education Institutes (grant no.
KLB09001).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
S-MY and D-NN designed the study, and analyzed and
interpreted the data. Y-QL and J-TC conducted the experiments and
contributed to writing the manuscript. All other authors, Z-YH,
S-FX, X-JW, Y-DW, JX, H-YL, J-YW, W-JY and L-PM made substantial
contributions to the experiments and the acquisition of data. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Roti G and Stegmaier K: New approaches to
target T-ALL. Front Oncol. 4:1702014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhao WL: Targeted therapy in T-cell
malignancies: dysregulation of the cellular signaling pathways.
Leukemia. 24:13–21. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qiu X, Cheng JC, Chang HM and Leung PC:
COX2 and PGE2 mediate EGF-induced E-cadherin-independent human
ovarian cancer cell invasion. Endocr Relat Cancer. 21:533–543.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pan J, Cheng L, Bi X, Zhang X, Liu S, Bai
X, Li F and Zhao AZ: Elevation of w-3 polyunsaturated fatty acids
attenuates PTEN-deficiency induced endometrial cancer development
through regulation of COX-2 and PGE2 production. Sci Rep.
5:149582015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Larsson K, Kock A, Idborg H, Henriksson
Arsenian M, Martinsson T, Johnsen JI, Korotkova M, Kogner P and
Jakobsson PJ: COX/mPGES-1/PGE2 pathway depicts an
inflammatory-dependent high-risk neuroblastoma subset. Proc Natl
Acad Sci U S A. 112:8070–8075. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Steinbach G, Lynch PM, Phillips RK,
Wallace MH, Hawk E, Gordon GB, Wakabayashi N, Saunders B, Shen Y,
Fujimura T, et al: The effect of celecoxib, a cyclooxygenase-2
inhibitor, in familial adenomatous polyposis. N Engl J Med.
342:1946–1952. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Howe LR and Dannenberg AJ: COX-2
inhibitors for the prevention of breast cancer. J Mammary Gland
Biol Neoplasia. 8:31–43. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yiannakopoulou E: Aspirin and NSAIDs for
breast cancer chemoprevention. Eur J Cancer Prev. 24:416–421. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fosbol EL, Kober L, Torp-Pedersen C and
Gislason GH: Cardiovascular safety of non-steroidal
anti-inflammatory drugs among healthy individuals. Expert Opin Drug
Saf. 9:893–903. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takeuchi K: Pathogenesis of NSAID-induced
gastric damage: Importance of cyclooxygenase inhibition and gastric
hypermotility. World J Gastroenterol. 18:2147–2160. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Larsson K and Jakobsson PJ: Inhibition of
microsomal prostaglandin E synthase-1 as targeted therapy in cancer
treatment. Prostaglandins Other Lipid Mediat. 120:161–165. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Olesch C, Sha W, Angioni C, Sha LK, Açaf
E, Patrignani P, Jakobsson PJ, Radeke HH, Grösch S, Geisslinger G,
et al: MPGES-1-derived PGE2 suppresses CD80 expression on
tumor-associated phagocytes to inhibit anti-tumor immune responses
in breast cancer. Oncotarget. 6:10284–10296. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Finetti F, Terzuoli E, Giachetti A, Santi
R, Villari D, Hanaka H, Radmark O, Ziche M and Donnini S: mPGES-1
in prostate cancer controls stemness and amplifies epidermal growth
factor receptor-driven oncogenicity. Endocr Relat Cancer.
22:665–678. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li YQ, Yin SM, Xie SF, Wang XJ, Ma LP, Nie
DN and Wu YD: Effect of mPGES-1 inhibitor MK886 on cell cycle of
leukemia HL-60 cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi.
20:1072–1076. 2012.(In Chinese). PubMed/NCBI
|
|
15
|
Li YQ, Yin SM, Nie DN, Xie SF, Ma LP, Wang
XJ and Wu YD: Effect of mPGES-1 inhibitor MK886 on apoptosis and
drug resistance of HL-60/A cells. Zhongguo Shi Yan Xue Ye Xue Za
Zhi. 20:829–834. 2012.(In Chinese). PubMed/NCBI
|
|
16
|
Li Y, Yin S, Nie D, Xie S, Ma L, Wang X,
Wu Y and Xiao J: MK886 inhibits the proliferation of HL-60 leukemia
cells by suppressing the expression of mPGES-1 and reducing
prostaglandin E2 synthesis. Int J Hematol. 94:472–478. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Munoz L and Ammit AJ: Targeting p38 MAPK
pathway for the treatment of Alzheimer's disease.
Neuropharmacology. 58:561–568. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Olajide OA, Bhatia HS, de Oliveira AC,
Wright CW and Fiebich BL: Anti-neuroinflammatory properties of
synthetic cryptolepine in human neuroblastoma cells: Possible
involvement of NF-κB and p38 MAPK inhibition. Eur J Med Chem.
63:333–339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Furuya H, Wada M, Shimizu Y, Yamada PM,
Hannun YA, Obeid LM and Kawamori T: Effect of sphingosine kinase 1
inhibition on blood pressure. FASEB J. 27:656–664. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Olajide OA, Velagapudi R, Okorji UP,
Sarker SD and Fiebich BL: Picralima nitida seeds suppress PGE2
production by interfering with multiple signalling pathways in
IL-1β-stimulated SK-N-SH neuronal cells. J Ethnopharmacol.
152:377–383. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Okorji UP, Velagapudi R, El-Bakoush A,
Fiebich BL and Olajide OA: Antimalarial drug artemether inhibits
neuroinflammation in BV2 microglia through Nrf2-dependent
mechanisms. Mol Neurobiol. 53:6426–6443. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hu H, Goltsov A, Bown JL, Sims AH, Langdon
SP, Harrison DJ and Faratian D: Feedforward and feedback regulation
of the MAPK and PI3K oscillatory circuit in breast cancer. Cell
Signal. 25:26–32. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bhatia HS, Baron J, Hagl S, Eckert GP and
Fiebich BL: Rice bran derivatives alleviate microglia activation:
Possible involvement of MAPK pathway. J Neuroinflammation.
13:1482016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fu P, Liang GJ, Khot SS, Phan R and Bach
LA: Cross-talk between MAP kinase pathways is involved in
IGF-independent, IGFBP-6-induced Rh30 rhabdomyosarcoma cell
migration. J Cell Physiol. 224:636–643. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
O'Callaghan G and Houston A: Prostaglandin
E2 and the EP receptors in malignancy: Possible therapeutic
targets? Br J Pharmacol. 172:5239–5250. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Diaz-Munoz MD, Osma-Garcia IC,
Cacheiro-Llaguno C, Fresno M and Iniguez MA: Coordinated
up-regulation of cyclooxygenase-2 and microsomal prostaglandin E
synthase 1 transcription by nuclear factor kappa B and early growth
response-1 in macrophages. Cell Signal. 22:1427–1436. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chabane N, Li X and Fahmi H: HDAC4
contributes to IL-1-induced mPGES-1 expression in human synovial
fibroblasts through up-regulation of Egr-1 transcriptional
activity. J Cell Biochem. 106:453–463. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Moon Y, Lee M and Yang H: Involvement of
early growth response gene 1 in the modulation of microsomal
prostaglandin E synthase 1 by epigallocatechin gallate in A549
human pulmonary epithelial cells. Biochem Pharmacol. 73:125–135.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Noma T, Takahashi-Yanaga F, Arioka M, Mori
Y and Sasaguri T: Inhibition of GSK-3 reduces prostaglandin E2
production by decreasing the expression levels of COX-2 and mPGES-1
in monocyte/macrophage lineage cells. Biochem Pharmacol.
116:120–129. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jakobsson PJ, Thoren S, Morgenstern R and
Samuelsson B: Identification of human prostaglandin E synthase: A
microsomal, glutathione-dependent, inducible enzyme, constituting a
potential novel drug target. Proc Natl Acad Sci U S A.
96:7220–7225. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tomiyasu H, Watanabe M, Sugita K,
Goto-Koshino Y, Fujino Y, Ohno K, Sugano S and Tsujimoto H:
Regulations of ABCB1 and ABCG2 expression through MAPK pathways in
acute lymphoblastic leukemia cell lines. Anticancer Res.
33:5317–5323. 2013.PubMed/NCBI
|
|
32
|
Liu Y, Ge J, Li Q, Guo X, Gu L, Ma ZG, Li
XH and Zhu YP: Low-dose anisomycin sensitizes
glucocorticoid-resistant T-acute lymphoblastic leukemia CEM-C1
cells to dexamethasone-induced apoptosis through activation of
glucocorticoid receptor and p38-MAPK/JNK. Leuk Lymphoma.
55:2179–2188. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Naci D and Aoudjit F: Alpha2beta1 integrin
promotes T cell survival and migration through the concomitant
activation of ERK/Mcl-1 and p38 MAPK pathways. Cell Signal.
26:2008–2015. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kawahara K, Hohjoh H, Inazumi T, Tsuchiya
S and Sugimoto Y: Prostaglandin E2-induced inflammation: Relevance
of prostaglandin E receptors. Biochim Biophys Acta. 1851:414–421.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xia S, Ma J, Bai X, Zhang H, Cheng S,
Zhang M, Zhang L, Du M, Wang Y, Li H, et al: Prostaglandin E2
promotes the cell growth and invasive ability of hepatocellular
carcinoma cells by upregulating c-Myc expression via EP4 receptor
and the PKA signaling pathway. Oncol Rep. 32:1521–1530. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Du M, Shi F, Zhang H, Xia S, Zhang M, Ma
J, Bai X, Zhang L, Wang Y, Cheng S, et al: Prostaglandin E2
promotes human cholangiocarcinoma cell proliferation, migration and
invasion through the upregulation of β-catenin expression via EP3-4
receptor. Oncol Rep. 34:715–726. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kim KM, Im AR, Kim SH, Hyun JW and Chae S:
Timosaponin AIII inhibits melanoma cell migration by suppressing
COX-2 and in vivo tumor metastasis. Cancer Sci. 107:181–188. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Qian JY, Leung A, Harding P and LaPointe
MC: PGE2 stimulates human brain natriuretic peptide expression via
EP4 and p42/44 MAPK. Am J Physiol Heart Circ Physiol.
290:H1740–H1746. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mendez M and LaPointe MC: PGE2-induced
hypertrophy of cardiac myocytes involves EP4 receptor-dependent
activation of p42/44 MAPK and EGFR transactivation. Am J Physiol
Heart Circ Physiol. 288:H2111–2117. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ryu WI, Lee H, Kim JH, Bae HC, Ryu HJ and
Son SW: IL-33 induces Egr-1-dependent TSLP expression via the MAPK
pathways in human keratinocytes. Exp Dermatol. 24:857–863. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jeong SH, Kim HJ, Ryu HJ, Ryu WI, Park YH,
Bae HC, Jang YS and Son SW: ZnO nanoparticles induce TNF-α
expression via ROS-ERK-Egr-1 pathway in human keratinocytes. J
Dermatol Sci. 72:263–273. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Naraba H, Yokoyama C, Tago N, Murakami M,
Kudo I, Fueki M, Oh-Ishi S and Tanabe T: Transcriptional regulation
of the membrane-associated prostaglandin E2 synthase gene.
Essential role of the transcription factor Egr-1. J Biol Chem.
277:28601–28608. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Khan KM, Kothari P, Du B, Dannenberg AJ
and Falcone DJ: Matrix metalloproteinase-dependent microsomal
prostaglandin E synthase-1 expression in macrophages: Role of TNF-α
and the EP4 prostanoid receptor. J Immunol. 188:1970–1980. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Galoian K, Temple HT and Galoyan A: mTORC1
inhibition and ECM-cell adhesion-independent drug resistance via
PI3K-AKT and PI3K-RAS-MAPK feedback loops. Tumor Biol. 33:885–890.
2012. View Article : Google Scholar
|
|
45
|
Mirzoeva OK, Das D, Heiser LM,
Bhattacharya S, Siwak D, Gendelman R, Bayani N, Wang NJ, Neve RM,
Guan Y, et al: Basal subtype and MAPK/ERK kinase
(MEK)-phosphoinositide 3-kinase feedback signaling determine
susceptibility of breast cancer cells to MEK inhibition. Cancer
Res. 69:565–572. 2009. View Article : Google Scholar : PubMed/NCBI
|