Introduction
Doxorubicin (DOX, also known as adriamycin), a
typical anthracycline, ranks as one of the most potent antitumor
agents available for breast cancer, lymphoma, and hematologic
malignancy (1). Unfortunately, the
clinical use of DOX is limited due to the life-threatening
cardiotoxicity, which may ultimately develop into cardiomyopathy
and congestive heart failure (2).
Dose limitation strategy is now most commonly used to reduce the
DOX-induced cardiotoxicity, as an association between the quantity
of DOX accumulated in the heart and the incidence of cardiac events
has been identified (3). However,
lower concentrations may preclude successful completion of
chemotherapy, causing a dilemma for oncologists and cardiologists
(4). Advances in the molecular basis
of anthracycline-induced cardiotoxicity indicate that multiple
mechanisms are involved, among which reactive oxygen species (ROS)
and oxidative stress are the most important factors (5). Furthermore, endogenous antioxidants
decreased following the DOX treatment. The administration of
antioxidants has been demonstrated to prevent cardiac injury
induced by DOX both in vitro and in vivo (6,7).
Nuclear factor (erythroid-derived 2)-like 2 (Nrf2),
a redox-sensitive transcription factor, is the central regulator of
cellular responses to electrophilic/oxidative stress. It has
previously been revealed that Nrf2 was essential for detoxification
gene activity in mammalian cardiac cells (8). Under physiological conditions,
Kelch-like ECH-associated protein 1 (Keap1), the cytosolic
regulatory protein, was tightly bound to Nrf2 to retain it in the
cytoplasm. Nrf2 was then released from Keap1 and translocated to
the nucleus in response to oxidants and electrophiles, which may
result in the subsequent binding to antioxidant responsive DNA
elements (AREs) and activate the transcription of downstream genes,
including antioxidants and phase II and phase III detoxification
enzymes (9). As DOX-induced
oxidative stress is the major cause of injury in cardiac cells
(4), enhancing the critical
biological functions of Nrf2 should be a safe and effective
strategy to counter DOX cardiotoxicity. On this basis, the
mechanisms of drug candidates against DOX-induced cardiotoxicity
focus on Nrf2, especially the bioactive natural products. For
instance, aringenin-7-O-glucoside, a flavonoid isolated from
Dracocephalum rupestre Hance, protected against DOX-induced
cardiomyocyte apoptosis via the nuclear translocation of Nrf2
(10). Similar results were also
observed in sulforaphane, a natural isothiocyanate compound in
cruciferous vegetables (7).
Furthermore, Nrf2 mediated the protective effect of a-Linolenic
acid on DOX-induced cardiotoxicity in rats (11).
Tanshinone IIA (Tan IIA), one of the major
components isolated from Radix Salvia miltiorrhiza, exhibits
potent antioxidant activity (12).
Emerging experimental studies and clinical trials have demonstrated
that Tan IIA prevents oxidative stress-triggered atherogenesis as
well as cardiac injury and hypertrophy (13). The protective effect of Tan IIA on
DOX-induced cardiotoxicity has been preliminarily investigated
(14), but the mechanism remains
unknown. In view of the critical role Nrf2 has in DOX-induced
cardiotoxicity, the present study aimed to determine whether the
Nrf2-dependent antioxidant response mediated the protective effect
of Tan IIA on DOX-induced cardiotoxicity in vivo and in
vitro.
Materials and methods
Chemicals and reagents
Sodium Tan IIA sulfonate injection was purchased
from Shanghai First Biochemical & Pharmaceutical Co., Ltd.
(Shanghai, China); DOX hydrochloride was purchased from Shenzhen
Main Luck Pharmaceuticals, Inc. (Shenzhen, China). The detection
kit for aspartate aminotransferase (AST) was purchased from Abbott
Laboratories Trading Shanghai Co., Ltd. (Shanghai, China). The kit
for measuring lactate dehydrogenase (LDH) was purchased from Ningbo
Meikang Biotechnology Co., Ltd. (Ningbo, China). The kit for
measuring creatine kinase (CK) and creatine kinase-muscle/brain
(CK-MB) from Beijing Strong Biotechnologies, Inc. (Beijing, China).
Malondialdehyde (MDA; cat. no. A003-1), reduced glutathione (GSH;
cat. no. A006-2), superoxide dismutase (SOD; cat. no. A001-3) and
catalase (CAT; cat. no. A007-2) assay kits were from Nanjing
Jiancheng Bioengineering Institute (Nanjing, China).
Dichloro-dihydro-fluorescein diacetate (DCFH-DA) was purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany; cat. no. D6883).
Tan IIA (purity, >98%) was purchased from
National Institutes for Food and Drug Control (Beijing, China).
DOX, tertbutyl hydroquinone (tBHQ), dimethyl sulfoxide (DMSO) and
MTT were purchased from Sigma-Aldrich; Merck KGaA. ROS Assay kit
(cat. no. S0033) and BCA protein assay kit (cat. no. P0009) was
purchased from Beyotime Institute of Biotechnology (Shanghai,
China). Small interfering (si)RNA was purchased from Ribobio Co.,
Ltd. (Guangzhou, China). Lipofectamine 2000 was purchased from
Invitrogen (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Anti-Nrf2 antibody were purchased from Santa Cruz Biotechnology,
Inc. (cat. no. sc-722; Dallas, TX, USA). Anti-proliferating cell
nuclear antigen (PCNA; cat. no. ab18197) and β-actin (cat. no.
ab8226) were purchased from Abcam (Cambridge, UK).
Animals and pharmacological
treatments
A total of 30 male Institute of Cancer Research mice
weighing 25–29 g and aged 6 weeks were purchased from Beijing Vital
River Laboratory Animal Technology Co., Ltd. (Beijing, China). The
mice were given rodent chow 5001 (Beijing Vital River Laboratory
Animal Technology Co., Ltd.) and distilled water ad libitum, and
housed at room temperature (24±2°C) under a humidity-controlled
(65±5%) condition on a regular 12-h light/dark cycle. The mice were
randomly assigned to 5 groups (n=6 in each group) as follows: The
control (normal saline) group, the DOX (18 mg/kg) group, the Tan
IIA (15 mg/kg) + DOX (18 mg/kg) group, the Tan IIA (30 mg/kg) + DOX
(18 mg/kg) group, and the Tan IIA (30 mg/kg) group. The dose of Tan
IIA (15 and 30 mg/kg) was selected based on the results of a
previous study (15). The mice were
treated with Tan IIA or 0.2 ml normal saline from day 1 to day 7,
and DOX was administered at day 5, the mice were then sacrificed at
day 8; all treatments were administered by intraperitoneal
injection. The mice in the control (normal saline) group did not
receive DOX. All animal use procedures were conducted according to
the Regulations of Experimental Animal Administration issued by the
State Committee of Science and Technology of the People's Republic
of China, with the approval of the Ethics Committee in The
Experimental Animal Center of the Second Xiangya Hospital
(Changsha, China).
Heart histopathological
examination
The mice hearts of each group were harvested
immediately following blood collection. Sections of the freshly
heart samples were used to analyze biochemical index. The rest of
samples were fixed with 4% paraformaldehyde for 24 h at 25°C, and
embedded in paraffin blocks. They were then sectioned 4-µm-thick
sections and stained with hematoxylin and eosin (HE) at room
temperature for 3 min. The structure was examined with a light
microscope (magnification, ×100) (CKX41; Olympus Corporation,
Tokyo, Japan).
Measurement of serum myocardial
enzymes
Blood were collected from the heart of anesthetized
mouse and centrifuged (1,006.2 × g, 10 min, 4°C) to obtain the
serum samples, which were assayed for the determination of serum
myocardial enzymes (AST, LDH, CK and CK-MB) using kits with an
automatic biochemical analyzer (Abbott Pharmaceutical Co., Ltd.,
Lake Bluff, IL, USA). The experiments were performed according to
the manufacturers' instructions.
Measurement of GSH, SOD, CAT and
MDA
The mice hearts were harvested immediately following
blood collection. The partial myocardial tissues from each heart
was minced and homogenized to prepare for GSH, SOD, CAT, and MDA
detection with corresponding kits. The experiments were performed
according to the manufacturers' protocol.
Cell culture and treatment
The H9c2 rat myoblast cell line obtained from
Xiangya Cell Bank (Central South University, Changsha, China) was
cultured in Dulbecco's modified Eagle's medium (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
FBS (Biological Industries, Kibbutz Beit Haemek, Israel) and
antibiotics (100 µg/ml streptomycin and 100 U/ml penicillin;
Hyclone; GE Healthcare Life Sciences, Logan, UT, USA) in a
humidified atmosphere with 5% CO2 at 37°C.
The cells were incubated with 1 µmol/l DOX for 24 h
with or without Tan IIA pretreatment for 4 h at 37°C, except where
indicated otherwise. Tan IIA was dissolved in DMSO and diluted with
cell culture media to achieve the final concentration (1, 3, 5 and
10 µM). The final concentration of Tan IIA was determined according
to previous reported study (16).
tBHQ (50 nM), a classical activator of Nrf2, was used to
pretreatment cells for 4 h and then treat with 1 µmol/DOX for 24
h.
siRNA transient transfection
The Nrf2 siRNA (sense: 5′-CGUGAAUCCCAAUGUGAAATT-3′,
antisense: 5′-UGUUUCACAUUGGGAUUCACGTT-3′) and negative control
siRNA (sense: 5′-CUUCCUCUCUUUCUCUCCCUUGUGA-3′, antisense:
5′-UCACAAGGGAGAGAAAGAGAGGAAGGA-3′) were synthesized and purchased
from Ribobio Co., Ltd. (Guangzhou, China). H9c2 cells were grown in
96-well plates and transiently transfected with 0.5 µg Nrf2 or
negative control siRNA constructs using Lipofectamine 2000
transfection reagent, according to the manufacturer's protocol.
After incubating at 33°C and 5% CO2 for 36 h, cells were
further treated with DOX and/or Tan II A prior to the MTT and
western blot analyses.
Cell viability analysis
Cell viability was determined by MTT assay according
to the manufacturer's protocol; DMSO was used to dissolve purple
formazan. Percent viability was defined as the relative absorbance
of the treated vs. control cells at the wavelength of 490 nm. The
morphological changes were detected with a light microscope
(magnification, ×200).
Measurement of intracellular ROS and
GSH
Intracellular accumulation of ROS was determined by
measuring the oxidative conversion of cell permeable DCFH-DA to
fluorescent dichlorofluorescein (DCF) in afluorospectrophotometer
(model F4000; Hitachi, Ltd., Tokyo, Japan). A total of
2×104 cells were incubated with 0.1 ml DCFH-DA (10 µM)
at 37°C for 20 min. DCF fluorescence distribution was detected by
fluorospectrophotometer analysis at an excitation wavelength of 488
nm and at an emission wavelength of 535 nm. Intracellular
accumulation of GSH was measured using the aforementioned assay kit
according to the corresponding manufacturer's protocol.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total mRNA was extracted from the heart tissue or
cells with TRIzol (Life Technologies; Thermo Fisher Scientific,
Inc.) and equal amounts of RNA were reverse-transcribed to cDNA
using a PrimeScript RT reagent kit with gDNA Eraser (Perfect Real
Time; Takara Bio, Inc., Otsu, Japan). cDNA amplification was
performed with an initial step for 15 sec at 95°C, followed by 40
cycles (15 sec at 95°C, then 31 sec at 60°C). Data analysis was
performed using the 7900HT Sequence Detection System (Applied
Biosystems; Thermo Fisher Scientific, Inc.) and the
2−ΔΔCq method (17). The
level of β-actin mRNA was used as the internal standard. The
primers for real-time PCR analysis are presented in Tables I and II.
 | Table I.Mice primers for quantitative
polymerase chain reaction. |
Table I.
Mice primers for quantitative
polymerase chain reaction.
| Gene | Forward | Reverse |
|---|
| Nrf2 |
5′-TAGTGCCCCTGGAAGTGTCA-3′ |
5′-TTGGGATTCACGCATAGGAG-3′ |
| HO-1 |
5′-AGCCCCACCAAGTTCAAACA-3′ |
5′-TGCCAACAGGAAGCTGAGAG-3′ |
| NQO1 |
5′-CAGCCAATCAGCGTTCGGTA-3′ |
5′-CTTCATGGCGTAGTTGAATGATGTC-3′ |
| GCLC |
5′-CAGTCAAGGACCGGCACAAG-3′ |
5′-CAAGAACATCGCCTCCATTCAG-3′ |
| MRP2 |
5′-CGCGTCCGGCAGTATATGA-3′ |
5′-ATAATCTTTGACTCAGTGTGGA-3′ |
| P-gp |
5′-CCCATCATTGCAATAGCAGG-3′ |
5′-GTTCAAACTTCTGCTCCTGA-3′ |
| β-actin |
5′-CATCCTGCGTCTGGACCTGG-3′ |
5′-TAATGTCACGCACGATTTCC-3′ |
| Table II.Rat primers for quantitative
polymerase chain reaction. |
Table II.
Rat primers for quantitative
polymerase chain reaction.
| Gene | Forward | Reverse |
|---|
| Nrf2 |
5′-CCATGCCTTCTTCCACGAA-3′ |
5′-AGGGCCCATGGATTTCAGTT-3′ |
| HO-1 |
5′-GCGAAACAAGCAGAACCCA-3′ |
5′-GCTCAGGATGAGTACCTCCCA-3′ |
| NQO1 |
5′-AACGTCATTCTCTGGCCAATTC-3′ |
5′-GCCAATGCTGTACACCAGTTGA-3′ |
| GCLC |
5′-GTCTTCAGGTGACATTCCAAGC-3′ |
5′-TGTTCTTCAGGGGCTCCAGTC-3′ |
| MRP2 |
5′-GCTGGTTGGAAACTTGGTCG-3′ |
5′-CAACTGCCACAATGTTGGTC-3′ |
| P-gp |
5′-ATCAACTCGCAAAAGCATCC-3′ |
5′-AATTCAACTTCAGGATCCGC-3′ |
| β-actin |
5′-TACAACCTCCTTGCAGCTCC-3′ |
5′-GGATCTTCATGAGGTAGTCAGTC-3′ |
Western blotting
The heart tissue of the mice and the cell extracts
were prepared in radioimmunoprecipitation assay buffer (Beyotime
Institute of Biotechnology). The nuclear and cytoplasmic extracts
were harvested with NE-PER nuclear and cytoplasmic extraction
reagents (Pierce; Thermo Fisher Scientific, Inc.) using the
manufacturer's protocols. The total mass of protein was determined
using a BCA assay. Western blotting was performed with some
modifications as described (15).
Total protein (20 µg/lane) was loaded and separated by 10% SDS-PAGE
electrophoresis and transferred to polyvinylidene difluroride
membranes. Following blocking in 5% non-fat milk in 0.05%
Tween-20/Tris-buffered saline for 1 h at room temperature, the
membranes were incubated overnight at 4°C with the following
primary antibodies: Nrf2 (1:1,000), PCNA and β-actin (both
1:2,000). The immunoblots were then incubated with horseradish
peroxidase-conjugated immunoglobulin G secondary antibodies (goat
anti-mouse, cat. no. sc-2005 and goat anti-rabbit, cat. no.
sc-2004; both 1:5,000; Santa Cruz Biotechnology, Inc.) at room
temperature for 1 h. The membranes were developed with
Pierce™ ECL Western Blotting Substrate (cat. no. 32106;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. The densities of the bands were determined by an imaging
densitometer and the grayscale value of the bands were quantified
by ImageJ analysis software (version 1.43; National Institutes of
Health, Bethesda, MD, USA). PCNA was used as the nuclear loading
control and β-actin was used as the cytosolic loading control.
Statistical analysis
All data were presented as the mean ± standard error
of the mean. Statistical analysis was performed by analysis of
variance followed by the Newman-Student-Keuls test for multiple
comparisons using SPSS 19.0 (IBM Corp., Armonk, NY, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
Tan IIA protects against DOX-induced
myocardial injury in mice
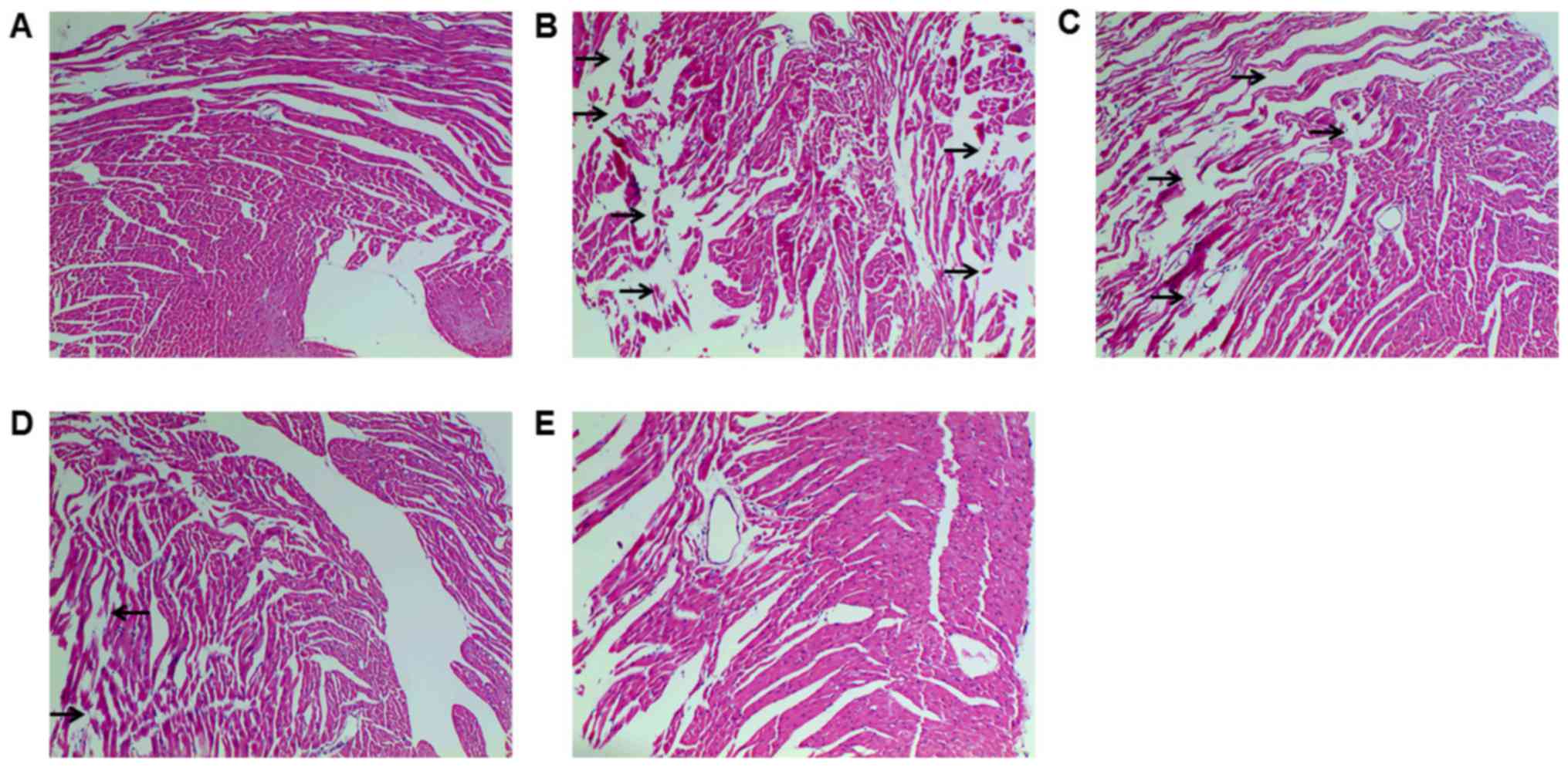
HE staining was conducted to detect
histopathological changes, which demonstrated myocardial fiber
fragmentation and gap enlargement in DOX group, which was
attenuated by Tan IIA pretreatment (Fig.
1). Serum myocardial enzymes (AST, LDH, CK and CK-MB) are the
important indexes that reflect the extent of myocardial injury
(18). As presented in Table III, DOX significantly increased the
activity of all enzymes compared with controls, indicating
cardiotoxicity. Conversely, Tan IIA significantly inhibited the
activity of AST, LDH and CK induced by DOX at 30 mg/kg, and
decreased the CK-MB activity at 15 and 30 mg/kg. These findings
suggest that Tan IIA can protect against DOX-induced myocardial
injury, in accordance with previous studies (13,14).
| Table III.Effect of Tan IIA pretreatment on the
serum myocardial enzymes. |
Table III.
Effect of Tan IIA pretreatment on the
serum myocardial enzymes.
| Group | AST (U/l) | LDH (U/l) | CK (U/l) | CK-MB (U/l) |
|---|
| Control | 127.6±14.3 | 698.7±60.0 | 1,866.3±279.4 | 390.3±71.4 |
| DOX (18 mg/kg) |
372.3±61.6a |
1,557.4±214.2a |
3,002.0±614.1a |
774.5±205.7a |
| D+T (15 mg/kg) | 333.4±110.9 | 1,249.5±518.8 | 2,159.7±749.7 |
471.0±171.0b |
| D+T (30 mg/kg) |
183.0±53.2c |
1,067.3±273.8b |
1,676.9±767.3b |
366.4±166.6c |
| Tan IIA (30
mg/kg) | 111.9±40.5 | 570.1±208.4 | 1,566.5±571.3 | 336.8±89.6 |
Antioxidant activity of Tan IIA is
associated with its protective effect against DOX-induced
cardiotoxicity in mice
The level of SOD, CAT, GSH and MDA were measured to
evaluate oxidative stress degree. DOX induced oxidative stress, as
indicated by significantly decreased SOD and CAT activities, GSH
content and increased MDA production compared with controls. Tan
IIA dose-dependently and significantly inhibited the effect of DOX
on SOD, CAT, GSH and MDA level in DOX+Tan IIA groups, and thus
exhibited potent antioxidant capacity (Table IV).
| Table IV.Effect of Tan IIA pretreatment on
antioxidant capacity in mice heart. |
Table IV.
Effect of Tan IIA pretreatment on
antioxidant capacity in mice heart.
| Group | GSH (µmol/g) | SOD (U/mg) | CAT (U/mg) | MDA (nmol/mg) |
|---|
| Control | 20.2±1.6 | 106.8±6.1 | 66.8±5.7 | 9.5±1.2 |
| DOX (18 mg/kg) |
15.0±1.6b |
79.0±16.8a |
52.0±4.3a |
14.4±1.7b |
| D+T (15 mg/kg) |
20.5±3.2d |
107.5±14.3d |
63.1±5.6c | 11.2±2.7 |
| D+T (30 mg/kg) |
23.5±3.8e |
118.6±12.3d |
71.0±9.6e |
9.2±2.0d |
| Tan IIA (30
mg/kg) | 21.4±2.6 | 108.8±12.7 | 68.5±8.3 | 9.0±4.4 |
Nrf2 signaling is associated with the
protective effect of Tan IIA on DOX-induced cardiotoxicity in
mice
To determine whether Tan IIA protected against
DOX-induced cardiotoxicity via induction of Nrf2 signaling, the
expression of Nrf2 and its downstream genes were measured by
western blotting and RT-qPCR. The mRNA results demonstrated that,
compared with controls, DOX alone had no significant influence on
Nrf2, heme oxygenase (HO)-1 and glutamate-cysteine ligase catalytic
subunit (GCLC), but significantly raised the mRNA expression of
NAD(P)H dehydrogenase (quinone) 1 (NQO1), multidrug
resistance-associated protein 2 (MRP2) and P-glycoprotein (P-gp).
Compared with DOX alone, Tan IIA pretreatment significantly
increased Nrf2, HO-1, NQO1 and GCLC in DOX+Tan IIA groups, but
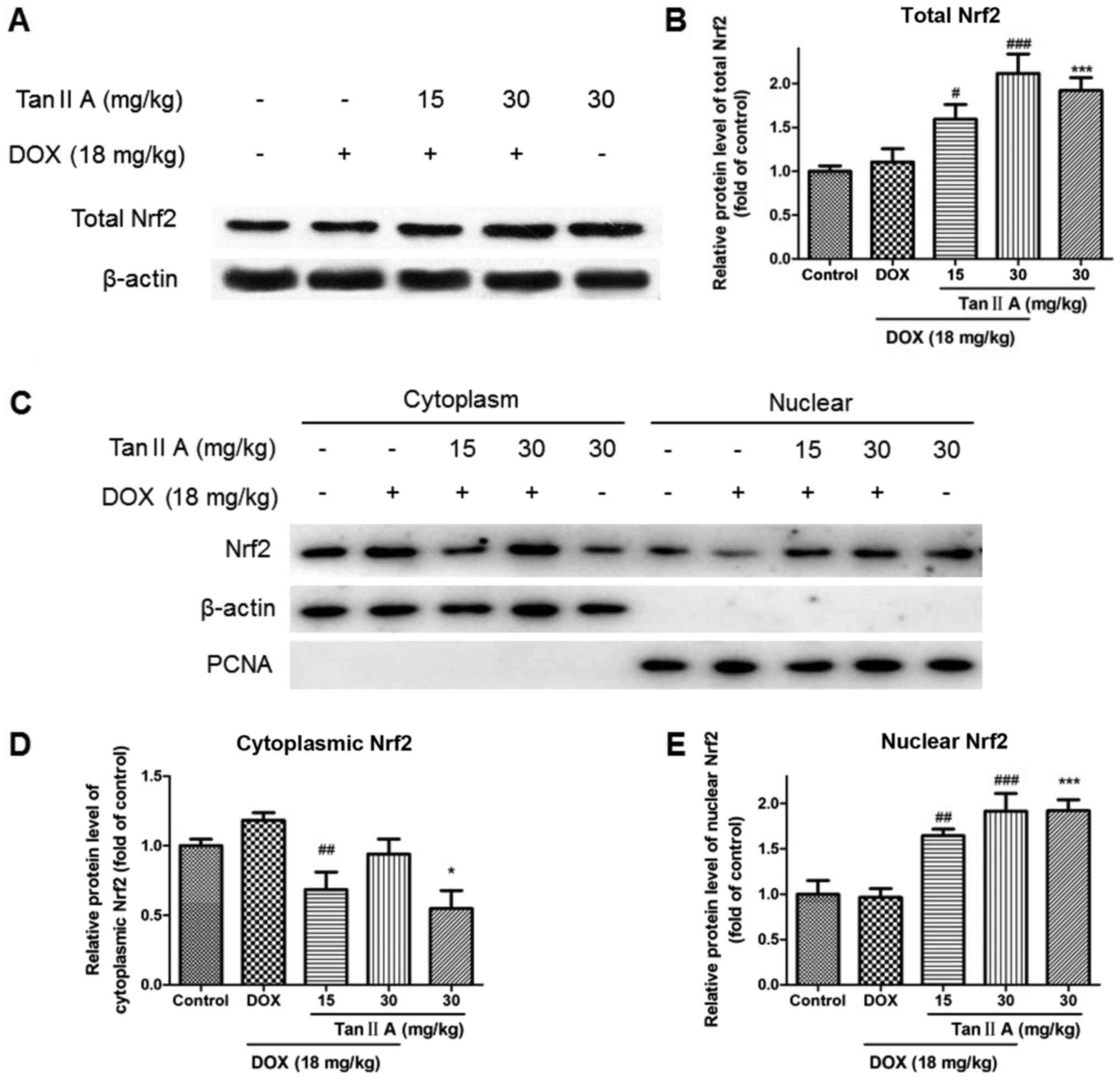
reduced the expression of MRP2 and P-gp (Fig. 2). As presented in Fig. 3A and B, the protein expression of
total Nrf2 was significantly increased in the DOX + Tan IIA groups
in a dose-dependent manner compared with DOX alone, and Tan IIA
alone also induced a significant increase in Nrf2 expression in the
heart tissue compared with controls. As nuclear translocation of
Nrf2 was the key to activating Nrf2-related pathway, nuclear
accumulation and cytoplasmic Nrf2 expression were evaluated.
Nuclear Nrf2 expression was similar to total Nrf2, whereas
cytoplasmic Nrf2 was decreased in the Tan IIA+DOX group and Tan IIA
alone group compared with the DOX or control group (Fig. 3C-E).
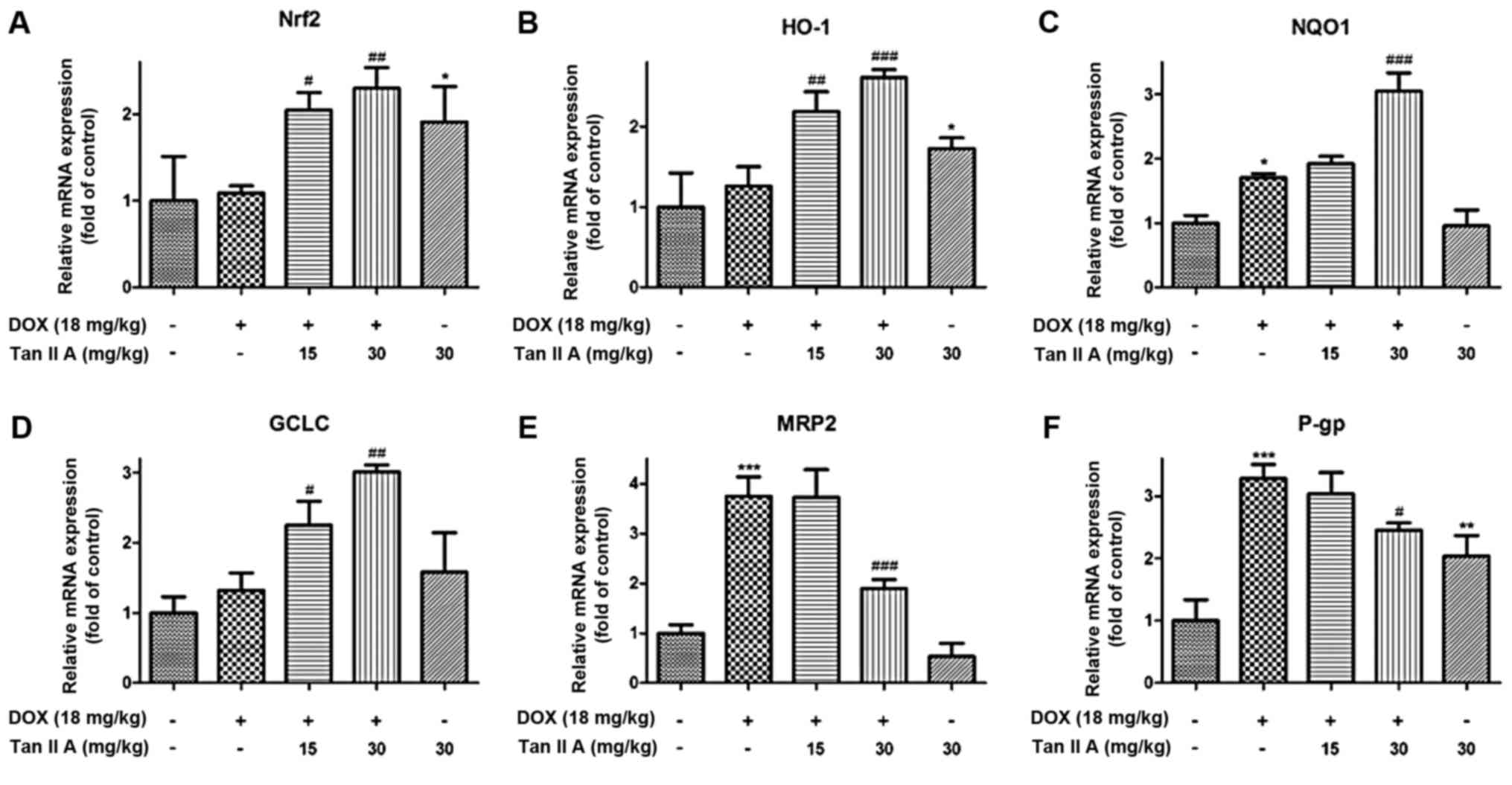 | Figure 2.Effect of Tan IIA on Nrf2 mRNA and
its downstream genes expression in DOX-treated mice heart. The mRNA
expression of (A) Nrf2, and its downstream genes, including (B)
HO-1, (C) NQO1, (D) GCLC, (E) MRP2 and (F) P-gp were determined by
reverse transcription-quantitative polymerase chain reaction. Data
are presented as the mean ± standard error of the mean (n=6).
*P<0.05, **P<0.01 and ***P<0.001 vs. the control;
#P<0.05, ##P<0.01 and
###P<0.001 vs. DOX. Tan IIA, tanshinone IIA; Nrf2,
nuclear factor (erythroid-derived 2)-like 2; DOX, doxorubicin;
HO-1, heme oxygenase-1; NQO1, NAD(P)H dehydrogenase (quinone) 1;
GCLC, glutamate-cysteine ligase catalytic subunit; MRP2, multidrug
resistance-associated protein 2; P-gp, P-glycoprotein. |
Tan IIA protects against DOX-induced
injury and oxidative stress in H9c2 cells
In vitro experiments were performed in H9c2
cells. The effect of DOX or Tan IIA treatment alone for 24 h on the
viability of H9c2 cells was investigated (Fig. 4A and B). DOX decreased the cell
viability in a dose-dependent manner (0.1–1 µM). DOX (1 µM) was
used in the subsequent experiments, which was consistent with
previous studies (5,11). Tan IIA (1–10 µM) treatment alone
exhibited no significant effect on cell viability, but the cell
viability was inhibited by Tan IIA at the concentration of 15 and
20 µM. Furthermore, Tan IIA (1–10 µM) pretreatment dose-dependently
reversed the inhibitive effect of DOX on cell viability (Fig. 4C). Additional experiments indicated
that DOX induced cell rounding and detachment, whereas Tan IIA
pretreatment protected the cells from such morphological changes
(Fig. 4D).
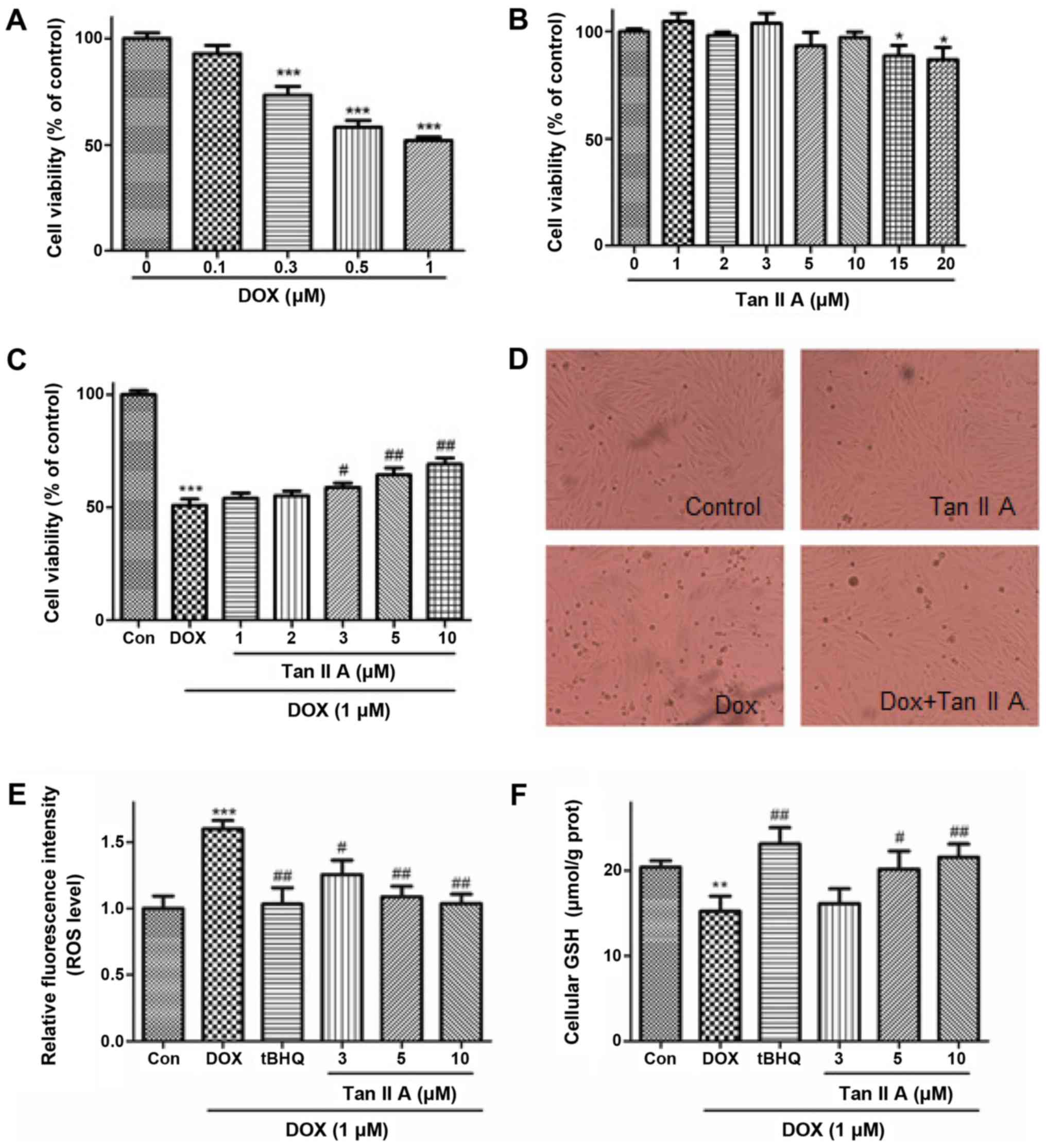 | Figure 4.Effect of Tan IIA on DOX-induced cell
injury and oxidative stress in H9c2 cells. Cells were incubated
with increasing concentrations of (A) DOX (0–1 µM) for 24 h or (B)
Tan IIA (0–20 µM) for 4 h, and cell viability was measured by MTT
assay. (C) H9c2 cells were pretreated with the indicated Tan IIA
concentrations for 4 h followed by DOX (1 µM, 24 h) treatment, and
cell viability was determined by MTT assay. (D) The cell
morphological changes were detected via light microscopy
(magnification, ×200). (E) Intracellular ROS levels were measured
with a fluorometric assay. (F) The content of intracellular GSH was
measured with a GSH assay kit. Data are presented as mean ±
standard error of the mean (n=3). *P<0.05, **P<0.01 and
***P<0.001 vs. the control; #P<0.05 and
##P<0.01 vs. DOX. Tan IIA, tanshinone IIA; DOX,
doxorubicin; ROS, reactive oxygen species; GSH, glutathione; tBHQ,
tertbutyl hydroquinone. |
To determine whether Tan IIA inhibited the
DOX-induced generation of ROS in H9c2 cells, the generation of ROS
in DOX-treated and Tan IIA + DOX-treated cells were measured via
fluorescence microscopy with DCFH-DA as a probe following 24 h
incubation (Fig. 4E), in which tBHQ,
a classical activator of Nrf2, was used as the positive control.
DOX significantly increased the ROS accumulation compared with
controls, and co-treatment with Tan IIA dose-dependently
ameliorated the ROS levels. As presented in Fig. 4F, DOX treatment resulted in
significant decreases of GSH levels, and this inhibition was
attenuated by Tan IIA pretreatment, which enhanced the GSH content
dose-dependently. These findings suggest that Tan IIA alleviated
the DOX-induced oxidative stress.
Nrf2 signaling is associated with the
protective effect of Tan IIA on DOX-induced H9c2 cells injury
The mRNA expression of Nrf2 was significantly and
dose-dependently increased following pretreatment with Tan IIA in
DOX+Tan IIA group compared with the DOX alone group, as were HO-1,
NQO1 and GCLC levels. Moreover, P-gp and MRP2 expression levels in
the DOX + Tan IIA groups were decreased with 5 and 10 µM Tan IIA,
but increased with 3 µM Tan IIA compared with DOX treatment alone,
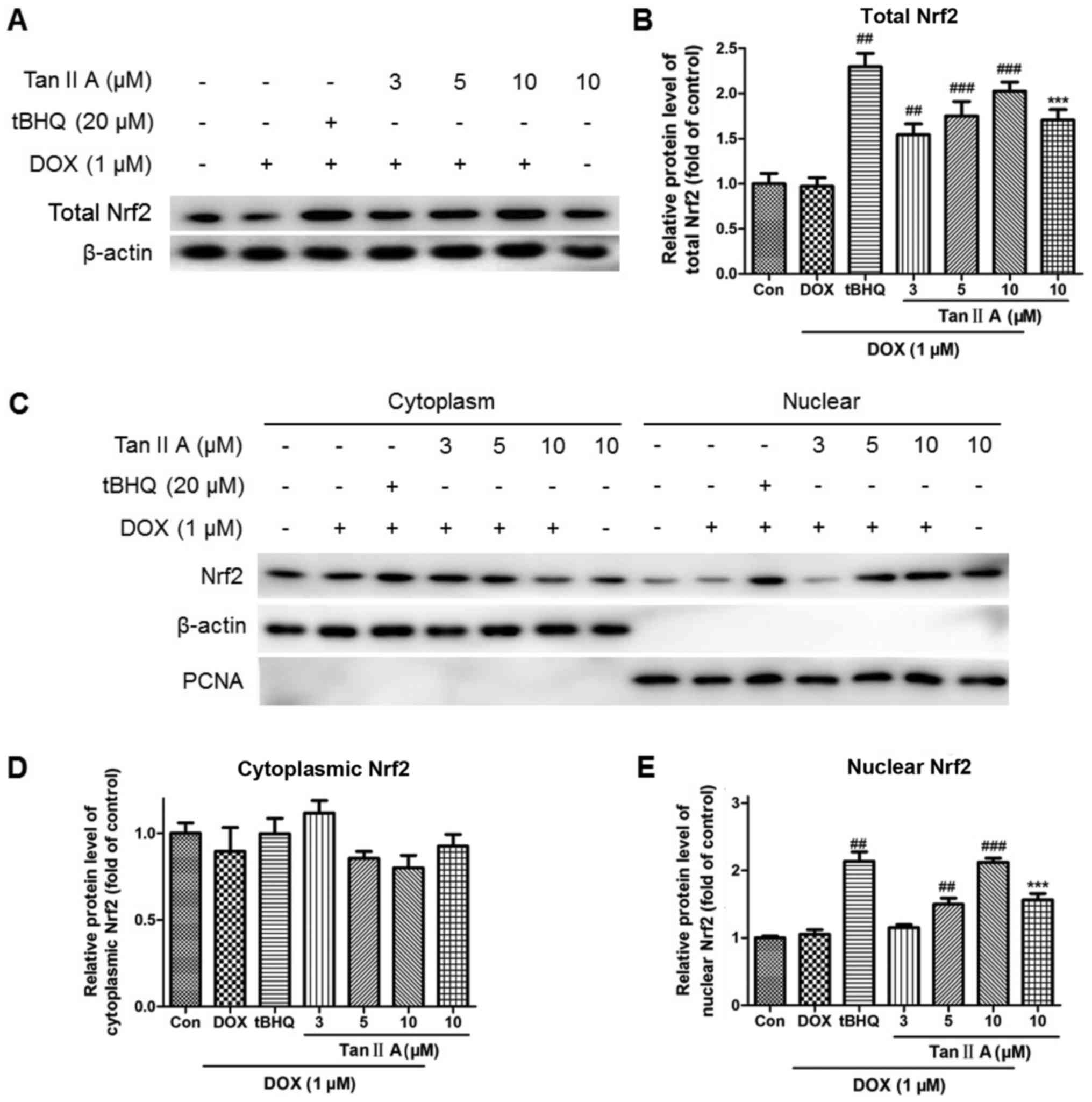
although they remained higher than those of the control (Fig. 5). Consistent with the mRNA data, Tan
IIA pretreatment dose-dependently increased the protein expression
of total Nrf2 compared with DOX alone (Fig. 6A and B). The nuclear and cytoplasmic
Nrf2 accumulations were also investigated. As presented in Fig. 6C-E, Tan IIA (5 and 10 µM)
pretreatment for 4 h prior to DOX treatment for 24 h significantly
increased the nuclear level of Nrf2, whereas no significant changes
were observed at the cytoplasmic level with all Tan IIA dose or at
the nuclear level with 3 µM Tan IIA.
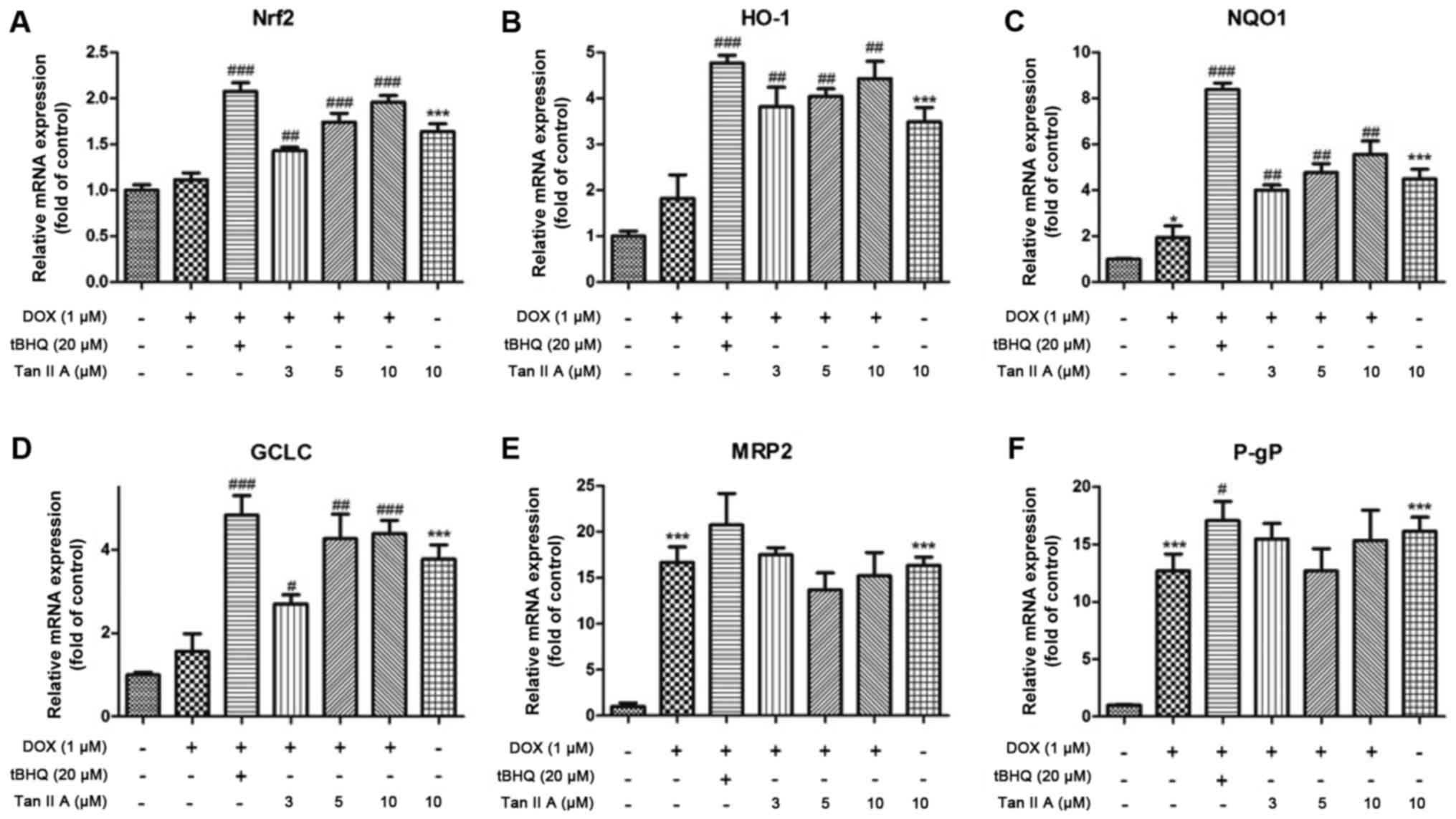 | Figure 5.Effect of Tan IIA on relative mRNA
expression of Nrf2 and its downstream genes in H9c2 cells. The mRNA
expression of (A) Nrf2, and its downstream genes expression
including (B) HO-1, (C) NQO1, (D) GCLC, (E) MRP2 and (F) P-gP were
determined by reverse transcription-quantitative polymerase chain
reaction. Data are presented as mean ± standard error of the mean
(n=3). *P<0.05 and ***P<0.001 vs. the control;
#P<0.05, ##P<0.01 and
###P<0.001 vs. DOX. Tan IIA, tanshinone IIA; Nrf2,
nuclear factor (erythroid-derived 2)-like 2; HO-1, heme
oxygenase-1; NQO1, NAD(P)H dehydrogenase (quinone) 1; GCLC,
glutamate-cysteine ligase catalytic subunit; MRP2, multidrug
resistance-associated protein 2; P-gp, P-glycoprotein; DOX,
doxorubicin; tBHQ, tertbutyl hydroquinone. |
Nrf2 knockdown inhibits the protective
effect of Tan IIA on DOX-induced H9c2 cells injury
To further determine whether Tan IIA protected H9c2
cells against DOX-induced toxicity in a Nrf2-dependent manner,
Nrf2-siRNA was transfected in H9c2 cells for 24 h to knockdown the
Nrf2 expression. As presented in Fig.
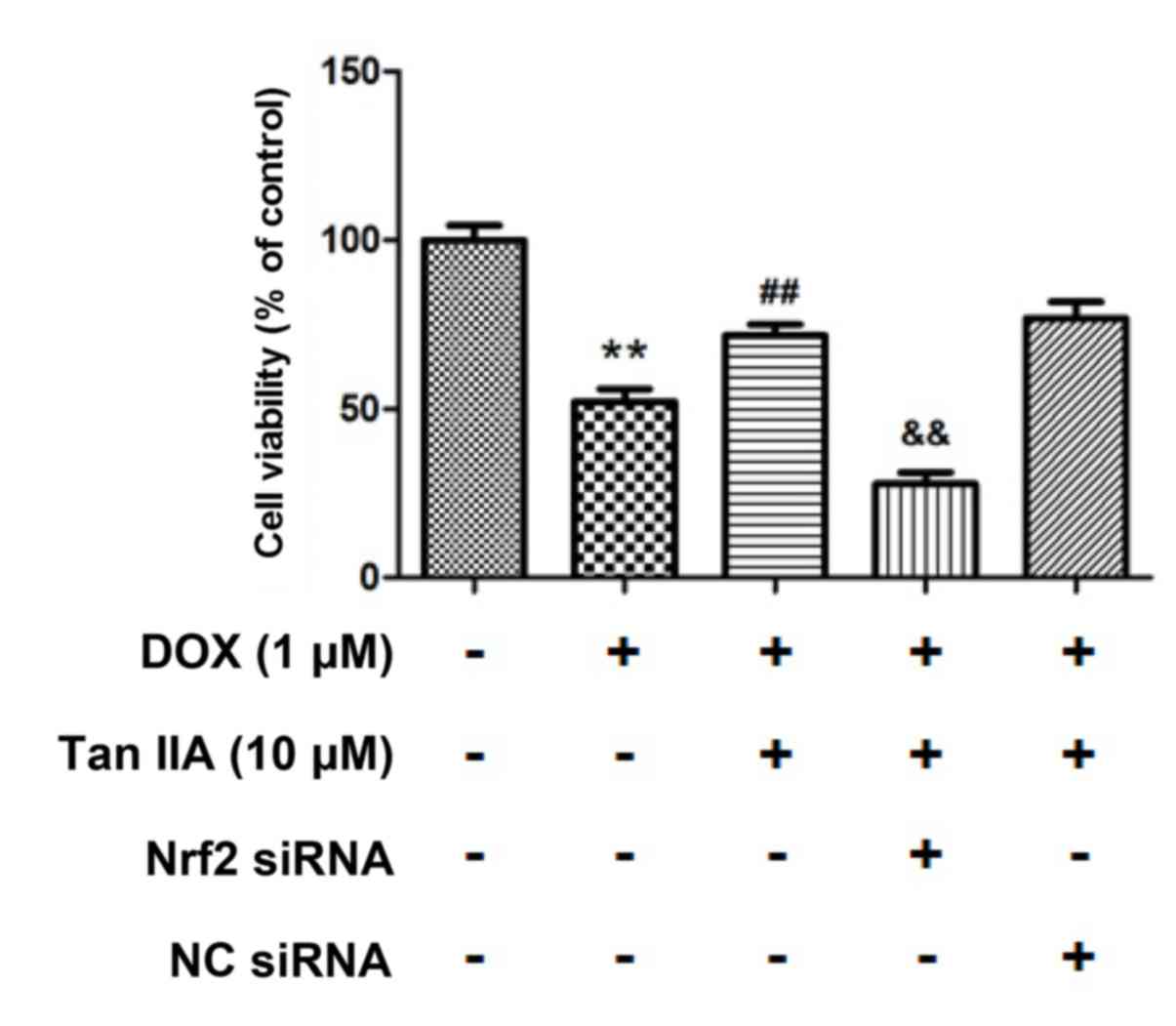
7, the protein level of Nrf2 was reduced to 38.4±9.0% following
Nrf2 siRNA transfection compared with that of the control. It was
also observed that nuclear Nrf2 accumulated in the DOX + Tan IIA
groups, and was significantly reduced in the Nrf2-siRNA + Tan IIA
groups. Furthermore, the MTT assay presented in Fig. 8 revealed that DOX treatment
significantly decreased the cell viability, while Tan IIA inhibited
the effect of DOX on H9c2 cells, which is consistent with the
results in Fig. 4C. Additionally,
pre-transfection with Nrf2-siRNA to knockdown the Nrf2 expression
reversed the effect of Tan IIA on the DOX-induced inhibition of
cell viability. These results suggest that Nrf2 mediates the
protective effect of Tan IIA on DOX-induced H9c2 cell injury.
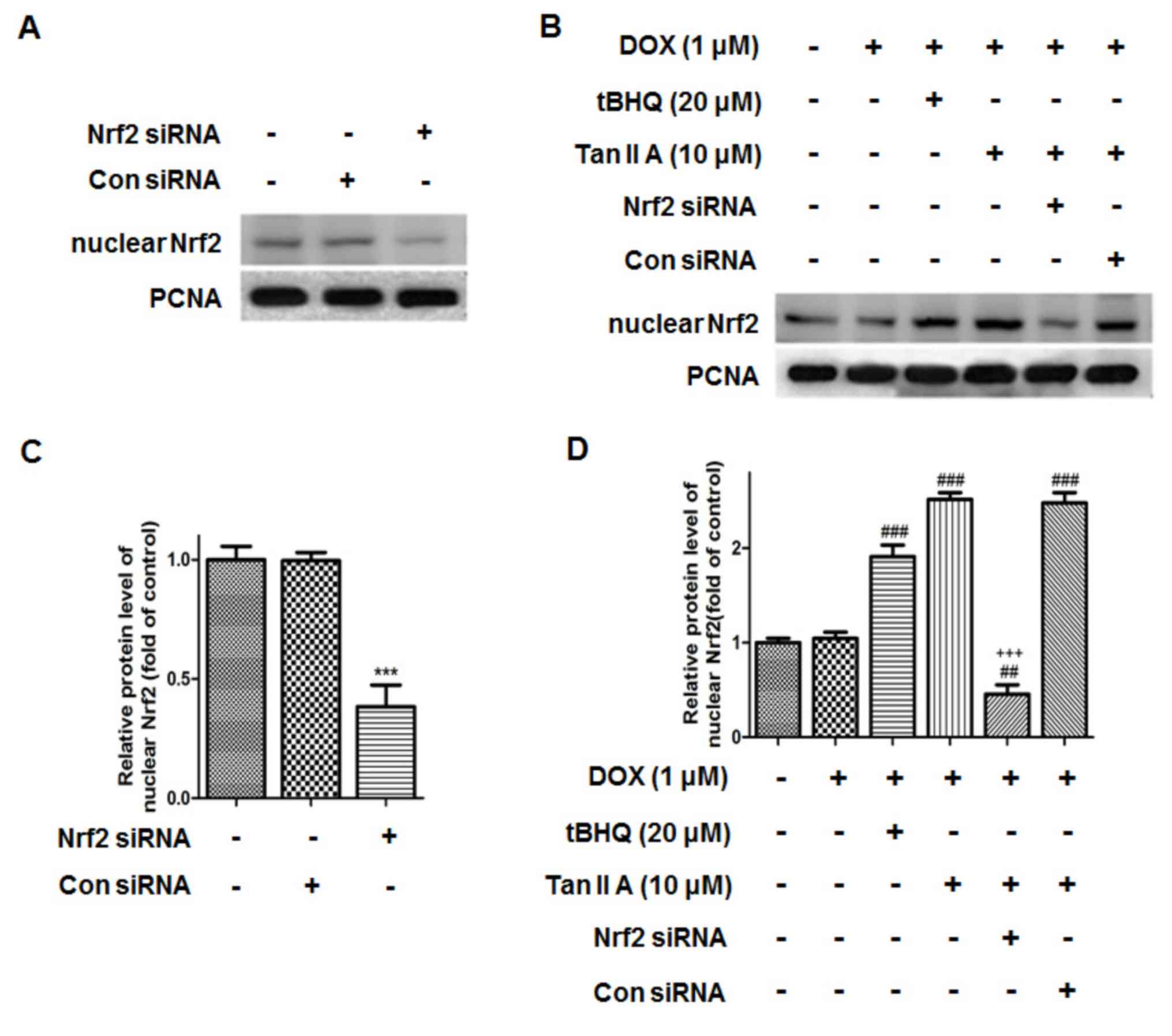 | Figure 7.Effect of Nrf2 siRNA on the protein
expression of nuclear Nrf2 in H9c2 cells. The protein expression of
nuclear Nrf2 upon (A) Nrf2 siRNA treatment, and (B) subsequent
treatment with DOX, tBHQ and Tan IIA was determined by western
blotting, (C and D) and quantitatively analysed. Data are presented
as mean ± standard error of the mean (n=3). ***P<0.001 vs. NC,
##P<0.01 and ###P<0.001 vs. DOX;
+++P<0.001 vs. DOX + Tan IIA (10 µM). Nrf2, nuclear
factor (erythroid-derived 2)-like 2; siRNA, small interfering RNA;
DOX, doxorubicin; tBHQ, tertbutyl hydroquinone; Tan IIA, tanshinone
IIA; NC, siRNA negative control; PCNA, proliferating cell nuclear
antigen. |
Discussion
Nrf2 is a transcription factor that serves a key
role in regulation of intracellular redox signaling (19). Under normal physiological conditions,
Nrf2 is bound to Keap1 and located in the cytosol (20). The Keap1 protein contains several
cysteine residues with sulfhydryl groups that can react with ROS
and electrophiles (9). When cells
are exposed to cellular stress, like ROS, the cysteine residues of
Keap1 are modified by oxidative/electrophilic molecules, which
results in breaking the bonds between Keap1 and Nrf2 (21). Once the bonds are broken, Nrf2
translocates to the cell nucleus and initiates transcription of
antioxidant genes including SOD, NQO1, GCLC, HO-1, MRP2 and P-gP
(22). Imbalance between free
radicals and anti-oxidant defense is associated with cellular
dysfunctions leading to the pathophysiology of various
cardiovascular diseases (23).
Therefore, Nrf2 may be considered as a key regulator in maintenance
of normal cardiovascular function.
DOX is a widely used anti-cancer agent associated
with irreversible cardiomyopathy (2). Although the precise cardiotoxicity
mechanisms have not been clearly documented, it is widely accepted
that overproduction of ROS and oxidative stress have vital roles
(5,24). In view of the potent antioxidant
effect of Nrf2, several studies have investigated the role of Nrf2
in DOX-induced cardiotoxicity in vivo and in vitro
(25,26). It has been reported that Nrf2
mediated the protective effect of sulforaphane, a-Linolenic acid
and naringenin-7-O-glucoside on DOX-induced cardiotoxicity
(7,10,11),
which indicated that Nrf2 may be a guided drug target for
DOX-induced cardiotoxicity.
Tan IIA, a major bioactive diterpene quinone of
Salva miltiorrhiza, is used in the treatment of coronary
heart disease, cerebrovascular disease, hepatitis and
hepatocirrhosis due to its multiple pharmacological activities
including anti-oxidant, anti-inflammatory and anti-neoplastic
effects (12). The potential
cardioprotective effects of Tan IIA on DOX-induced cardiotoxicity
have been confirmed in animal models by decreasing the ST-interval
and QRS interval, and improving the occurrence of myocardium
fibrosis (14). On this basis, the
present study further elucidated the inherent mechanism. Another
previous study demonstrated that Tan IIA modulated Nrf2 expression
in JB6 cells through epigenetic regulations (16). Others reported that Tan IIA inhibited
atherosclerosis in endothelial cells via an Nrf2-related pathway
(27). In the present study, it was
not only discovered that Nrf2 nuclear accumulation was increased
when treated with Tan IIA alone, but also that Tan IIA induced Nrf2
activity significantly in the Tan IIA + DOX-treated groups in mice
and in H9c2 cells, and enhanced the expression of HO-1, NQO1 and
GCLC. The essential role of Nrf2 in the protective effects of Tan
IIA was further supported by the knock-down experiments. Silencing
the expression of Nrf2 by siRNA effectively suppressed the Tan
IIA-induced activation of Nrf2 and at the same time reversed the
increase in cell survival rate in the Tan IIA pretreatment groups.
These data strongly suggest that the protective effect of Tan IIA
on DOX-induced cardiotoxicity was at least partly mediated by the
activation of the Nrf2 signaling pathway.
As Nrf2 induced downstream genes including
antioxidants, phase II metabolism enzymes, and phase III drug
transporters, HO-1, NQO1, GCLC, MRP2 and P-gp were focused on as
they had been reported most in previous studies. HO-1 and NQO1 are
known as important intracellular antioxidants to defend oxidative
stress both inside and outside cells (28). GSH is vital in the detoxification of
xenobiotics, and DOX treatment resulted in the depletion of GSH
levels (29). GCLC is the catalytic
subunit of GCL that catalyzes the rate-limiting step in the
biosynthesis of GSH (30). Tan IIA
activated these genes to decrease the oxidative burden by
antioxidants and phase II conjugation reactions. However, although
MRP2 and P-gp were also downstream targets of Nrf2, their
expression exhibited no significant changes compared with Tan IIA
pretreatment groups with DOX alone. MRP2 and P-gp are phase III
drug transporters which can remove xenobiotics and metabolites that
accumulate in the tissues and lead to toxicity. However, such
transporters are more likely to be studied in metabolism organs
like the liver and the kidney (31).
Few studies on transporters in the heart have been published, to
the best of our knowledge, suggesting that transporters are not the
main target of drugs or toxins in the heart. Conversely, Tan IIA
has mostly been demonstrated to have anti-oxidant and
anti-inflammation properties, and the ability of Tan IIA to
modulate drug transporters, particularly MRP2 and P-gp, may be
quite weak (12). In addition, there
may be other signal pathways that can modulate MRP2 and P-gp other
than Nrf2.
Notably, it was observed that DOX-treatment induced
oxidative stress but did not activate Nrf2 in the present study. In
addition to Nrf2, many other factors are associated with
DOX-treatment induce oxidative stress, including NADPH and NQO1
(32,33). This may be the cause of the present
result. To the best of our knowledge, previous studies have
investigated the effect of DOX-treatment on Nrf2 nuclear
accumulation, however, yet received inconsistent results. Li et
al (7) and Han et al
(10) demonstrated that
DOX-treatment can inhibit the Nrf2 nuclear transport, whereas Yu
et al (11) received the
opposite result. These inconsistent results may be due to different
treatment times and concentrations.
Although several natural or synthetic compounds may
be used to prevent DOX-induced cardiotoxicity (34–36), one
of the major advantages of Tan IIA is that it has already been used
in clinic settings to treat coronary heart disease and angina
pectoris as Tan IIA sodium sulfonate injection with acceptable
bioavailability (37). The dose of
Tan IIA used in humans was 1.5 mg/kg, whereas 30 mg/kg in mice
(15), as used in the present study,
can be converted to 4.95 mg/kg in human. This suggests that the
dose of Tan IIA for treating DOX-induced cardiotoxicity in clinical
settings may need to be raised to >3 times as much as the dose
that patients are currently treated with. Furthermore, Tan IIA was
previously demonstrated to be able to suppress the proliferation of
cancer cells and exhibit anticancer activity (38–40),
which may improve anti-cancer potency when used together with
DOX.
A previous study demonstrated the protective effect
of Tan IIA against DOX-induced cardiotoxicity (14), but there were several limitations. It
has previously been suggested that H9c2 cells were undifferentiated
myoblasts, which may not be considered as cardiomyocytes (41). Furthermore, the study by Jiang et
al (14) demonstrated that cell
viability was not sufficient to indicate the protective effects of
Tan IIA against DOX-induced cardiotoxicity. More functional assays
may be required in future studies in primary cardiomyocytes to
verify the protective effects of Tan IIA, such as alteration of
cell cycle and apoptosis-related protein expressions.
In conclusion, the present study suggests that the
Nrf2-dependent antioxidant response mediates the protective effect
of Tan IIA on DOX-induced cardiotoxicity, indicating that Tan IIA
may be a promising therapeutic adjuvant that may prevent the heart
from serious side effects of DOX. Furthermore, these findings
emphasize the vital role of Nrf2 signaling as promising targets to
identify more potent pharmacological agents against DOX-induced
cardiotoxicity.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81703518, 81202985
and 81573686), Scientific Research Project of Hunan Provincial
Health and Family Planning Commission (grant no. B20180253), and
Open-End Fund for the Valuable and Precision Instruments of Central
South University (grant no. CSUZC201837).
Availability of data and materials
The datasets used or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
MY, WL and BZ designed the experiments. ZG, LC, PF,
ZL, ZW, SC, ZH and SW performed the experiments. ZG and WL analyzed
the data. WL and BZ wrote the manuscript.
Ethics approval and consent to
participate
All animal use procedures were conducted according
to the Regulations of Experimental Animal Administration issued by
the State Committee of Science and Technology of the People's
Republic of China, with the approval of the Ethics Committee of The
Experimental Animal Center of the Second Xiangya Hospital
(Changsha, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gorini S, De Angelis A, Berrino L, Malara
N, Rosano G and Ferraro E: Chemotherapeutic drugs and mitochondrial
dysfunction: Focus on doxorubicin, trastuzumab, and sunitinib. Oxid
Med Cell Longev. 2018:75827302018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vejpongsa P and Yeh ET: Prevention of
anthracycline-induced cardiotoxicity: Challenges and opportunities.
J Am Coll Cardiol. 64:938–945. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
van Dalen EC, van der Pal HJ, Caron HN and
Kremer LC: Different dosage schedules for reducing cardiotoxicity
in cancer patients receiving anthracycline chemotherapy. Cochrane
Database Syst Rev: CD005008. 2006. View Article : Google Scholar
|
|
4
|
Pugazhendhi A, Edison TNJI, Velmurugan BK,
Jacob JA and Karuppusamy I: Toxicity of doxorubicin (Dox) to
different experimental organ systems. Life Sci. 200:26–30. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Carvalho FS, Burgeiro A, Garcia R, Moreno
AJ, Carvalho RA and Oliveira PJ: Doxorubicin-induced
cardiotoxicity: From bioenergetic failure and cell death to
cardiomyopathy. Med Res Rev. 34:106–135. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen CT, Wang ZH, Hsu CC, Lin HH and Chen
JH: In vivo protective effects of diosgenin against
doxorubicin-induced cardiotoxicity. Nutrients. 7:4938–4954. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li B, Kim DS, Yadav RK, Kim HR and Chae
HJ: Sulforaphane prevents doxorubicin-induced oxidative stress and
cell death in rat H9c2 cells. Int J Mol Med. 36:53–64. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Howden R: Nrf2 and cardiovascular defense.
Oxid Med Cell Longev. 2013:1043082013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma Q and He X: Molecular basis of
electrophilic and oxidative defense: Promises and perils of Nrf2.
Pharmacol Rev. 64:1055–1081. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Han X, Pan J, Ren D, Cheng Y, Fan P and
Lou H: Naringenin-7-O-glucoside protects against
doxorubicin-induced toxicity in H9c2 cardiomyocytes by induction of
endogenous antioxidant enzymes. Food Chem Toxicol. 46:3140–3146.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yu X, Cui L, Zhang Z, Zhao Q and Li S:
α-Linolenic acid attenuates doxorubicin-induced cardiotoxicity in
rats through suppression of oxidative stress and apoptosis. Acta
Biochim Biophys Sin (Shanghai). 45:817–826. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu S and Liu P: Tanshinone II-A: New
perspectives for old remedies. Expert Opin Ther Pat. 23:149–153.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gao S, Liu Z, Li H, Little PJ, Liu P and
Xu S: Cardiovascular actions and therapeutic potential of
tanshinone IIA. Atherosclerosis. 220:3–10. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang B, Zhang L, Wang Y, Li M, Wu W, Guan
S, Liu X, Yang M, Wang J and Guo DA: Tanshinone IIA sodium
sulfonate protects against cardiotoxicity induced by doxorubicin in
vitro and in vivo. Food Chem Toxicol. 47:1538–1544. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang W, Guan C, Sun X, Zhao Z, Li J, Fu X,
Qiu Y, Huang M, Jin J and Huang Z: Tanshinone IIA protects against
acetaminophen-induced hepatotoxicity via activating the Nrf2
pathway. Phytomedicine. 23:589–596. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang L, Zhang C, Guo Y, Su ZY, Yang Y, Shu
L and Kong AN: Blocking of JB6 cell transformation by tanshinone
IIA: Epigenetic reactivation of Nrf2 antioxidative stress pathway.
AAPS J. 16:1214–1225. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Alam MF, Khan G, Safhi MM, Alshahrani S,
Siddiqui R, Sivagurunathan Moni S and Anwer T: Thymoquinone
ameliorates doxorubicin-induced cardiotoxicity in swiss albino mice
by modulating oxidative damage and cellular inflammation. Cardiol
Res Pract. 2018:14830412018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sies H, Berndt C and Jones DP: Oxidative
stress. Annu Rev Biochem. 86:715–748. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bellezza I, Giambanco I, Minelli A and
Donato R: Nrf2-Keap1 signaling in oxidative and reductive stress.
Biochim Biophys Acta. 1865:721–733. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Silva-Islas CA and Maldonado PD: Canonical
and non-canonical mechanisms of Nrf2 activation. Pharmacol Res.
134:92–99. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jiang S, Yang Y, Li T, Ma Z, Hu W, Deng C,
Fan C, Lv J, Sun Y and Yi W: An overview of the mechanisms and
novel roles of Nrf2 in cardiovascular diseases. Expert Opin Ther
Targets. 20:1413–1424. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ramprasath T, Vasudevan V, Sasikumar S,
Puhari SS, Saso L and Selvam GS: Regression of oxidative stress by
targeting eNOS and Nrf2/ARE signaling: A guided drug target for
cardiovascular diseases. Curr Top Med Chem. 15:857–871. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tacar O, Sriamornsak P and Dass CR:
Doxorubicin: An update on anticancer molecular action, toxicity and
novel drug delivery systems. J Pharm Pharmacol. 65:157–170. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li S, Wang W, Niu T, Wang H, Li B, Shao L,
Lai Y, Li H, Janicki JS, Wang XL, et al: Nrf2 deficiency
exaggerates doxorubicin-induced cardiotoxicity and cardiac
dysfunction. Oxid Med Cell Longev. 2014:7485242014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang LF, Su SW, Wang L, Zhang GQ, Zhang R,
Niu YJ, Guo YS, Li CY, Jiang WB, Liu Y and Guo HC:
Tert-butylhydroquinone ameliorates doxorubicin-induced
cardiotoxicity by activating Nrf2 and inducing the expression of
its target genes. Am J Transl Res. 7:1724–1735. 2015.PubMed/NCBI
|
|
27
|
Liu Z, Wang J, Huang E, Gao S, Li H, Lu J,
Tian K, Little PJ, Shen X, Xu S and Liu P: Tanshinone IIA
suppresses cholesterol accumulation in human macrophages: Role of
heme oxygenase-1. J Lipid Res. 55:201–213. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sarkar S, Mukherjee S, Chattopadhyay A and
Bhattacharya S: Low dose of arsenic trioxide triggers oxidative
stress in zebrafish brain: Expression of antioxidant genes.
Ecotoxicol Environ Saf. 107:1–8. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cheraghi M, Namdari M, Daraee H and
Negahdari B: Cardioprotective effect of magnetic hydrogel
nanocomposite loaded N,α-L-rhamnopyranosyl vincosamide isolated
from Moringa oleifera leaves against doxorubicin-induced cardiac
toxicity in rats: In vitro and in vivo studies. J Microencapsul.
34:335–341. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhu H, Long MH, Wu J, Wang MM, Li XY, Shen
H, Xu JD, Zhou L, Fang ZJ, Luo Y and Li SL: Ginseng alleviates
cyclophosphamide-induced hepatotoxicity via reversing disordered
homeostasis of glutathione and bile acid. Sci Rep. 5:175362015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kawase A, Norikane S, Okada A, Adachi M,
Kato Y and Iwaki M: Distinct alterations in ATP-binding cassette
transporter expression in liver, kidney, small intestine, and brain
in adjuvant-induced arthritic rats. J Pharm Sci. 103:2556–2564.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Park J, Park E, Ahn BH, Kim HJ, Park JH,
Koo SY, Kwak HS, Park HS, Kim DW, Song M, et al: NecroX-7 prevents
oxidative stress-induced cardiomyopathy by inhibition of NADPH
oxidase activity in rats. Toxicol Appl Pharmacol. 263:1–6. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hajra S, Patra AR, Basu A and Bhattacharya
S: Prevention of doxorubicin (DOX)-induced genotoxicity and
cardiotoxicity: Effect of plant derived small molecule
indole-3-carbinol (I3C) on oxidative stress and inflammation.
Biomed Pharmacother. 101:228–243. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Al-Harthi SE, Alarabi OM, Ramadan WS,
Alaama MN, Al-Kreathy HM, Damanhouri ZA, Khan LM and Osman AM:
Amelioration of doxorubicininduced cardiotoxicity by resveratrol.
Mol Med Rep. 10:1455–1460. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Thandavarayan RA, Giridharan VV, Arumugam
S, Suzuki K, Ko KM, Krishnamurthy P, Watanabe K and Konishi T:
Schisandrin B prevents doxorubicin induced cardiac dysfunction by
modulation of DNA damage, oxidative stress and inflammation through
inhibition of MAPK/p53 signaling. PLoS One. 10:e01192142015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen RC, Xu XD, Zhi Liu X, Sun GB, Zhu YD,
Dong X, Wang J, Zhang HJ, Zhang Q and Sun XB: Total flavonoids from
clinopodium chinense (Benth.) O. Ktze protect against
doxorubicin-induced cardiotoxicity in vitro and in vivo. Evid Based
Complement Alternat Med. 2015:4725652015.PubMed/NCBI
|
|
37
|
Chen Y, Tu JH, He YJ, Zhang W, Wang G, Tan
ZR, Zhou G, Fan L and Zhou HH: Effect of sodium tanshinone II A
sulfonate on the activity of CYP1A2 in healthy volunteers.
Xenobiotica. 39:508–513. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Munagala R, Aqil F, Jeyabalan J and Gupta
RC: Tanshinone IIA inhibits viral oncogene expression leading to
apoptosis and inhibition of cervical cancer. Cancer Lett.
356:536–546. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Su CC: Tanshinone IIA potentiates the
efficacy of 5-FU in Colo205 colon cancer cells in vivo through
downregulation of P-gp and LC3-II. Exp Ther Med. 3:555–559. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lin C, Wang L, Wang H, Yang L, Guo H and
Wang X: Tanshinone IIA inhibits breast cancer stem cells growth in
vitro and in vivo through attenuation of IL-6/STAT3/NF-kB signaling
pathways. J Cell Biochem. 114:2061–2070. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Branco AF, Sampaio SF, Moreira AC, Holy J,
Wallace KB, Baldeiras I, Oliveira PJ and Sardão VA:
Differentiation-dependent doxorubicin toxicity on H9c2
cardiomyoblasts. Cardiovasc Toxicol. 12:326–340. 2012. View Article : Google Scholar : PubMed/NCBI
|