Introduction
Gastric cancer is one of the most common human
malignancies among all clinical types of cancer and remains the
second leading cause of cancer-related death worldwide (1). Statistics have indicated that more than
70% of new cases and mortalities of gastric cancer occur in
developing countries (2). Gastric
cancer has demonstrated a higher morbidity and mortality rate than
other carcinomas of the digestive system (3,4).
Previous research has indicated that the 5-year survival rate was
<80% (5). Resistance to apoptosis
of gastric cancer cells in patients with gastric cancer has
previously been reported, and various reports have demonstrated
that apoptosis resistance of gastric cancer was inevitable in
cancer progression (6,7). At present, resistance to apoptosis has
become the greatest challenge in cancer therapy due to fierce
resistance of tumor cells though various kinds of molecular
mechanisms (8–10). In addition, despite the great
progress made in the treatment of gastric cancer, patients often
miss the opportunity for a surgical cure as the cancer has already
developed into the advanced stage by the time of diagnosis, which
leads to a reduced overall survival for gastric cancer patients
(11). A previous study has
suggested that targeted therapies for advanced gastric cancer are
efficient for patients with gastric cancer (12). Therefore, exploring more efficient
targeted molecular therapies has attracted increasing interest by
researchers and clinicians in the field of cancer research and
clinical therapy.
Mixed-lineage kinases (MLKs) are a class of
serine-threonine kinases that belong to the superfamily of
mitogen-activated protein kinase kinase kinases (MAP3Ks) and are
believed to control multiple intracellular signaling pathways in
cells (13). All MLK family members
are characterized by a signature Tyr kinase domain and a Ser/Thr
label in the catalytic domain in the amino-terminal SRC-homology
domain, as well as a Cdc42/Rac-interactive binding motif and a
leucine-zipper region (14). Proline
is abundant in the carboxyl terminus, with different forms in
different members of the family among all MLKs, which suggests that
this region serves in different and essential regulatory functions
in cells (15). Previous research
has indicated that MLK-4 is activated in colorectal cancer, where
it synergistically cooperates with activated Ras signaling to drive
tumorigenesis (16). Therefore, we
assumed that MLK-4 may have an important role in gastric carcinoma
tumorigenesis.
MLK-4 is the second most frequently mutated protein
kinase and has been identified in microsatellites in various human
tumor cells (17). However, the
function and pathobiological importance of MLK-4 is not fully
understood. MLK-4 is an important member of the MLK family that
regulates the extracellular signal-regulated kinase (ERK) and c-Jun
N-terminal kinase (JNK) signaling pathways (18). In addition, a study by Kim et
al (19) indicated that
serine/threonine kinase MLK-4 determined mesenchymal identity in
glioma stem cells through nuclear factor (NF)-κB signaling.
However, previous research has also reported that MLK-4 regulated
the JNK, p38, and ERK signaling pathways in colorectal cancer cells
(20). The function of MLK-4 has not
been well elucidated and the important implications of MLK-4 in the
apoptosis, development and treatment are equivocal in gastric
carcinoma.
The present study demonstrated that MLK-4 was
overexpressed in gastric cancer cells and tumors. The results
indicated that neutralizing MLK-4 expression using anti-MLK-4
antibody decreased viability, self-renewal, motility, metastasis,
invasion and radioresistance of gastric cancer cells through
modulation of the JNK signaling pathway. The present results also
demonstrated that MLK-4 induced JNK activation through regulation
of mitogen-activated protein kinase kinase (MKK)4 and JNK kinase
(JNKK)2/MKK7 phosphorylation, which may be involved in gastric
cancer cell apoptosis in response to anti-cancer drug treatments.
Collectively, the present results suggested that MLK-4 serves as an
upstream regulator in the JNK signaling pathway and may be a
potential molecular target for gastric cancer therapy.
Materials and methods
Cells and reagents
Gastric tumor cell lines, HGC-27 and BGC-823, and
human gastric mucosa epithelial cells, GES-1, were purchased from
the American Type Culture Collection (Manassas, VA, USA). All tumor
cells were cultured in Dulbecco's modified Eagle's medium (DMEM;
Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (Invitrogen; Thermo Fisher
Scientific, Inc.). GES-1 cells were cultured in Eagle's minimal
essential medium supplemented with 10% fetal calf serum (FCS; both
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). All cells were
cultured in a 37°C humidified atmosphere of 5% CO2.
MTT cytotoxicity and colony formation
assays
HGC-27 and BGC-823 cells (1×106) were
incubated with MLK-4 (2 mg/ml; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) in 96-well plates for 96 h at 37°C in
triplicate, and phosphate-buffered saline (PBS) was added instead
of MLK-4 as a control. Subsequently, 20 µl MTT (5 mg/ml) in PBS
solution was added to each well and the cells were further
incubated for 4 h at 37°C. Following this, all medium was removed
and 100 µl dimethyl sulfoxide was added into the wells to
solubilize the crystals. The optical density was measured by a
Bio-Rad (ELISA) reader (Bio-Rad Laboratories, Inc., Hercules, CA,
USA) at a wavelength of 450 nm. In addition, HGC-27 and BGC-823
cells (1×106/well) in 6 well plates cultured in DMEM
with 10% fetal bovine serum were transfected with 50 nM small
interfering (si)RNA-MLK-4 (5′-CAUCUACGAUCCGACUAUU-3′) using
Lipofectamine® 2000 Transfection Reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol and colony formation was observed in a 6 day culture.
Cells were further analyzed 48 h after transfection.
ELISA
The affinity of MLK-4 for MLK receptor was
determined by the Human MLK4 ELISA kit (cat. no. EH10132; Wuhan
Fine Biotech Co., Ltd., Wuhan, China). The procedures were
performed according to the manufacturer's protocol. The final
results were recorded at 450 nm on an ELISA plate reader (Spectra
Max 190; Molecular Devices, LLC, Sunnyvale, CA, USA).
Cell invasion and migration
assays
MLK-knockdown HGC-27 and BGC-823 cells
(1×105) were cultured in Eagle's minimal essential
medium with 5% FCS. For migration assays, HGC-27 and BGC-823 cells
were transfected with 50 nM siRNA-MLK-4 and incubated for in the
upper chamber for 96 h at 37°C using a control Transwell insert (BD
Biosciences, Franklin Lakes, NJ, USA). Eagle's minimal essential
medium with 5% FCS was added to the upper and lower chambers. For
invasion assays, HGC-27 and BGC-823 cells were suspended at a
density of 1×105 cells in 500 µl of serum-free DMEM. The
cells were treated with 50 nM siRNA-MLK-4 for 48 h at 37°C and then
subjected to the tops of BD BioCoat Matrigel invasion chambers (BD
Biosciences), according to the manufacturer's protocol. The cells
were then washed with PBS, fixed with 4% paraformaldehyde for 20
min at 37°C and stained with Giemsa stain at 37°C for 20 min. The
number of tumor cells that had invaded and migrated were counted in
at least three randomly stained fields using a fluorescence
microscope for every membrane (BZ-9000; Keyence Corporation, Osaka,
Japan).
Targeted deletion of the MLK-4 locus in gastric
tumor cells. Disruption of MLK-4 exon 1 in gastric tumor HGC-27 and
BGC-823 cells was conducted to knockdown MLK-4 according to a
previous study (21). The purpose
clones were screened after a 12-day growth period under 0.4 mg/ml
geneticin (Invitrogen; Thermo Fisher Scientific, Inc.).
Subsequently, the clones were propagated for 10 generations.
Homologous recombination clones were screened and confirmed using
locus-specific polymerase chain reaction (PCR).
Overexpession of MLK-4 in gastric
tumor cells
Human MLK-4 cDNA plasmids (2.5 µg) were transfected
into 293T cells (1×106; both Cell Biology Laboratory,
Zhejiang Chinese Medical University, Zhejiang, China) for 48 h to
generate a lentivirus using Lipofectamine 2000 according to the
manufacturer's protocol. The viral supernatant was subsequently
collected and used to infect the HGC-27 and BGC-823 cells using
Lipofectamine 2000 and the MLK-4 lentivirus (5 µg). Further
analysis was performed 72 h post-transfection.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was obtained from HGC-27, BGC-823 and
GES-1 cells using TRIzol reagent (Thermo Fisher Scientific, Inc.)
according to the manufacturers' protocol. For DNase treatment, 2
units of DNase I polymerase (Invitrogen; Thermo Fisher Scientific,
Inc.) were used per µg of total RNA at 37°C for 30 min.
Approximately 5 µg RNA for each sample was reverse transcribed
using an oligo-(dT) primer and M-MLV reverse transcriptase
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
according to the manufacturer's protocol. qPCR analysis was
performed in a final volume of 10 µl, which contained 5 µl
SsoFast™ EvaGreen Supermix (Bio-Rad Laboratories, Inc.),
1 µl cDNA (1:50 dilution) and 2 µl forward and reverse primers (1
mM) with the ABI Prism 7500 sequence detection system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). All forward and
reverse primers (MLK-4, sense 5′-ACGACAGCCATATCGAGACA-3′ and
antisense 5′-CGAGATGACGAGGATTGCAG-3′; β-actin, sense
5′-GTGGGCGCCCAGGCACCA-3′ and antisense
5′-CTCCTTAATGTCACGCACGATTT-3′) were synthesized by Invitrogen
(Thermo Fisher Scientific, Inc.). PCR cycling was performed under
the following conditions: 94°C for 30 sec, and 45 cycles of 95°C
for 5 sec, 54°C for 10 sec and 72°C for 10 sec. Relative mRNA
expression levels were calculated using the 2−ΔΔCq
method (22). The results were
expressed as the n-fold of the control. β-actin was used as the
endogenous control.
Mutation screening by
semi-quantification RT-PCR analysis
Total RNA from cultured cells was extracted with
RNeasy Mini kit (Qiagen Sciences, Inc., Gaithersburg, MD, USA)
according to the manufacturer's protocol. A volume of 20 µl DNase
(Invitrogen; Thermo Fisher Scientific, Inc.) was added to remove
the genomic DNA according to the manufacturer's protocol. Following
this, a total of 1 µg RNA was reverse transcribed into cDNA using a
High Capacity cDNA reverse transcription kit (Qiagen Sciences,
Inc.) at 37°C for 30 min. cDNA (10 ng) was subjected to qPCR using
a SYBR Green Master Mix system (Bio-Rad Laboratories, Inc.) using
the primers (Takara Biotechnology Co., Ltd., Dalian, China) in
Table I. The PCR conditions included
an initial denaturation step of 94°C for 2 min, followed by 30
cycles of 94°C for 30 sec, 59°C for 30 sec, 72°C for 2 min and a
final elongation step at 72°C for 10 min. PCR products were mixed
with an equal volume of gel-loading buffer (95% formamide, 20 mM
EDTA and 0.05% bromophenol blue, 0.05% xylene cyanol), denatured at
95°C for 5 min and immediately placed on ice for 5 min. The samples
(1 µg) were loaded directly onto 8% polyacrylamide gels and run for
8 h at room temperature and 40 W in a solution of 0.5X TBE (Takara
Biotechnology Co., Ltd.). Bands were visualized using 1 µg/µl
ethidium bromide, which was added to the gel. GAPDH was used as an
internal control to normalize gene expression. The relative gene
expression levels were calculated using Quantity-One software
(version 1.0; Bio-Rad Laboratories, Inc.) and the 2−ΔΔCq
method (23). All experiments were
repeated ≥3 times.
 | Table I.Sequences of primers pairs mixed
lineage kinase-4 used for reverse transcription-quantitative
polymerase chain reaction analysis. |
Table I.
Sequences of primers pairs mixed
lineage kinase-4 used for reverse transcription-quantitative
polymerase chain reaction analysis.
| Gene | Sequence |
|---|
| H261Q | F:
5′-ACGACAGCCATATCGAGACA-3′ |
|
| R:
5′-ACAGTCCTCCTTCATTCAGT-3′ |
| G291E | F:
5′-TGGATGTATGAAGGGTTGAA-3′ |
|
| R:
5′-GAAAATATAAGGGGGCAGAT-3′ |
| A293E | F:
5′-TCTCCCCTGTAAACCCTAAC-3′ |
|
| R:
5′-GATGGAGGACAAGGGTATGC-3′ |
| W296E | F:
5′-TCTCCCCTGTAAACCCTAAC-3′ |
|
| R:
5′-GCCAGCCGGCTTTTACAAT-3′ |
| R338H | F:
5′-TCTCCCCTGTAAACCCTAAC-3′ |
|
| R:
5′-GCCAGCCGGCTTTTACAAT-3′ |
| Wild type | F:
5′-GAGGGCAGAATCATCACGAAGT-3′ |
|
| R:
5′-GGTGAGCATTATCACCCAGAA-3′ |
| β-actin | F:
5′-AGAAAATCTGGCACCACACC-3′ |
|
| R:
5′-TAGCACAGCCTGGATAGCAA-3′ |
Co-immunoprecipitation assay
HGC-27 (1×106) cells grown in DMEM
supplemented with 10% FCS were transfected with 5 µg MLK-4 vector
(Shanghai Zeye Biotechnology Co., Ltd., Shanghai, China) using
Lipofectamine 2000. After 24 h, the cells were chilled to 4°C and
lysed by incubating for 15 min in lysis buffer (Sigma-Aldrich;
Merck KGaA). Nuclei were removed by centrifugation at 10,000 × g
for 15 min at 37°C. Flag- or V5-tagged proteins were purified from
the cell lysate by immunoprecipitation using EZ-Magna ChIP kit
(cat. no. 17-409; EMD Millipore, Billerica, MA, USA) according to
manufacturer's protocol. The immunoprecipitates were subjected to
western blotting analysis with different antibodies.
Colony formation assay
MLK-4-slienced HGC-27 and BGC-823 cells were
cultured for 5 days and transferred to 6-well plates at a density
of 500 cells/well. After 7 days of culturing, the cells were fixed
with 4% polyformaldehyde (Sigma-Aldrich; Merck KGaA) and then
stained with diluted Giemsa stain (1:20) for 20 min at 37°C.
Following the rinsing of the cells with distilled water, colonies
of cells were detected by a BZ-9000 fluorescence microscope.
Western blotting
HGC-27 and BGC-823 cells were homogenized in lysate
buffer containing protease-inhibitor (Sigma-Aldrich; Merck KGaA)
and were centrifuged at 6,000 × g (4°C) for 10 min. Subsequently,
the supernatant was used for analysis of protein levels. Protein
concentration was measured by a BCA protein assay kit (Thermo
Fisher Scientific, Inc.). SDS-PAGE assays were performed, as
previously described (24). Protein
samples (20 µg/lane) were resolved by 15% SDS-PAGE and transferred
to polyvinylidene fluoride membranes (EMD Millipore). The membranes
were blocked with 5% skimmed milk for 1 h at 37°C and subsequently
incubated with primary antibodies: MLK-4 (1:1,000; cat. no.
ab93798), Bcl-2 (1:1,000; cat. no. ab692), P53 (1:1,000; cat. no.
ab26), Bax (1:1,000; cat. no. ab32503), MMP-3 (1:1,000; cat. no.
ab53015), CT-1 (1:500; cat. no. ab34710), Fibronectin (1:1,000;
cat. no. ab6328), PPAR-γ (1:1,000; cat. no. ab45036), STAT-3
(1:1,000; cat. no. ab119352), NF-κBp65 (1:1,000; cat. no. ab16502)
and β-actin (1:1,000; cat. no. ab124721) (all Abcam, Cambridge, UK)
for 12 h at 4°C. Following this, membranes were incubated with
horseradish peroxidase-conjugated rabbit anti-mouse IgG mAb (cat.
no. PV-6001; OriGene Technologies, Inc., Beijing, China) for 1 h at
37°C. Following the washing of the membranes with 0.1% Tween 20 in
Tris-buffer solution, the membranes was developed using a
chemiluminescence assay system (Roche Diagnostics, Basel,
Switzerland) and exposed to Kodak exposure films (Kodak, Rochester,
NY, USA). Densitometric quantification of the western blotting data
was performed using Quantity-One software (version 1.0; Bio-Rad
Laboratories, Inc.).
Animal study
All animal procedures were approved by the Ethics
Committee of the First Affiliated Hospital, Shihezi University
School of Medicine (Shihezi, China). A total of 60
immunocompromised CD1-nude athymic male mice (6–8 weeks old; 30–35
g) were purchased from Shanghai SLAC Laboratory Animal Co., Ltd.
(Shanghai, China). The mice were housed in a temperature-controlled
facility at 23±1°C with a relative humidity of 50±5% and 12 h
light/dark cycle with free access to food and water. MLK-4
knockdown HGC-27 or HGC-27 cells (1×107) were injected
subcutaneously in posterior flanks of 30 mice. Gastric tumor
diameters were measured every 2 days by using a caliper. Tumor
volumes were calculated according to a previous study (25).
Statistical analysis
All data are presented as the mean ± standard error
of the mean of triplicate experiments. All data were analyzed using
SPSS Statistics software (version 19.0; IBM Corp., Armonk, NY,
USA). Unpaired data were compared using Student's t-tests and
comparisons of data between multiple groups were analyzed using
one-way analysis of variance followed by Dunnett's test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Expression and function of MLK-4 in
gastric cancer cells
In order to analyze MLK-4 expression levels, RT-qPCR
and western blotting were used to evaluate the expression level of
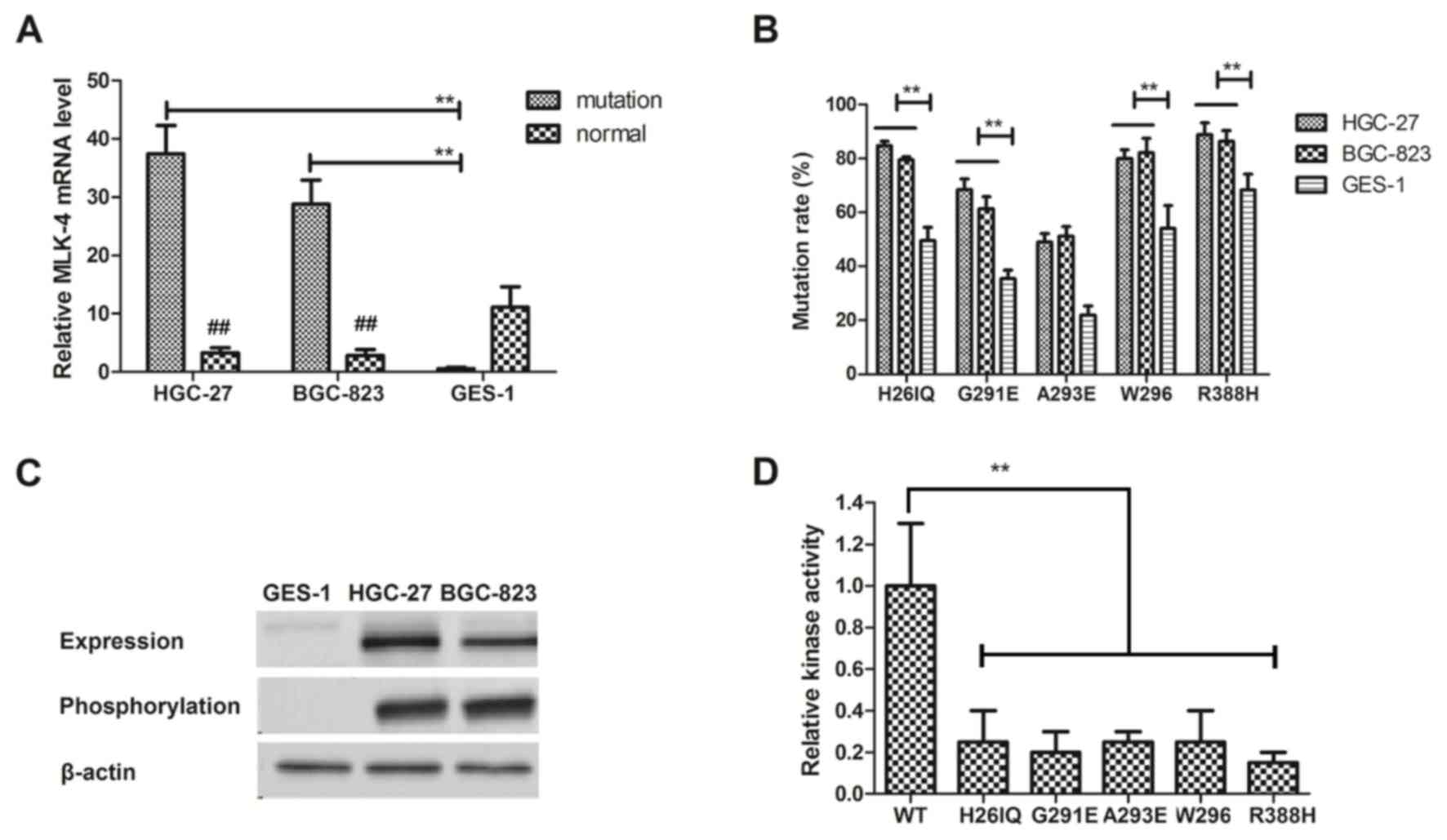
mutational MLK-4 in gastric cancer cells. The results in Fig. 1A demonstrated that the expression
level of mutated MLK-4 was significantly higher in gastric cancer
cell lines, HGC-27 and BGC-823, compared with normal GES-1 cells
(P<0.01). The results in Fig. 1B
revealed that H26IQ, G291E, A293E, W296 and R388H had higher
mutation rates located within the kinase catalytic domain compared
with normal gastric GES-1 cells. The results in Fig. 1C indicated that these mutations
(H26IQ, G291E, A293E, W296 and R388H) markedly decreased MKK4
phosphorylation levels. The results in Fig. 1D demonstrated that MLK-4 functions as
an oncogene with gain-of-function mutations in gastric cancer
cells. Overall, the data suggested that the MLK-4 mutations located
within the kinase domain in gastric cancer cells.
Reintroduction of MLK-4 decreases cell
migration and tumor viability in vitro
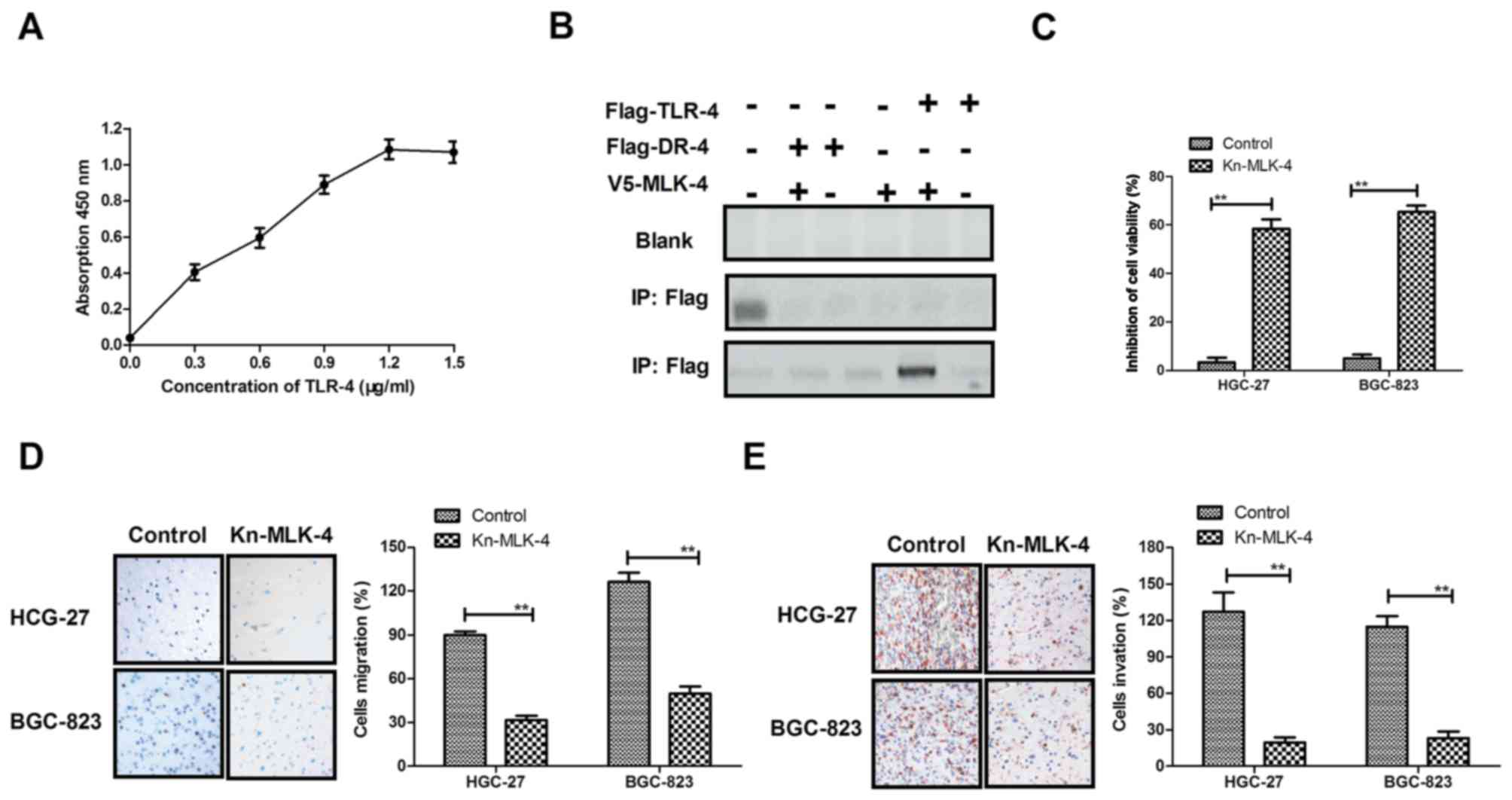
To confirm the binding receptor of MLK-4, an ELISA
system was used to determine the intracellular binding receptor of
MLK-4. The results in Fig. 2A
demonstrated that MLK-4 interacted with the Toll-like receptor
(TLR)-4 intracellular domain in HGC-27 and BGC-823 cells. In
addition, co-immunoprecipitation by Flag-labeled TLR-4 with
V5-tagged MLK-4 was performed to confirm the interaction between
MLK-4 and TLR-4 in human gastric mucosa epithelial GES-1 cells.
Flag-labeled death receptor-4 (DR-4) was used as a control for
TLR-4. The results in Fig. 2B
demonstrated that MLK-4 co-precipitated with Flag-TLR-4, but not
with Flag-DR-4 for homogenates of HGC-27 or BGC-823 cells. In
addition, the inhibitory effects of MLK-4 on gastric cancer cell
lines, HGC-27 and BGC-823, were analyzed. As demonstrated in
Fig. 2C, the viability of HGC-27 and
BGC-823 cells was significantly inhibited in the MLK-4-treated
group compared with control cells (P<0.01). The migration and
invasion assays (Fig. 2D and E)
indicated that the migratory and invasive abilities of gastric
cancer cells were significantly suppressed following MLK-4
treatment compared with the control cells (P<0.01). These data
suggested that MLK-4 had an important role in suppressing
viability, migration and apoptosis-resistance in gastric cancer
cells.
Inhibition of the JNK signaling
pathway regulated by MLK-4 in gastric cancer cells
To evaluate the biological mechanism of MLK-4
loss-of-function mutations in gastric cancer cells, JNK expression
and phosphorylation in MLK-4 knockdown in HGC-27 and BGC-823 cells
were analyzed. The results in Fig.
3A demonstrated that MLK-4 knockdown markedly decreased JNK
expression and phosphorylation in HGC-27 and BGC-823 cells compared
with the control. P53, peroxisome proliferator (PPAR)-γ, signal
transducer and activator of transcription (STAT)-3 and nuclear
factor (NF)-κB expression levels in MLK-4-silenced gastric tumor
cells were also analyzed. As demonstrated in Fig. 3B, upregulation of MLK-4 expression
markedly increased P53, PPAR-γ, STAT-3 and NF-κ5 expression levels
in gastric tumor cells. Subsequently, apoptosis-related gene
expression was analyzed in gastric tumor cells overexpressing
MLK-4. The results in Fig. 3C
indicated that expression levels of B-cell lymphoma (Bcl)-2 and
Bcl-2-assocaitated X protein were downregulated in gastric tumor
cells overexpressing MLK-4. Furthermore, migration-related protein
expression in gastric tumor cells was evaluated. It was
demonstrated that matrix metalloproteinase-3, collagen type I and
fibronectin expression levels were upregulated following MLK-4
treatment (Fig. 3D). Collectively,
these data suggested an inhibitory role of MLK-4 on the
TLR-4-mediated JNK signaling pathway.
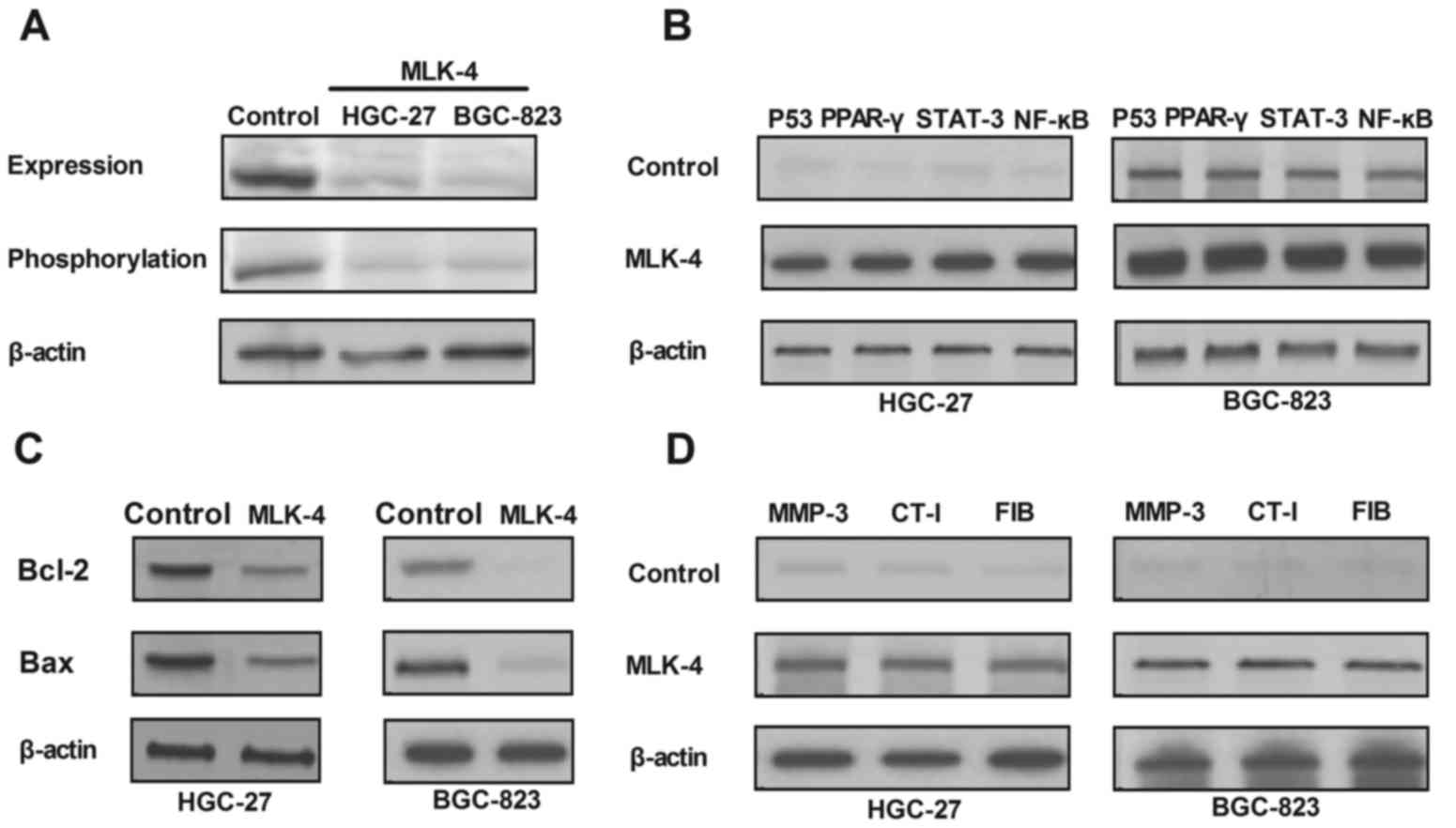 | Figure 3.Regulation of JNK signaling pathway
induced by MLK-4 in gastric cancer cells. (A) Expression and
phosphorylation levels of JNK in HGC-27 and BGC-823 cells following
transduction with MLK-4. (B) Upregulation of P53, PPAR-γ, STAT-3
and NF-κB expression in MLK-4-transduct gastric tumor cells. (C)
Downregulation of Bcl-2 and Bax in gastric tumor cells
overexpressing MLK-4. (D) Expression levels of MMP-3, CT-I and FIB
following MLK-4 treatment. MLK-4, mixed lineage kinase-4; JNK,
c-Jun N-terminal kinase; PPAR-γ, peroxisome proliferator-γ; STAT-3,
signal transducer and activator of transcription-3; NF-κB, nuclear
factor-κB; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-acssociated X
protein; MMP-3, matrix metalloproteinase-3; CT-I, collagen type I;
FIB, fibronectin. |
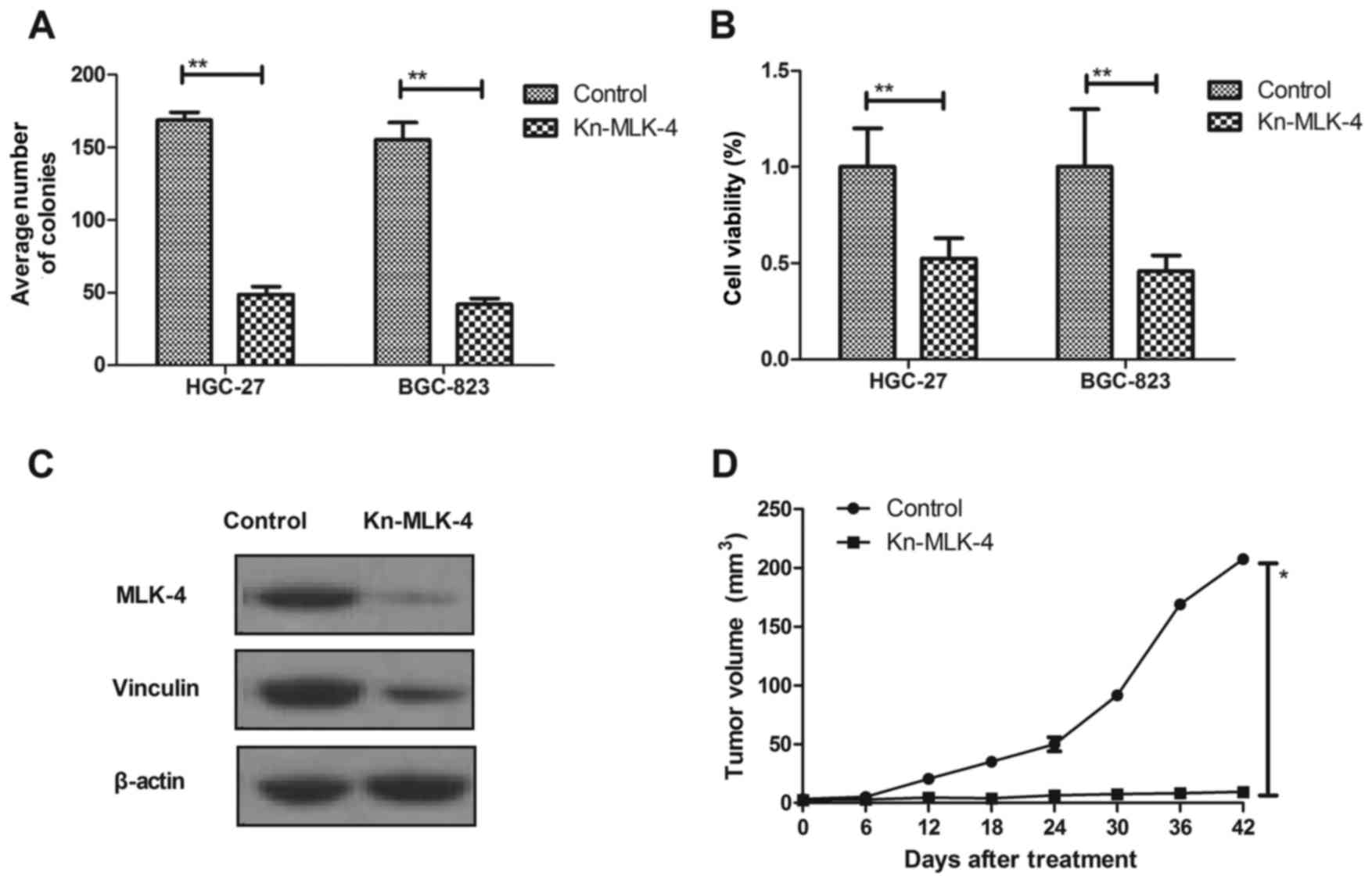
Gene silencing or genetic inactivation of mutational
MLK-4 blocks the tumorigenic properties of cancer cells. The
regulatory effects of MLK-4 on transcriptional downregulation in
gastric cancer were investigated. The results in Fig. 4A demonstrated that knockdown of MLK-4
expression in gastric cancer cells significantly decreased the
number of colonies formed compared with control cells (P<0.01).
It was observed that cell viability with wild type MLK-4 was
significantly higher compared with MLK-4-knockdown cells
(P<0.01; Fig. 4B). In addition,
it was demonstrated that downregulation of MLK-4 markedly decreased
MLK-4 and vinculin protein expression levels in tumors (Fig. 4C). Furthermore, the present study
demonstrated the inhibitory effects of reduced or abrogated
mutational MLK-4 expression or injected normal MLK-4 on
tumor-bearing mice in vivo. The MLK-4 knockdown HGC-27 cells
and the parental control were injected into immunocompromised mice.
Notably, it was observed that tumor volume in mice with MLK-4
knockdown was significantly reduced compared with those injected
with control HGC-27 cells at day 24, 30, 36 and 42 (P<0.01;
Fig. 4D). These results suggested
that knockdown of MLK-4 may contribute to the treatment of gastric
cancer.
Discussion
The aim of the present study was to investigate the
function of MLK-4 in gastric carcinoma growth and tumorigenesis. A
study by Martini et al (16)
suggested that MLK-4 was frequently mutated in colon cancer, which
has an important role in cancer tumorigenesis, local invasion and
long distance metastasis. Therefore, understanding the role of
MLK-4 is essential for tumor therapy in human tumorigenesis and
metastasis. The present study investigated the expression and
mutation of MLK-4 in gastric cancer cells and subcutaneous tumors,
as well as the normal function of MLK-4. Results demonstrated that
H26IQ, G291E, A293E, W296 and R388H mutations occur at a high rate
and all of them are located within the kinase catalytic domain,
which is critical for kinase activity of DFG and HRD motifs
(26). These findings were
consistent with previous research (26,27). The
mutational locations in the kinase catalytic domain affected the
biological function of MLK-4 and influence protein catalytic
function, which provided the impetus to cancer cell viability and
invasion. The in vivo experiments confirmed our hypothesis
and indicated that MLK-4 mutation contributed to the formation of
subcutaneous tumors, which suggested that mutated MLK-4 may serve
as a potential target in cancer therapy.
MLKs are a class of serine/threonine protein kinases
that regulate JNK and p38 mitogen-activated protein kinase (MAPK)
signaling pathways in cells (28).
MLK-4 is a member of the MLK family of kinases and is an
understudied protein kinase; however, understanding of this protein
is essential to gain insight into the role of this kinase in
tumorigenesis (29). A previous
report indicated that MLK1-4 have regulatory effects on MAP3Ks and
may regulate the activities MKK3/6 and MKK4/7, which further
induces JNK and p38 signaling pathway activation (28). In addition, previous research has
also identified that MAPK/ERK kinase (MEK) function, endowed by
MLK-1-4, are able to directly activate and reactivate the MEK/ERK
pathway in a kinase-dependent manner in the presence of RAF
inhibitors (13). Therefore, MLK-4
serves as a direct activator of the JNK, p38 MAPK and MEK/ERK
signaling pathways in cancer cells. The results of the present
study focused on the JNK signaling pathway regulated by MLK-4 and
indicated that silencing mutational MLK-4 expression significantly
inhibited tumor cell viability, promoted apoptotic sensibility and
prevented the growth of subcutaneous tumors.
Apoptosis resistance is the most serious obstacle in
cancer clinical treatment (30,31).
Decreasing apoptosis resistance of cancer cells and/or tumors will
be beneficial for patients undergoing oncotherapy in clinics. MLK-4
is associated with apoptosis resistance in different cancer cells
(32). MLK-4 is involved in the
regulation of apoptosis in mammalian cells, and presents a
potential molecular target in the treatment of human cancer with
higher MLK-4 mutation rates. A study by Wang et al (33) indicated that proliferation, survival,
migration, invasion and apoptosis of estrogen receptor-positive
breast cancer cells were highly associated with MLK-4 activities,
and suggested that silencing MLK-4 activities may serve as a novel
target therapy for breast cancer. A study by Müller et al
(34) demonstrated that MLK was able
to protect granule cells against colchicine-induced apoptosis
through the JNK/MLK signaling pathway. In addition, MLKs have been
evidenced as a physiological element of nerve growth factor
induction of the JNK signaling pathway, which suggests that MLKs
may be potential therapeutic targets in the majority of
apoptosis-related diseases (35).
Furthermore, essential roles of MLKs in the tumor necrosis
factor-induced programmed necrosis pathway has been indicated in
tumor cells (36). However, the
apoptosis resistance function of MLK-4 in cancer cells is seldom
reported and the signaling pathway remains unclear.
The present study aimed to understand the molecular
mechanisms of MLK-4 in regulation of protein kinase-based apoptosis
resistance in the context of gastric tumor tissue.
Apoptosis-related gene expression in the JNK signaling pathway was
investigated in vivo. The results indicated that knockdown
of mutational MLK-4 was beneficial for blocking subcutaneous tumor
formation in gastric tumor-bearing mice as MLK-4 mutation promoted
gastric carcinoma tumorigenesis and normal MLK-4 expression was
downregulated (37). Therefore,
MLK-4 may be a potential biomarker and may be used to diagnose and
provide prognostic information for gastric cancer.
In conclusion, the present study provided insight on
genetic mutation and the normal function of MLK-4 in gastric cancer
cells and tissues. Activation of the JNK signaling pathway through
mutation of MLK-4 induced metastasis and apoptosis-resistance in
gastric cancer. Due to the present molecular study of gastric
cancer, MLK-4 may serve as a potential molecular target for the
treatment of gastric cancer.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YX, JN and DL performed the experiments and
organized the data. JH, LQ and XP assisted in the analysis of data.
YX wrote the manuscript.
Ethics approval and consent to
participate
All animal procedures were approved by the Ethics
Committee of the First Affiliated Hospital, Shihezi University
School of Medicine (Shihezi, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Areia M, Carvalho R, Cadime AT, Rocha
Goncalves F and Dinis-Ribeiro M: Screening for gastric cancer and
surveillance of premalignant lesions: A systematic review of
cost-effectiveness studies. Helicobacter. 18:325–337. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hossain MM and Chen Z: Comments on
‘Application of an adaptive design to a randomized phase II
selection trial in gastric cancer: A report of the study design’ by
Satoshi Morita and Junichi Sakamoto. Pharm Stat. 11:267–268. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pimenta-Melo AR, Monteiro-Soares M,
Libânio D and Dinis-Ribeiro M: Missing rate for gastric cancer
during upper gastrointestinal endoscopy: A systematic review and
meta-analysis. Eur J Gastroenterol Hepatol. 28:1041–1019. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Veisani Y and Delpisheh A: Survival rate
of gastric cancer in Iran; a systematic review and meta-analysis.
Gastroenterol Hepatol Bed Bench. 9:78–86. 2016.PubMed/NCBI
|
|
5
|
Nakajima T, Inokuchi K, Hattori T, Inoue
K, Taguchi T, Kondou T, Abe O, Kikuchi K, Tanabe T and Ogawa N:
Multi-institutional cooperative study of adjuvant
immunochemotherapy in gastric cancer-five-year survival rate. Gan
To Kagaku Ryoho. 16:799–806. 1989.(In Japanese). PubMed/NCBI
|
|
6
|
Jung SA, Park YM, Hong SW, Moon JH, Shin
JS, Lee HR, Ha SH, Lee DH, Kim JH, Kim SM, et al: Cellular
inhibitor of apoptosis protein 1 (cIAP1) stability contributes to
YM155 resistance in human gastric cancer cells. J Biol Chem.
290:9974–9985. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Izawa M, Mori T, Satoh T, Teramachi K and
Sairenji T: Identification of an alternative form of caspase-9 in
human gastric cancer cell lines: A role of a caspase-9 variant in
apoptosis resistance. Apoptosis. 4:321–325. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Moody SE, Schinzel AC, Singh S, Izzo F,
Strickland MR, Luo L, Thomas SR, Boehm JS, Kim SY, Wang ZC and Hahn
WC: PRKACA mediates resistance to HER2-targeted therapy in breast
cancer cells and restores anti-apoptotic signaling. Oncogene.
34:2061–2071. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shiota M, Yokomizo A and Naito S:
Pro-survival and anti-apoptotic properties of androgen receptor
signaling by oxidative stress promote treatment resistance in
prostate cancer. Endocr Relat Cancer. 19:R243–R253. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yamato I, Sho M, Shimada K, Hotta K, Ueda
Y, Yasuda S, Shigi N, Konishi N, Tsujikawa K and Nakajima Y:
PCA-1/ALKBH3 contributes to pancreatic cancer by supporting
apoptotic resistance and angiogenesis. Cancer Res. 72:4829–4839.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Coccolini F, Catena F, Glehen O, Yonemura
Y, Sugarbaker PH, Piso P, Ceresoli M, Montori G and Ansaloni L:
Effect of intraperitoneal chemotherapy and peritoneal lavage in
positive peritoneal cytology in gastric cancer. Systematic review
and meta-analysis. Eur J Surg Oncol. 42:1261–1267. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li K and Li J: Current molecular targeted
therapy in advanced gastric cancer: A comprehensive review of
therapeutic mechanism, clinical trials, and practical application.
Gastroenterol Res Pract. 2016:41056152016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marusiak AA, Edwards ZC, Hugo W, Trotter
EW, Girotti MR, Stephenson NL, Kong X, Gartside MG, Fawdar S,
Hudson A, et al: Mixed lineage kinases activate MEK independently
of RAF to mediate resistance to RAF inhibitors. Nat Commun.
5:39012014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Handley ME, Rasaiyaah J, Barnett J,
Thakker M, Pollara G, Katz DR and Chain BM: Expression and function
of mixed lineage kinases in dendritic cells. Int Immunol.
19:923–933. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jaeschke A and Davis RJ: Metabolic stress
signaling mediated by mixed-lineage kinases. Mol Cell. 27:498–508.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Martini M, Russo M, Lamba S, Vitiello E,
Crowley EH, Sassi F, Romanelli D, Frattini M, Marchetti A and
Bardelli A: Mixed lineage kinase MLK4 is activated in colorectal
cancers where it synergistically cooperates with activated RAS
signaling in driving tumorigenesis. Cancer Res. 73:1912–1921. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Seit-Nebi A, Cheng W, Xu H and Han J: MLK4
has negative effect on TLR4 signaling. Cell Mol Immunol. 9:27–33.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Soung YH, Lee JW, Kim SY, Nam SW, Park WS,
Lee JY, Yoo NJ and Lee SH: Kinase domain mutation of MLK4 gene is
uncommon in gastric and hepatocellular carcinomas. Dig Liver Dis.
38:2832006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim SH, Ezhilarasan R, Phillips E,
Gallego-Perez D, Sparks A, Taylor D, Ladner K, Furuta T, Sabit H,
Chhipa R, et al: Serine/Threonine kinase MLK4 determines
mesenchymal identity in glioma stem cells in an NF-κB-dependent
manner. Cancer Cell. 29:201–213. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Marusiak AA, Stephenson NL, Baik H,
Trotter EW, Li Y, Blyth K, Mason S, Chapman P, Puto LA, Read JA, et
al: Recurrent MLK4 loss-of-function mutations suppress JNK
signaling to promote colon tumorigenesis. Cancer Res. 76:724–735.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kohli M, Rago C, Lengauer C, Kinzler KW
and Vogelstein B: Facile methods for generating human somatic cell
gene knockouts using recombinant adeno-associated viruses. Nucleic
Acids Res. 32:e32004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xiao S, Wang J and Xiao N: MicroRNAs as
noninvasive biomarkers in bladder cancer detection: A diagnostic
meta-analysis based on qRT-PCR data. Int J Biol Markers.
31:e276–e285. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wai-Hoe L, Wing-Seng L, Ismail Z and
Lay-Harn G: SDS-PAGE-based quantitative assay for screening of
kidney stone disease. Biol Proced Online. 11:145–160. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhuang T, Djemil T, Qi P, Magnelli A,
Stephans K, Videtic G and Xia P: Dose calculation differences
between Monte Carlo and pencil beam depend on the tumor locations
and volumes for lung stereotactic body radiation therapy. J Appl
Clin Med Phys. 14:40112013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ruan J, Mei L, Zhu Q, Shi G and Wang H:
Mixed lineage kinase domain-like protein is a prognostic biomarker
for cervical squamous cell cancer. Int J Clin Exp Pathol.
8:15035–15038. 2015.PubMed/NCBI
|
|
27
|
Wang H, Sun L, Su L, Rizo J, Liu L, Wang
LF, Wang FS and Wang X: Mixed lineage kinase domain-like protein
MLKL causes necrotic membrane disruption upon phosphorylation by
RIP3. Mol Cell. 54:133–146. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gallo KA and Johnson GL: Mixed-lineage
kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol.
3:663–672. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lotharius J, Falsig J, van Beek J, Payne
S, Dringen R, Brundin P and Leist M: Progressive degeneration of
human mesencephalic neuron-derived cells triggered by
dopamine-dependent oxidative stress is dependent on the
mixed-lineage kinase pathway. J Neurosci. 25:6329–6342. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chinchar E, Makey KL, Gibson J, Chen F,
Cole SA, Megason GC, Vijayakumar S, Miele L and Gu JW: Sunitinib
significantly suppresses the proliferation, migration, apoptosis
resistance, tumor angiogenesis and growth of triple-negative breast
cancers but increases breast cancer stem cells. Vasc Cell.
6:122014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guidicelli G, Chaigne-Delalande B,
Dilhuydy MS, Pinson B, Mahfouf W, Pasquet JM, Mahon FX, Pourquier
P, Moreau JF and Legembre P: The necrotic signal induced by
mycophenolic acid overcomes apoptosis-resistance in tumor cells.
PLoS One. 4:e54932009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vacratsis PO and Gallo KA: Zipper-mediated
oligomerization of the mixed lineage kinase SPRK/MLK-3 is not
required for its activation by the GTPase cdc 42 but is necessary
for its activation of the JNK pathway. Monomeric SPRK L410P does
not catalyze the activating phosphorylation of Thr258 of murine
MITOGEN-ACTIVATED protein kinase kinase 4. J Biol Chem.
275:27893–27900. 2000.PubMed/NCBI
|
|
33
|
Wang L, Gallo KA and Conrad SE: Targeting
mixed lineage kinases in ER-positive breast cancer cells leads to
G2/M cell cycle arrest and apoptosis. Oncotarget. 4:1158–1171.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Müller GJ, Geist MA, Veng LM, Willesen MG,
Johansen FF, Leist M and Vaudano E: A role for mixed lineage
kinases in granule cell apoptosis induced by cytoskeletal
disruption. J Neurochem. 96:1242–1252. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mota M, Reeder M, Chernoff J and Bazenet
CE: Evidence for a role of mixed lineage kinases in neuronal
apoptosis. J Neurosci. 21:4949–4957. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fang W, Jia ZS and Tong ZS: Research
progress in mixed lineage kinase domain-like. Sheng Li Ke Xue Jin
Zhan. 46:265–268. 2015.PubMed/NCBI
|
|
37
|
Garlena RA, Gonda RL, Green AB, Pileggi RM
and Stronach B: Regulation of mixed-lineage kinase activation in
JNK-dependent morphogenesis. J Cell Sci. 123:3177–3188. 2010.
View Article : Google Scholar : PubMed/NCBI
|