Introduction
Coronary artery spasm (CAS) serves an important role
in the pathogenesis of numerous myocardial ischemic disease,
including stable angina, unstable angina, acute myocardial
infarction and sudden cardiac death (1,2). The
pathogenesis of CAS occurs through a complex mechanism that
involves endothelial dysfunction, inflammation, hyperactivity of
smooth muscle cells and other factors (3,4).
However, the specific regulation of CAS remains to be
elucidated.
Nitric oxide (NO) is generated from L-arginine by
constitutive endothelial NO synthase (eNOS) (5). In the vasculature, the bioactivity of
eNOS and NO is regulated by the caveolins, which are
scaffolding/regulatory proteins that are particularly abundant in
the endothelial cell plasma membrane (6,7).
Caveolin-1 (Cav-1) is the most important caveolar coat protein
involved in the control of vascular reactivity by combining with
eNOS (8). eNOS binds to Cav-1 via
the caveolin scaffolding domain, which is located at amino acids
350–358 (9). This interaction leads
to the inhibition of eNOS activity, resulting in a decrease in the
basal release of NO (10) and
increase in the cardiovascular tone (11).
Endothelial dysfunction is characterized mainly by a
reduction in eNOS-derived NO production and bioactivity (12), as well as an imbalance between the
endothelium-derived relaxation factor and vasoconstrictors.
Previous studies have demonstrated endothelial dysfunction at the
site of a CAS (13). Therefore,
maintenance of the release and bioactivity of NO in the endothelial
environment is important in preventing CAS. Previous studies have
demonstrated that eNOS becomes hyperactivated in the absence of
Cav-1, leading to marked vascular relaxation (12,14,15).
Thus, CAS may be caused by decreased NO release as a result of eNOS
inhibition by Cav-1 in certain pathological conditions. This
negative regulation is particularly important since eNOS activation
has been associated with a protective effect against the
development of CAS. However, the potential of Cav-1 knockdown to
alleviate or inhibit the development of CAS remains to be
investigated. It can be speculated that Cav-1 knockdown in
endothelial cells may produce high levels of bioactive NO, which
regulates the vascular tone in order to inhibit the occurrence of
CAS.
In the current study, an in vitro model of
endothelial damage stimulated by lipopolysaccharide (LPS) treatment
was established, and then the CAS environment was mimicked by the
addition of acetylcholine (ACh) to investigate the effects of Cav-1
knockdown. It was hypothesized that Cav-1 knockdown in this
LPS-induced model of endothelial cell dysfunction may increase
ACh-stimulated NO production. It was observed that the high levels
of NO induced by Cav-1 knockdown serve a vital role in the
inhibition of CAS development. Therefore, targeting the interaction
between CAS and Cav-1 represents a potential therapeutic strategy
for CAS.
Materials and methods
Culture and identification of
HUVECs
Primary human umbilical vein endothelial cells
(HUVECs; ScienCell Research Laboratories, Inc., Carlsbad, CA, USA)
were maintained in endothelial cell medium (ECM; ScienCell Research
Laboratories, Inc.) containing 5% fetal bovine serum (FBS), 1%
endothelial cell growth supplement and 1% penicillin/streptomycin.
The cells were incubated at 37°C in a humidified atmosphere with 5%
CO2. In all experiments, cells at 90% confluence and
between passages 3 and 5 were used. The cells were characterized as
endothelial cells according to their morphology and factor VIII
staining using an anti-factor VIII antibody (cat. no. 21458–1-AP;
ProteinTech Group, Inc., Wuhan, China), which is a well-recognized
marker of endothelial cells. The medium was aspirated; the cells
were washed with PBS seeded on clean glass coverslips. The cells
were fix with freshly made 4% paraformaldehyde (Servicebio, Wuhan,
China) in PBS at room temperature for 1 h. The coverslips were
rinsed with PBS three times (5 min/wash) and treated with 0.1%
TritonX-100 (cat. no. T8200; Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China) in PBS for 15 min at room
temperature. The coverslips were rinsed with PBS three times (5
min/wash). The cells were then blocked in 10% normal blocking goat
serum in PBS at room temperature for 30 min. The blocking solution
was aspirated and the cells were incubated with the rabbit
anti-factor VIII antibodies (1:100) for 4 h at 4°C. The coverslips
were rinsed with PBS four times (5 min/wash). Then the cells were
incubated with fluorescein isothiocyanate-conjugated goat
anti-rabbit immunoglobulin G (1:100; cat. no. ZF-0311; ZSGB-Bio;
OriGene Technologies, Inc., Beijing, China) for 30 min at room
temperature in a dark, moist environment. The coverslips were
rinsed with PBS three times (5 min/wash). The coverslip were then
incubated with DAPI (cat. no. C1005; Beyotime Institute of
Biotechnology, Jiangsu, China) to stain the nuclei and cells were
examined using a fluorescence microscope (magnification, ×200).
Experimental groups
Preliminary experiments demonstrated that
significant cellular damage is caused by pretreatment of HUVECs
with 75 µg/ml LPS (serotype 026:B6 from Escherichia coli;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 24 h at 37°C.
Subsequently, these cells were treated with 10 µM ACh
(Sigma-Aldrich; Merck KGaA) for 1 h at 37°C. The cells were then
divided into the following experimental groups: i) HUVECs/si-NC,
which was a negative control for Cav-1 downregulation; ii)
HUVECs/si-NC + LPS; iii) HUVECs/si-NC + ACh; iv) HUVECs/si-NC + LPS
+ ACh; v) HUVECs/siCav-1, in which Cav-1 downregulation was
performed; vi) HUVECs/siCav-1 + LPS; vii) HUVECs/siCav-1 + ACh; and
viii) HUVECs/siCav-1 + LPS + ACh. Subsequently, following all the
treatments, the cell culture supernatants were centrifuged at 1,800
× g for 10 min at 4°C to remove any cell debris.
Transfection and Lipofectamine
assay
Small interfering RNA (siRNA) duplex
oligonucleotides that were specific for Cav-1 were synthesized by
Suzhou GenePharma Co., Ltd. (Suzhou; China). The sequences were as
follows: siCav-1 (1),
5′-GCAUUUGGAAGGCCAGCUUTT-3′; siCav-1 (2), 5′-CCCACUCUUUGAAGCUGUUTT-3′; siCav-1
(3), 5′-GCAGUUGUACCAUGCAUUATT-3′;
and si-NC, 5′-UUCUCCGAACGUGUCACGUTT-3′. For transfection, cells
(2×105 cells/well) were seeded into 6-well culture
plates at 50–60% confluence. After 24 h, siRNA (30 pmol) mixed with
5 µl Lipofectamine RNAiMAX reagent (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) in 250 µl serum-free Opti-MEM
(Gibco; Thermo Fisher Scientific, Inc.) was added to each well and
incubated for 15 min at room temperature, followed by addition of
2.5 ml ECM without serum. After 4–6 h, the transfection solution
was replaced with the complete culture medium and incubated for a
further 48 h at 37°C. Subsequently, the cells were harvested and
the knockdown efficiency was evaluated by western blotting.
Western blot analysis of siRNA
transfection efficiency
Cells were lysed in radioimmunoprecipation assay
(RIPA) buffer and phenylmethylsulfonyl fluoride (Beyotime Institute
of Biotechnology, Jiangsu, China) for 15 min on ice. The total cell
lysates were centrifuged at 12,000 × g for 15 min at 4°C, and the
protein concentration was measured using the BCA Protein Assay
reagent (Beyotime Institute of Biotechnology). Proteins were
denatured by adding 1X sodium dodecyl sulfate (SDS; Beyotime
Institute of Biotechnology) loading buffer and boiled for 10 min.
Next, the protein samples (~10 µg) were separated by 12%
SDS-polyacrylamide gel electrophoresis and transferred to
nitrocellulose membranes (Pall Corporation, Dreieich, Germany). The
blots were then blocked for 2 h at room temperature with 5% non-fat
dried milk, and subsequently incubated overnight at 4°C with the
following primary detection antibodies: Rabbit polyclonal
anti-CAV-1 (1:1,500; cat. no. sc-894; Santa Cruz Biotechnology
Inc., Dallas, TX, USA) and rabbit polyclonal anti-GAPDH antibody
(1:2,000; cat. no. BS60630; Bioworld Technology, Co, Ltd., Nanjing,
China). Subsequent to washing three times (15 min per wash) with
Tris-buffered saline and 0.05% Tween-20, the blots were incubated
with a goat anti-rabbit secondary detection antibody (1:4,000; cat.
no. ZDR-530612; ZSGB-Bio; OriGene Technologies, Inc.) for 60 min at
room temperature. Following further washing, the immunoreactivity
was visualized with an enhanced chemiluminescence kit (Advansta,
Inc., Menlo Park, CA, USA). Relative quantification of proteins was
performed using ImageLab software (version 4.0; Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Three independent
experiments were performed for each analysis.
Cell counting kit-8 (CCK-8) assay of
LPS-treated HUVEC viability
In order to induce endothelial cell injury, HUVECs
were incubated with bacterial LPS, and the cell viability was
assessed using the CCK-8 assay (Dojindo Molecular Technologies,
Inc., Kumamoto, Japan). The HUVECs/si-NC and HUVECs/siCav-1 were
plated in flat-bottom 96-well plates (5×103 cells/well)
and allowed to adhere for 24 h at 37°C. Cells were then treated
with various concentrations of LPS (0, 10, 25, 50, 75 and 100
µg/ml) for 24 h at 37°C in ECM containing 2.5% FBS, while PBS was
added to cells in the control group. After 24 h, the medium was
removed, and the CCK-8 solution was added (10 µl CCK-8 + 90 µl ECM)
to each well. The plates were then incubated for 2 h and the
absorbance was detected at 450 nm in a microplate reader.
Measurement of superoxide dismutase
(SOD) inhibition
SOD inhibition was measured using the SOD Assay
kit-WST kit (Dojindo Molecular Technologies, Inc.) according to
manufacturer's protocol. Briefly, HUVECs were seeded in 6-well
plates (1×105 cells/well) for 24 h prior to the addition
of LPS (75 µg/ml). After 24 h, the cells were lysed in RIPA buffer
and centrifuged at 10,000 × g for 15 min at 4°C. The supernatants,
enzyme-working solution and WST solution were added to 96-well
plates according to the manufacturer's protocol. Finally, the
absorbance was measured at 450 nm with a microplate reader.
Measurement of a fluorescent molecular
probe for analysis of intracellular Ca2+
[(Ca2+)i]
To monitor alterations in the [Ca2+]i
levels in response to ACh treatment (10 µM), cells were loaded with
the fluorescent dye Fluo4-acetoxymethyl ester (Fura4-AM; Dojindo
Molecular Technologies, Inc.). Briefly, cells were seeded into a
culture chamber for 24 h at 37°C, and LPS was added for a further
24 h, followed by incubation with Fura4-AM (5 µM) for 30 min at
37°C. Subsequent to removing the medium, cells were covered with
Hank's balanced salt solution with Ca2+ (cat. no. CC014;
Macgene, Biotechnology Ltd., Beijing, China), and visualized under
a fluorescence microscope (IX 70; Olympus Corp., Hamburg, Germany)
at an excitation wavelength of 488 nm and emission wavelength of
512 nm. Images were acquired at 1 sec intervals for 300 sec.
Alterations in the fluorescence reflecting changes in
[Ca2+]i were presented as the integrated optical density
and were quantified using Image-Pro Plus version 6.0 software
(Media Cybernetics, Inc., Rockville, MD, USA).
Nitrate reductase method for the
detection of the NO content in cells
The NO content in HUVECs/si-NC and HUVECs/siCav-1
treated with or without LPS and/or Ach was also investigated. The
culture supernatants were collected from all eight experimental
groups following treatment. Next, NO concentrations were measured
using a nitrate reductase method with an NO detection kit (Nanjing
Jiancheng Bioengineering Institute, Nanjing, China) according to
the manufacturer's protocol.
Statistical analysis
All data are presented as the mean ± standard
deviation. Relative Cav-1/GAPDH protein expression levels, HUVEC
viability and SOD inhibition were analyzed by one-way analysis of
variance for multiple-group comparisons. When a statistical
difference was observed, the data were further analyzed using
Bonferroni's post-hoc test with GraphPad Prism version 5 software
(GraphPad Software, Inc., La Jolla, CA, USA). P≤0.05 was considered
to indicate differences that were statistical significance.
Results
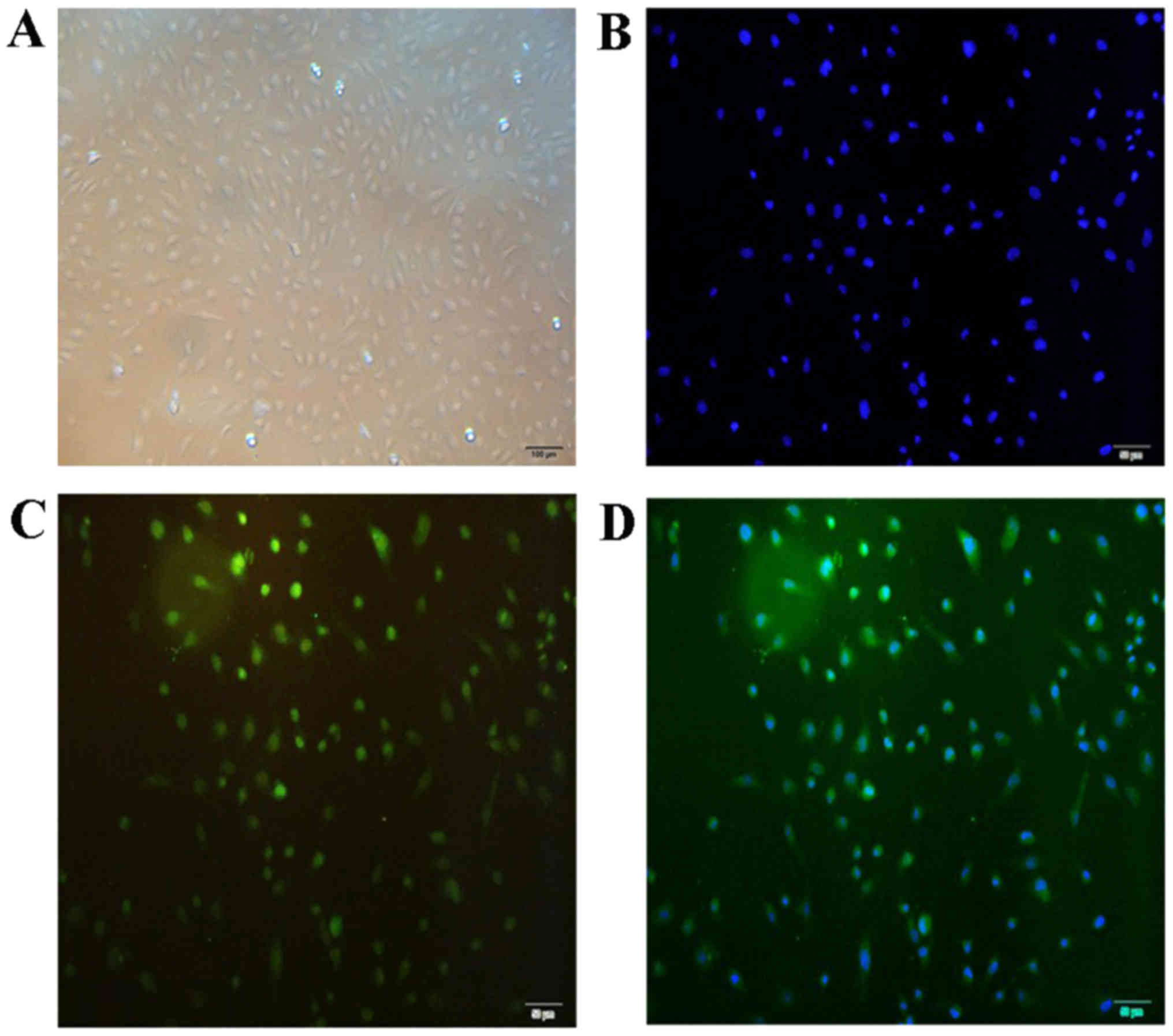
Characterization of cultured
HUVECs
The primary HUVECs started to attach to the plates
subsequent to culture for ~1 h, demonstrating a typical
cobblestone-like appearance under the inverted microscope and
positive immunofluorescent staining with anti-factor VIII antibody
(Fig. 1). Thus, these cells were
confirmed to be HUVECs.
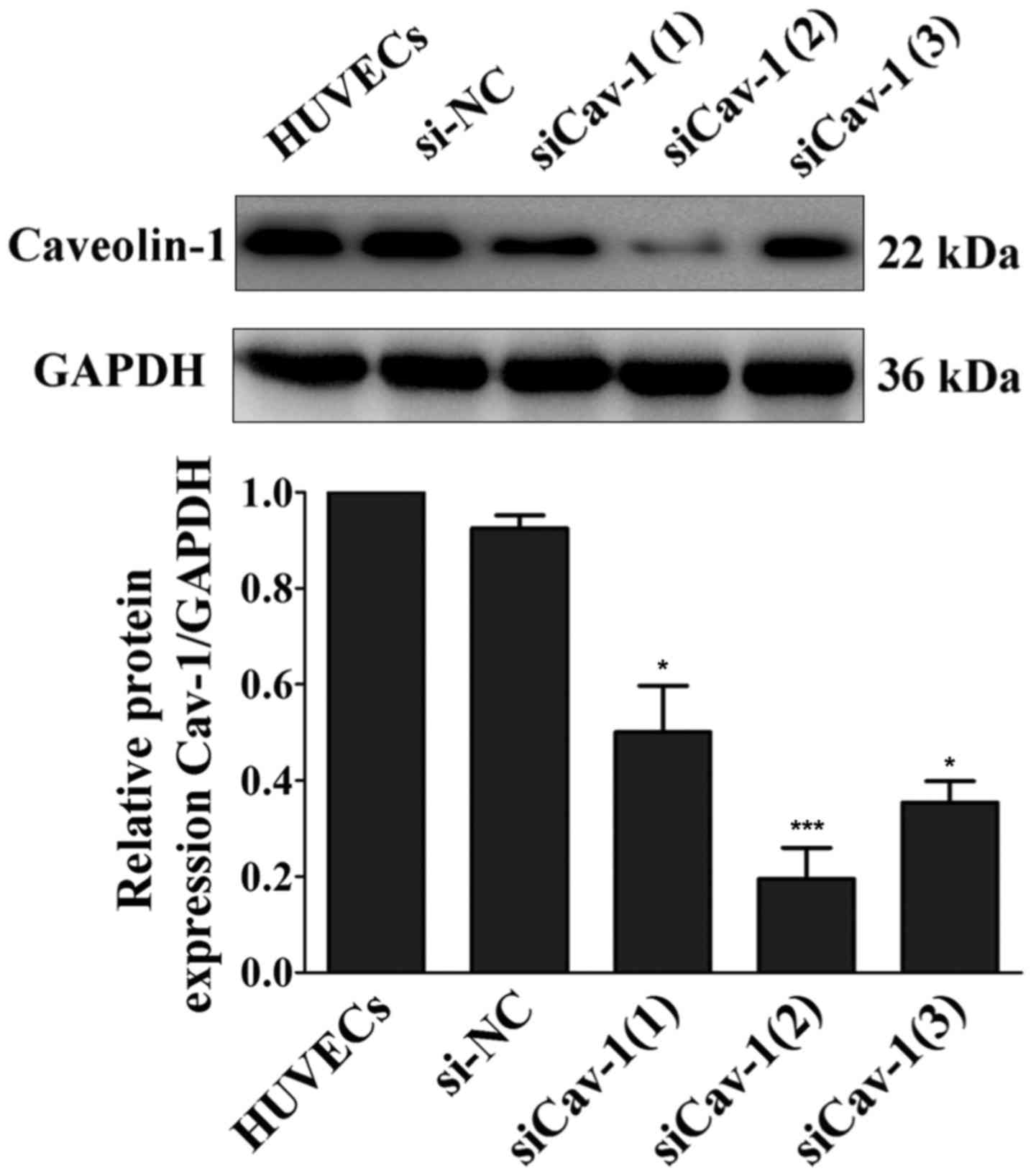
siRNA-mediated downregulation of Cav-1
expression in HUVECs
Western blot analysis revealed that, among the three
double-stranded siRNAs investigated, siCav-1(2) transfection
presented the best efficiency at inducing a reduction in Cav-1
protein expression compared with the with HUVECs group (~70%
reduction; P<0.001; Fig. 2).
Thus, siCav-1(2) was used in all subsequent experiments in the
present study.
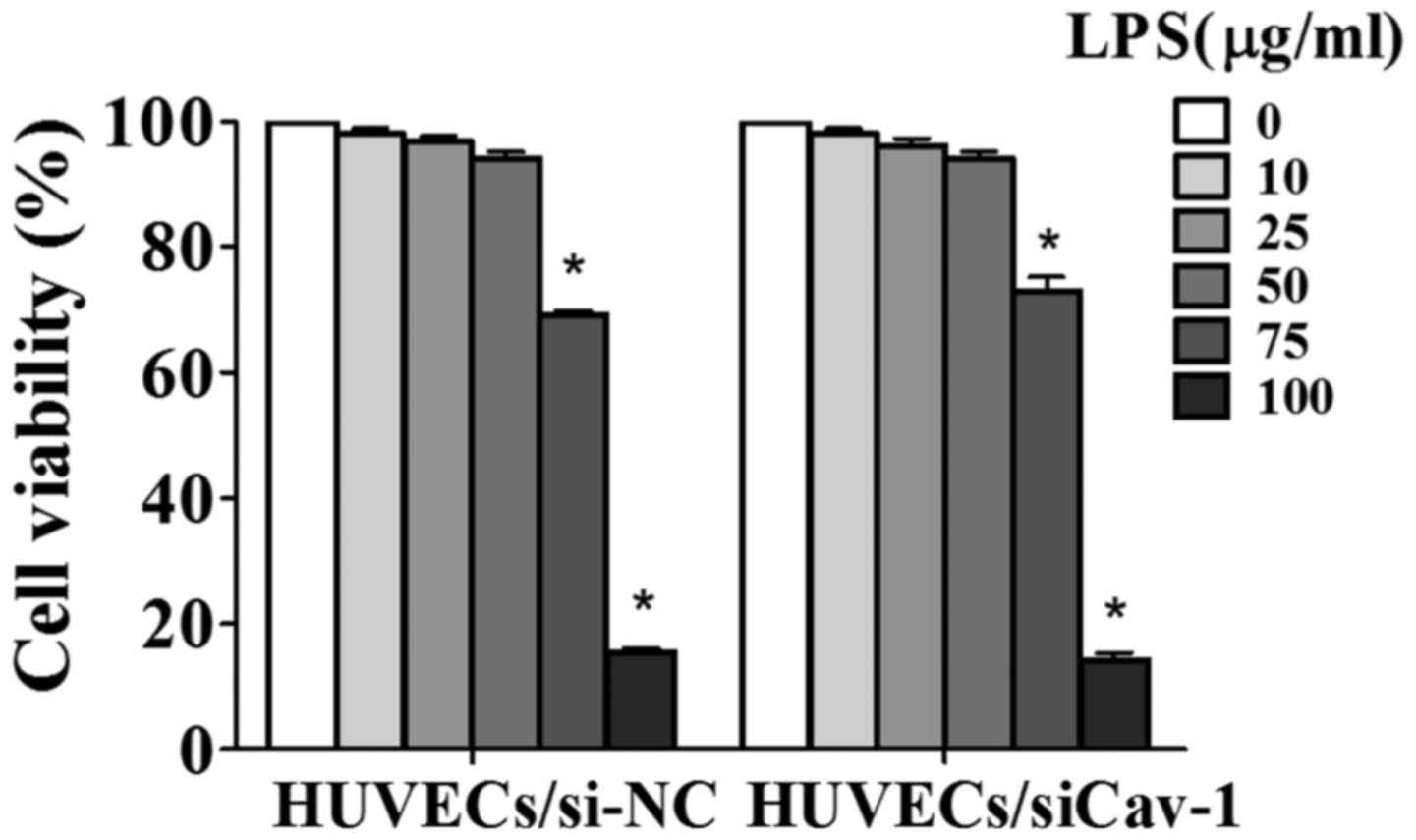
LPS-induced a reduction in HUVEC
viability
Following the incubation of HUVECs/si-NC and
HUVECs/siCav-1 with different concentrations of LPS for 24 h, CCK8
analysis demonstrated that the LPS-induced cell viability was
decreased in a dose-dependent manner. There were no significant
alterations in the viability of HUVECs/si-NC and HUVECs/siCav-1
following treatment with LPS at the lower concentrations (10, 25
and 50 µg/ml). However, compared with the untreated cells, a
significant decrease in cell viability was observed following
treatment of HUVECs/si-NC and HUVECs/siCav-1 with LPS at 75 and 100
µg/ml (P<0.001). Treatment with LPS at 75 and 100 µg/ml for 24 h
resulted in respective cell viabilities of 69.06 and 15.56% in the
HUVECs/si-NC, and 72.90 and 13.02% in the HUVECs/siCav-1. When 100
µg/ml was used, the cell viabilities were so the subsequent
experiments would have been detrimentally affected, therefore this
concentration was not selected. There was no significant difference
in the viability of the HUVECs/si-NC and HUVECs/siCav-1 groups when
treated with LPS at 75 µg/ml; thus, 75 µg/ml LPS was used in all
subsequent experiments (Fig. 3).
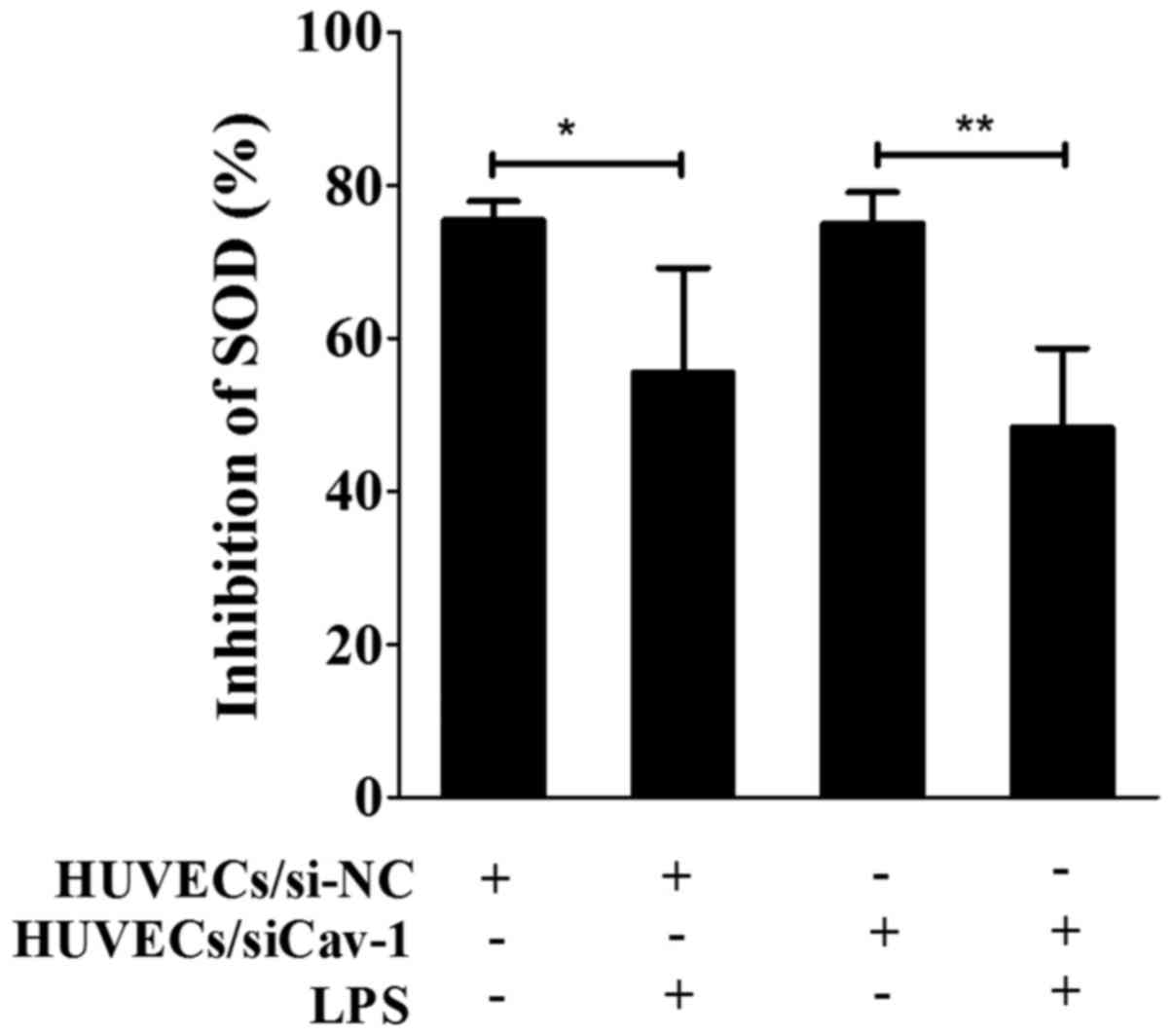
LPS promoted HUVEC damage through SOD
inhibition
SOD inhibition, which reflects cell damage, was
determined in order to evaluate the effects of LPS on HUVECs
(Fig. 4). SOD inhibition of 55.45
and 75.36% was observed in HUVECs/si-NC following treatment with
and without LPS, respectively. Similarly, SOD inhibition of 48.28
and 75.05% was observed in HUVECs/siCav-1 following treatment with
and without LPS, respectively. Thus, SOD inhibition was
significantly decreased in the presence of LPS (P<0.05),
indicating that LPS (75 µg/ml) caused injury in HUVECs/siCav-1 and
HUVECs/siCav-1.
[Ca2+]i levels in response
to ACh
Alterations in [Ca2+]i were measured in
HUVECs/si-NC with or without LPS treatment prior to and following
the addition of Ach, used as a marker of the CAS environment
mimicked in vitro. In the HUVECs/si-NC group without LPS
treatment, a rapid increase in [Ca2+]i was induced,
reaching a maximum followed by a sustained plateau after the
addition of Ach (Fig. 5A). Certain
cells also demonstrated a [Ca2+]i oscillation phenomenon
(16). In the HUVECs/si-NC group
treated with LPS, a smaller peak and plateau of [Ca2+]i
was observed, with certain cells presenting no response to
treatment with Ach (Fig. 5B). Thus,
the [Ca2+]i was increased in the untreated HUVECs/si-NC
group, but was unaltered in the LPS-treated cells, indicating that
the LPS-induced injury influenced the level of [Ca2+]i
following ACh stimulation and that a CAS environment was
successfully mimicked in vitro.
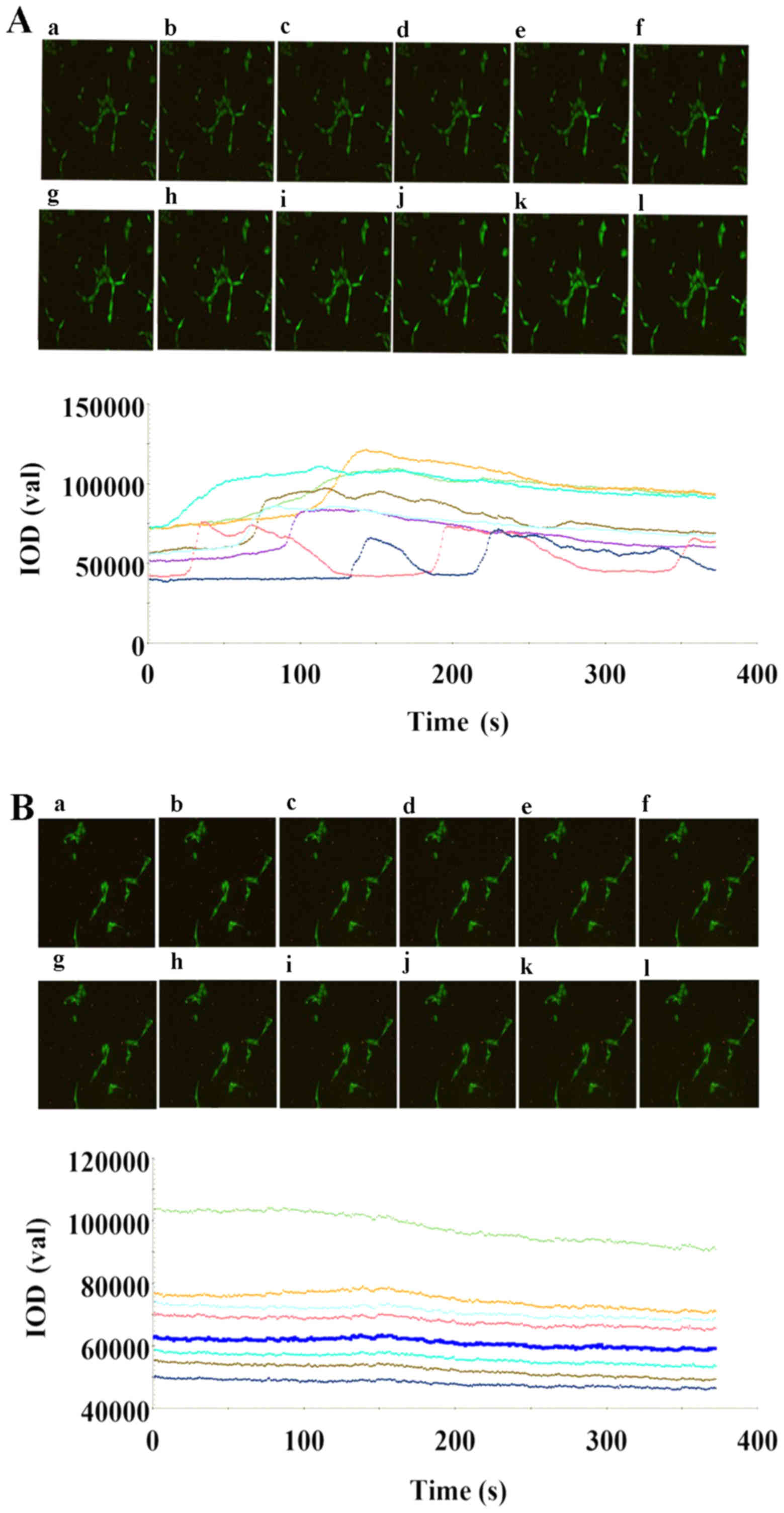 | Figure 5.Effects of 10 µM Ach added at 10 sec
on the [Ca2+]i responses in Fluo4-acetoxymethyl
ester-loaded HUVECs/si-NC with or without LPS treatment. The
responses in the eight cell groups were evaluated. The
ACh-stimulated changes in the [Ca2+]i fluorescence of
HUVECs/si-NC are shown in the (A) absence and (B) presence of LPS,
at the following time points: (a) 1 sec; (b) 17 sec; (c) 35 sec;
(d) 70 sec; (e) 99 sec; (f) 122 sec; (g) 158 sec; (h) 199 sec; (i)
212 sec; (j) 232 sec; (k) 268 sec; and (l) 334 sec. In HUVECs/si-NC
without LPS treatment, a characteristic biphasic [Ca2+]i
response was observed following the addition of ACh (10 µM) at 10
sec, with an initial rapid increase in [Ca2+]i, reaching
a maximum level at a different time-points for every cell, followed
by a sustained plateau in [Ca2+]i that declined slowly
toward the baseline. In addition, a [Ca2+]i oscillation
phenomenon was observed with peak and plateau levels occurring
regularly. In the HUVECs/si-NC group with LPS treatment, the
addition of ACh (10 µM) at 10 sec resulted in a smaller peak and
plateau compared with those observed in the group without LPS
treatment, with certain cells not presenting any response.
[Ca2+]i, intracellular Ca2+; LPS,
lipopolysaccharide; ACh, acetylcholine; HUVECs, human umbilical
vein endothelial cells; Cav-1, caveolin-1; si, small interfering
RNA; NC, negative control. |
Downregulated Cav-1 expression
enhances NO production in LPS-treated HUVECs stimulated with
Ach
A significant difference in NO content was observed
between the ACh-stimulated HUVECs/si-NC groups with and without LPS
treatment (P<0.01). In the group without LPS treatment, the
expression of NO was significantly higher in comparison with that
in the LPS-treated group (4.161±1.224 vs. 1.679±1.049 µmol/l,
respectively; P<0.05; Fig. 6).
Similarly, in the HUVECs/siCav-1 group, the content of NO in the
ACh-stimulated group without LPS treatment was significantly higher
compared with that in the group treated with LPS (P<0.001). In
the absence of LPS pretreatment, there was a significant difference
in the NO expression between the HUVECs/si-NC and HUVECs/siCav-1
groups following ACh stimulation (P<0.001). Following ACh
stimulation, the NO content in LPS-treated HUVECs/siCav-1 was
significantly higher when compared with that in the LPS-induced
HUVECs/si-NC cells (3.869±0.7679 vs. 1.241±0.3674 µmol/l,
respectively; P<0.05; Fig. 6). No
significant difference in NO content was identified in LPS-treated
HUVECs/si-NC with or without ACh. Additionally, no significant
difference in NO content was identified in LPS-treated
HUVECs/siCav-1 with or without ACh.
 | Figure 6.Production of NO with or without ACh
exposure in HUVECs/si-NC and HUVECs/siCav-1 with or without LPS
pretreatment. ACh stimulated the production of NO in HUVECs/si-NC
and HUVECs/siCav-1. The NO level was lower when cells were treated
with LPS compared with the levels observed without LPS treatment.
ACh stimulated the LPS-induced HUVECs/siCav-1 to produce
significantly higher NO levels compared with those produced by
LPS-induced HUVECs/si-NC. *P≤0.05, **P≤0.01, ***P≤0.001. NO, nitric
oxide; HUVECs, human umbilical vein endothelial cells; LPS,
lipopolysaccharide; ACh, acetylcholine; Cav-1, caveolin-1; si,
small interfering RNA; NC, negative control; IOD, integrated
optical density. |
Discussion
In the current study, a CAS microenvironment model
for endothelial cells was successfully established as observed by
the alterations in [Ca2+]i, which was used as an
indicator. ACh stimulation increased the [Ca2+]i levels
in cells that were not damaged by LPS pretreatment, while there was
little or no change in [Ca2+]i following ACh exposure in
LPS-treated HUVECs. These results demonstrated that Cav-1 served an
important regulatory role in this process and that downregulation
of Cav-1 promotes the release of NO in LPS-treated HUVECs
stimulated with ACh. In the CAS microenvironment the production of
NO was increased by knocking down Cav-1.
The presence of an intact endothelium is essential
for ACh to induce dilation of isolated arteries by NO release. It
has been observed that ACh stimulation induces arterial
constriction in the absence of the endothelium, due to a reduced
bioavailability of NO resulting from its decreased formation or
accelerated degradation (5,17). The current study demonstrated that
when the endothelium was damaged, ACh treatment did not increase
the [Ca2+]i; however, the inhibitory role of the
Cav-1/eNOS complex cannot be dismissed. The endothelium-dependent
agonists, ACh and bradykinin, increase [Ca2+]i and then
increase the Ca2+/calmodulin complex, which in turn,
activates eNOS (18). The elevation
of [Ca2+]i in endothelial cells induced by ACh is the
result of Ca2+ release from intracellular stores and
transmembrane Ca2+ influx (19,20). The
endothelial target for ACh is widely expressed in the vascular
endothelium, and the endothelium-dependent relaxation mediated by
the receptor is considered to be the classical indicator of the
endothelial dysfunction (21,22). It
has been demonstrated that the expression and bioactivity of the
endothelial target for ACh is decreased by endothelial damage. In
addition, Cav-1/eNOS binding can be reversed by calcium influx and
increased Ca2+/calmodulin complex formation (23). Therefore, ACh can initiate
endothelial cell signaling transduction, leading to decreased NO
formation via numerous pathways, including the
Ca2+/calmodulin signaling pathway (24). In vitro models of spasm in
arteries exposed to ACh and pharmacological provocation tests using
ACh or ergonovine are well established and have been widely used in
the diagnosis of coronary spastic angina (25). However, there are few reports that
describe the use of endothelial cells in a model of the CAS
microenvironment.
Numerous studies have generated models of coronary
spasm using isolated arteries, which contain endothelial and smooth
cells; thus, these models are not suitable for investigating the
roles of the different cell types. Therefore, for improved
investigation of the role of Cav-1 in endothelial cells, it is
hypothesized that the alterations in [Ca2+]i in
LPS-damaged cells stimulated with ACh can be considered as a marker
the reflects the CAS microenvironment in vitro. In the
present study, ACh stimulation induced a less marked increase in
[Ca2+]i in LPS-damaged HUVECs when compared with that
induced in cells without LPS treatment. This observation supports
the hypothesis that the changes in [Ca2+]i are a
suitable marker for the status of the CAS microenvironment.
In the present study, it was observed that the
siRNA-mediated downregulation of Cav-1 increased the production of
NO when LPS-damaged HUVECs were exposed to ACh. This highlighted
Cav-1 inhibition as a potential therapeutic target that may promote
NO levels in CAS. These effects are consistent with a previous
study that revealed significantly higher basal NO release in Cav-1
knockout mice when compared with that in wild-type mice (26). Furthermore, in the absence of Cav-1,
arteries exhibited a lack of steady contractile tone as well as
increased relaxation with high NO generation following ACh
stimulation (26).
The current study demonstrated that Cav-1
downregulation increased the release of NO when LPS-damaged HUVECs
were stimulated by ACh. However, the limitations of the study
should be noted. The molecular mechanism of signal transduction is
complicated and the marker used in the model employed in the
present study requires further investigation. Although the results
of the study indicated the regulatory influence of Cav-1 on NO, a
more mechanistic understanding is necessary to fully clarify how
Cav-1 influences eNOS and NO following ACh stimulation.
Furthermore, the present study was based on an in vitro
system and since Cav-1 serves a vital role in cells, the degree of
Cav-1 knockdown requires further investigation to prevent the
occurrence of CAS without affecting other Cav-1 functions.
In conclusion, the present study revealed that LPS
and ACh stimulation downregulated Cav-1 expression in a CAS
microenvironment, which may serve a key role in NO production.
Therefore, Cav-1 may be a potential therapeutic target in an
efficient management of coronary spasm.
Acknowledgements
The abstract of the current study was accepted by
the 22nd World conference of 2017 International Academy of
Cardiology (Vancouver, BC, Canada) as an oral presentation.
Funding
The current study was funded by the National Natural
Science Foundation of China (grant no. 81670324).
Availability of data and materials
The datasets used and/or analyzed during the study
are available from the corresponding authors on reasonable
request.
Authors' contributions
XC and ZY designed the study, performed experiments
and analyzed the data. XC was a major contributor in writing the
manuscript. MJ and SY analyzed the data and revised the article. XS
and RH designed the study and revised the article. All authors
participated in the design of the experiments. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
An Zhen Hospital of Capital Medical University (Beijing,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Takagi Y, Yasuda S, Takahashi J, Tsunoda
R, Ogata Y, Seki A, Sumiyoshi T, Matsui M, Goto T, Tanabe Y, et al:
Clinical implications of provocation tests for coronary artery
spasm: Safety, arrhythmic complications and prognostic impact:
Multicentre registry study of the Japanese coronary spasm
association. Eur Heart J. 34:258–267. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yasue H, Nakagawa H, Itoh T, Harada E and
Mizuno Y: Coronary artery spasm-clinical features, diagnosis,
pathogenesis, and treatment. J Cardiol. 51:2–17. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lanza GA, Careri G and Crea F: Mechanisms
of coronary artery spasm. Circulation. 124:1774–1782. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Satoh S, Omura S, Inoue H, Mori T,
Takenaka K, Numaguchi K, Mori E, Aso A, Nakamura T and Hiyamuta K:
Clinical impact of coronary artery spasm in patients with no
significant coronary stenosis in acute coronary syndromes. J
Cardiol. 61:404–409. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Furchgott RF and Zawadzki JV: The
obligatory role of endothelial cells in the relaxation of arterial
smooth muscle by acetylcholine. Nature. 288:373–376. 1980.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yamada E: The fine structure of the gall
bladder epithelium of the mouse. J Biophys Biochem Cytol.
1:445–458. 1955. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Osugi T, Saitoh S, Matumoto K, Muto M,
Aikawa K, Ohkawara H, Sugimoto K, Kamioka M, Ishibashi T and
Maruyama Y: Preventive effect of chronic endothelin type A receptor
antagonist on coronary microvascular spasm induced by repeated
epicardial coronary artery endothelial denudation in pigs. J
Atheroscler Thromb. 17:54–63. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shiroto T, Romero N, Sugiyama T,
Sartoretto JL, Kalwa H, Yan Z, Shimokawa H and Michel T: Caveolin-1
is a critical determinant of autophagy, metabolic switching, and
oxidative stress in vascular endothelium. PLoS One. 9:e878712014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Okamoto T, Schlegel A, Scherer PE and
Lisanti MP: Caveolins, a family of scaffolding proteins for
organizing ‘preassembled signaling complexes’ at the plasma
membrane. J Biol Chem. 273:5419–5422. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grayson TH, Chadha PS, Bertrand PP, Chen
H, Morris MJ, Senadheera S, Murphy TV and Sandow SL: Increased
caveolae density and caveolin-1 expression accompany impaired
NO-mediated vasorelaxation in diet-induced obesity. Histochem Cell
Biol. 139:309–321. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bucci M, Gratton JP, Rudic RD, Acevedo L,
Roviezzo F, Cirino G and Sessa WC: In vivo delivery of the
caveolin-1 scaffolding domain inhibits nitric oxide synthesis and
reduces inflammation. Nat Med. 6:1362–1367. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Weller R, Dykhuizen R, Leifert C and
Ormerod A: Nitric oxide release accounts for the reduced incidence
of cutaneous infections in psoriasis. J Am Acad Dermatol.
36:281–282. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Saitoh S, Takeishi Y and Maruyama Y:
Mechanistic insights of coronary vasospasm and new therapeutic
approaches. Fukushima J Med Sci. 61:1–12. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gratton JP, Bernatchez P and Sessa WC:
Caveolae and caveolins in the cardiovascular system. Circ Res.
94:1408–1417. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maniatis NA, Shinin V, Schraufnagel DE,
Okada S, Vogel SM, Malik AB and Minshall RD: Increased pulmonary
vascular resistance and defective pulmonary artery filling in
caveolin-1-/-mice. Am J Physiol Lung Cell Mol Physiol.
294:L865–L873. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ohi Y, Takai N, Muraki K, Watanabe M and
Imaizumi Y: Ca2+-images of smooth muscle cells and endothelial
cells in one confocal plane in femoral artery segments of the rat.
Jpn J Pharmaclo. 86:106–113. 2001. View Article : Google Scholar
|
|
17
|
Furchgott RF: The 1996 albert lasker
medical research awards. The discovery of endothelium-derived
relaxing factor and its importance in the identification of nitric
oxide. JAMA. 276:1186–1188. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Smiljić S, Nestorović V and Savić S:
Modulatory role of nitric oxide in cardiac performance. Med Pregl.
67:345–352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nilius B: Permeation properties of a
non-selective cation channel in human vascular endothelial cells.
Pflugers Arch. 416:609–611. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Newby AC and Henderson AH:
Stimulus-secretion coupling in vascular endothelial cells. Annu Rev
Physiol. 52:661–674. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Furchgott RF: Role of endothelium in
responses of vascular smooth muscle. Circ Res. 53:557–573. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shan LM and Wang H: Pharmacological
characteristics of the endothelial target for acetylcholine induced
vascular relaxation. Life Sci. 70:1285–1298. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Egashira K, Suzuki S, Hirooka Y, Kai H,
Sugimachi M, Imaizumi T and Takeshita A: Impaired
endothelium-dependent vasodilation of large epicardial and
resistance coronary arteries in patients with essential
hypertension. Different responses to acetylcholine and substance P.
Hypertension. 25:201–206. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Haga T: Molecular properties of muscarinic
acetylcholine receptors. Proc Jpn Acad Ser B Phys Biol Sci. 89:pp.
226–256. 2013; View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hung MJ: Current advances in the
understanding of coronary vasospasm. World J Cardiol. 2:34–42.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Drab M, Verkade P, Elger M, Kasper M, Lohn
M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, et al:
Loss of caveolae, vascular dysfunction, and pulmonary defects in
caveolin-1 gene-disrupted mice. Science. 293:2449–2452. 2001.
View Article : Google Scholar : PubMed/NCBI
|