Introduction
Alzheimer's disease (AD) is a chronic
neurodegenerative disease that frequently occurs in the elderly
population and is characterized by progressive learning dysfunction
and memory impairment (1).
Pathologically, accumulation of senile plaques, neurofibrillary
tangles (NFTs) and neuronal loss have been observed in the brain
cortex and hippocampus of patients with AD (2). Although the underlying mechanisms have
not been fully elucidated, β-amyloid (Aβ) aggregation and tau
hyperphosphorylation have been revealed to exhibit key roles in the
pathogenesis of AD (3,4), as they are primary components of senile
plaques and NFTs, respectively (5,6) and are
associated with neuronal loss and memory deficits (7,8).
Therefore, potential pharmacological agents targeting the
inhibition of these pathological processes may be promising
therapeutic approaches in treating AD.
Ellagic acid (EA) is a natural polyphenol present in
several types of fruits and nuts, including strawberries,
pomegranates and walnuts (9). EA has
been reported to possess multiple pharmacological effects,
including antioxidant, anti-inflammatory, antitumor and
antifibrotic properties (10,11). A
previous study demonstrated the neuroprotective role of EA in
several central nervous system diseases (12). Farbood et al (13) reported that EA alleviates brain
injury-induced cognitive dysfunction via inhibition of
neuroinflammation. Notably, Sarkaki et al (14) demonstrated that EA mitigates the
symptoms of Parkinson's disease induced by 6-hydroxydopamine, and
this effect may be attributed to its antioxidant properties
(9). Furthermore, Feng et al
(15) indicated that EA reduces
Aβ42-induced neurotoxicity, which suggested a beneficial role of EA
in AD. In accordance with these results, Kiasalari et al
(16) further revealed that EA
ameliorated learning and memory deficits in a rat model of AD
induced by Aβ25–35 and suggested the underlying mechanisms were
associated with its inhibition of oxidative stress and the nuclear
factor (NF)-κB/NF erythroid 2 like 2/Toll-like receptor 4 signaling
pathway. However, whether EA affects Aβ production and tau
hyperphosphorylation remains unclear.
In the present study, the APP/PS1 double-transgenic
mouse model was employed to evaluate the efficacy of EA in the
treatment of AD. Brain hippocampi were examined for alterations in
neuronal apoptosis, Aβ plaque formation and tau
hyperphosphorylation. In addition, the RAC-α
serine/threonine-protein kinase (AKT)/glycogen synthase kinase
(GSK) 3β signaling pathway, which is involved in the regulation of
tau hyperphosphorylation, was also investigated.
Materials and methods
Animals and treatment
Male APP/PS1 double-transgenic and wild-type (WT)
C57BL/6 mice (6-months-old, weighing 24–28 g, n=12 per group) were
purchased from Nanjing Biomedical Research Institute of Nanjing
University (Nanjing, China). Prior to the experiment, all the mice
were acclimatized for 1 week and were kept at a 12-h light/dark
cycle at room temperature (21–23°C) with 55% humidity and access to
food and water ad libitum. Mice were randomly divided into
four groups (n=12 mice per group), namely the WT, WT+EA, APP/PS1
and APP/PS1+EA groups. The mice were intragastrically administered
EA (50 mg/kg/day, cat. no. E102710; Shanghai Aladdin Bio-Chem
Technology Co., Ltd., Shanghai, China) or the same volume of 10%
DMSO for 60 consecutive days. The dosage of EA used was based on
previous data (14,17), which demonstrated that EA at this
dose exerted protective effects in a model of Parkinson's disease.
The animal experiments were performed in accordance with the
guidelines for the Care and Use of Laboratory Animals and were
approved by the Institutional Animal Care and Use Committee of
Heilongjiang University of Chinese Medicine (Harbin, China).
Morris water maze (MWM) test
Following 60 days of EA treatment, the spatial
learning and memory abilities of the mice were assessed using the
MWM test, as previously described (18). The apparatus included a black
circular tank (180 cm in diameter) filled with water (temperature,
22–24°C). A platform (9 cm in diameter) was placed at the targeted
quadrant 1.5 cm below the water surface. Mice were randomly placed
into one of the four quadrants facing the maze wall and allowed to
search for the platform. If the mouse did not find the platform
within 120 sec, it was guided to the platform for 30 sec. The
training lasted for 5 consecutive days, with four sessions per day.
The time required to find the hidden escape platform was recorded.
The probe trial was performed following 5 days of hidden platform
trials. Mice were allowed to swim freely in the water without the
platform for 120 sec. The frequency of crossing the platform
location, the time spent in the target quadrant and the swimming
tracks were monitored.
Tissue preparation
Following the behavioral tests, the mice were
euthanized. Part of the resected hippocampal tissues were fixed in
10% formalin at 4°C for 24 h, embedded in paraffin and sliced into
5-µm sections. The remaining tissues were homogenized for western
blot analysis and ELISA.
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) assay
TUNEL staining (In Situ Cell Death Detection kit;
Roche Diagnostics, Indianapolis, IN, USA) was used to assess
neuronal apoptosis in the hippocampus. The TUNEL assay was
performed according to standard procedures. Hippocampal slices
(5-µm thick) were deparaffinized, rehydrated, permeabilized and
blocked in 3% H2O2 for 10 min at room
temperature. Following washing, the slices were incubated in TUNEL
reaction mixture for 60 min at 37°C and processed with 50 µl
converter-peroxidase for 30 min at 37°C. Sections were stained
using the diaminobenzidine substrate kit (cat no. DA1010; Beijing
Solarbio Science & Technology Co., Ltd., Beijing, China) for 3
min at room temperature for color development and counterstained
with hematoxylin for 3 min at room temperature. Apoptotic cells
were assessed under a light microscope (magnification, ×400). The
number of apoptotic cells was calculated in six different visual
fields of each section.
Thioflavin-S staining
Amyloid plaque formation was measured by
thioflavin-S staining (cat no. T1892; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) according to the manufacturer's instructions.
Hippocampal slices (5-µm thick) were deparaffinized, rehydrated and
stained in thioflavin-S solution at room temperature for 8 min.
Subsequently, the slices were successively rinsed in 50% alcohol
and distilled water. Amyloid deposition was evaluated under a
fluorescent microscope (magnification, ×100 and ×400).
Immunohistochemical analysis
The expression of tau protein in the hippocampus was
determined using immunohistochemical analysis. The procedures were
performed as previously described (19) with slight modifications. Briefly,
hippocampal sections were subjected to heat-induced epitope
retrieval by immersing slides in a boiling solution for 10 min,
followed by 3% H2O2 treatment to block
endogenous peroxidases for 15 min at room temperature. The sections
were then blocked with 10% goat serum (cat no. SL038; Beijing
Solarbio Science & Technology Co., Ltd.) for 15 min at room
temperature, followed by incubation with anti-tau antibody (cat no.
D155045; 1:50; Sangon Biotech, Co., Ltd., Shanghai, China) at 4°C
overnight. Following washing three times in PBS, the sections were
incubated with a biotinylated anti-rabbit IgG antibody (cat no.
A0277, 1:200; Beyotime Institute of Biotechnology, Shanghai, China)
for 30 min at 37°C, followed by incubation with the avidin
biotinylated horseradish peroxidase (cat no. A0303; 1:200; Beyotime
Institute of Biotechnology) for 30 min at 37°C. Color was developed
using the diaminobenzidine substrate kit (cat no. DA1010; Beijing
Solarbio Science & Technology Co., Ltd.) for 3 min at room
temperature and counterstained with hematoxylin for 3 min at room
temperature. Images of the stained sections were analyzed under a
light microscopy (magnification, ×400).
ELISA
The concentration of Aβ40 and Aβ42 in the
hippocampus was quantified using commercial ELISA kits (cat no.
CEA864Mu and cat no. CEA946Mu; USCN Business Co., Ltd., Wuhan,
China). ELISA was performed according to the manufacturer's
protocol.
Western blot analysis
Western blot analysis was performed according to
standard protocols with some modifications (20). Briefly, total protein from each
tissue sample were lysed with radioimmunoprecipitation assay lysis
buffer (Beyotime Institute of Biotechnology) following the
manufacturer's instructions. Protein concentrations were measured
using the BCA method. A total of 20 µg proteins were separated by
SDS-PAGE (8, 11 or 15% gels) and transferred to polyvinylidene
difluoride membranes. Following blocking with 5% skimmed milk at
room temperature for 1 h, the membrane was incubated with a
specific primary antibody at 4°C overnight. The primary antibodies
used in the study included anti-cleaved caspase-3 antibody (cat no.
ab2302; 1:1,000), anti-pSer199-tau antibody (cat no. ab81268;
1:10,000) (both from Abcam, Cambridge, UK), anti-amyloid precursor
protein (APP) antibody (cat no. D260097; 1:500; Sangon Biotech,
Co., Ltd.), anti-pThr668-APP antibody (cat no. D155082; 1:500;
Sangon Biotech, Co., Ltd.), anti-β secretase (BACE) 1 (cat no.
D220305; 1:500; Sangon Biotech, Co., Ltd.), anti-pSer396-tau
antibody (cat no. D155045; 1:500; Sangon Biotech, Co., Ltd.),
anti-pSer473-AKT antibody (cat no. KG11054; 1:500; Nanjing KenGen
Biotech, Co., Ltd., Nanjing, China), anti-AKT antibody (cat no.
KG21054; 1:500; Nanjing KenGen Biotech, Co., Ltd.),
anti-pTyr216-GSK3β (cat no. KG11301-2; 1:500; Nanjing KenGen
Biotech, Co., Ltd), anti-GSK3β antibody (cat no. KG21002-2; 1:500;
Nanjing KenGen Biotech, Co., Ltd.), anti-tau antibody (cat no.
KG21099-2; 1:500; Nanjing KenGen Biotech, Co., Ltd.) and
anti-β-actin antibody (cat no. bsm-33139M; 1:500; Bioss Antibodies,
Beijing, China). The membrane was subsequently incubated with goat
anti-rabbit IgG horseradish peroxidase-conjugated antibody (cat no.
A0208; 1:5,000; Beyotime Institute of Biotechnology) at 37°C for 45
min. An enhanced chemiluminescence detection reagent (Beyotime
Institute of Biotechnology) was used for enhanced blot detection.
The band intensity was assessed and analyzed with Gel-Pro-Analyzer
software 4.0 (Media Cybernetics, Inc., Rockville, MD, USA).
Statistical analysis
Data are presented as the mean ± standard deviation.
Analyses were performed using one-way analysis of variance and the
Bonferroni post-hoc test with GraphPad Prism 5.0 software (GraphPad
Software, Inc., La Jolla, CA, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
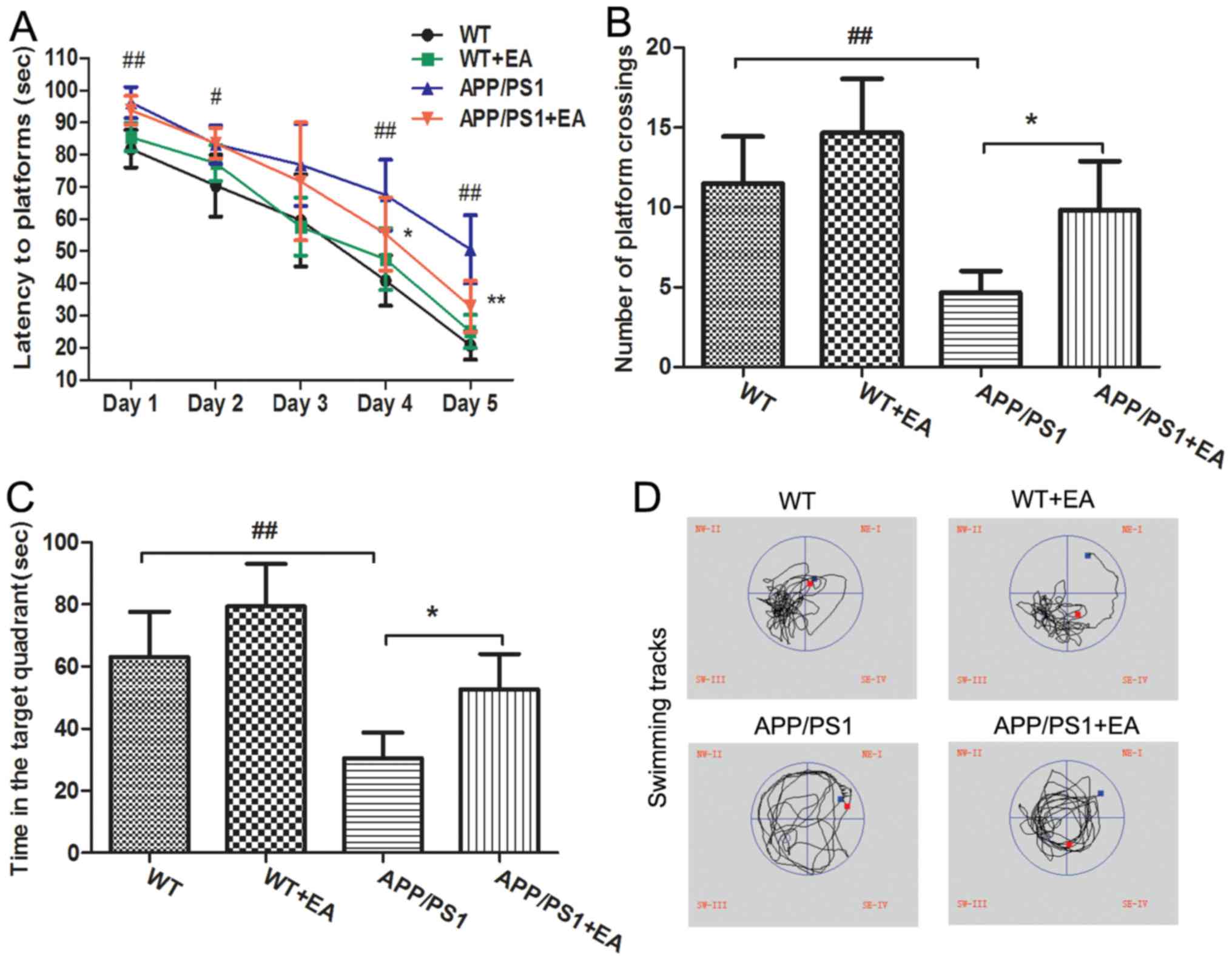
EA improves learning and memory
deficits in APP/PS1 transgenic mice
APP/PS1 transgenic mice begin to exhibit memory
impairment and amyloid plaque deposition at ~6 months of age
(21). In the present study, the MWM
test was performed to evaluate the learning and memory abilities of
the mice. In the hidden platform trials, the APP/PS1 model group
exhibited significantly longer escape latencies at days 1, 2, 4 and
5 of the trial compared with the WT group (P<0.01; Fig. 1A). EA treatment significantly
decreased the escape latencies at day 4 and 5 of the trial compared
with the APP/PS1 group (P<0.05), reflecting the improved
learning ability with EA treatment. In the probe trials, following
removal of the platform, the mice randomly swam in the pool for 120
sec. In addition, the number of platform crossings and the time
spent in the targeted quadrant for the APP/PS1-vehicle treated mice
was significantly decreased compared with the WT group (P<0.01;
Fig. 1B and C). By contrast, the
EA-treated APP/PS1 mice crossed the position of the platform more
frequently and spent more time searching for the platform in the
target quadrant compared with the APP/PS1-vehicle treated mice
(P<0.05). Furthermore, the swimming tracks of EA-treated APP/PS1
mice were similar to those of the WT group (Fig. 1D). Taken together, the MWM test
results suggested that EA treatment improved the spatial learning
and memory abilities in APP/PS1 transgenic mice.
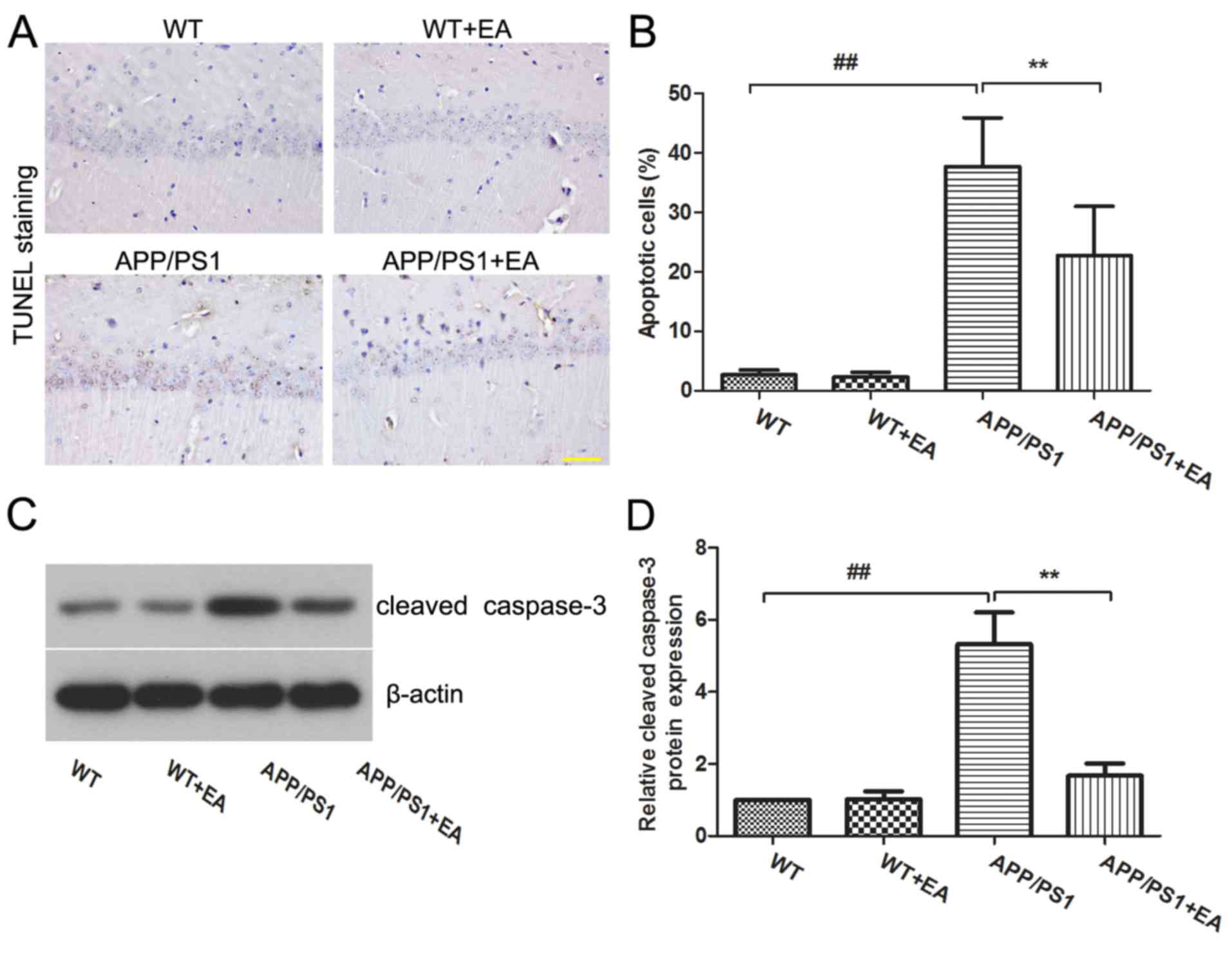
EA reduces neuronal cell apoptosis in
the hippocampus of APP/PS1 transgenic mice
Neuronal loss is responsible for the learning and
memory deficits in patients with AD (22). The present study further evaluated
the effects of EA on neuronal apoptosis in the hippocampus. TUNEL
staining indicated that the APP/PS1 group exhibited an increased
proportion of apoptotic cells in the hippocampus compared with the
WT group, whereas EA treatment reduced the numbers of apoptotic
cells compared with the APP/PS1-vehicle group (Fig. 2A). Furthermore, the differences
between the percentage of apoptotic cells exhibited in the
hippocampus of mice in these groups were statistically significant
(P<0.01; Fig. 2B). Western blot
analysis also revealed that the expression of cleaved caspase-3 was
upregulated in APP/PS1 mice, and EA treatment reduced the
expression level of caspase-3 in the hippocampus compared with that
in the APP/PS1-vehicle group (Fig.
2C). Statistical analysis indicated these differences were
significant (P<0.01; Fig.
2D).
EA reduces Aβ deposition and Aβ
protein expression levels in the hippocampus of APP/PS1 transgenic
mice
Accumulation of Aβ in the brain is considered to be
a primary hallmark of AD (23).
Deposited amyloid plaques in the hippocampus may further induce
neuronal loss and cognitive impairment deficits (20,24). The
present study further evaluated the effect of EA on Aβ deposition.
Aβ plaques were stained with thioflavin-S (Fig. 3A). Compared with the WT group, the
APP/PS1 group exhibited increased amyloid staining in the
hippocampus, whereas EA treatment reduced the amyloid aggregates in
the hippocampus. ELISA also revealed significantly increased levels
of Aβ40 and Aβ42 in the APP/PS1 group, whereas EA treatment
effectively reversed this increase (P<0.01; Fig. 3B and C).
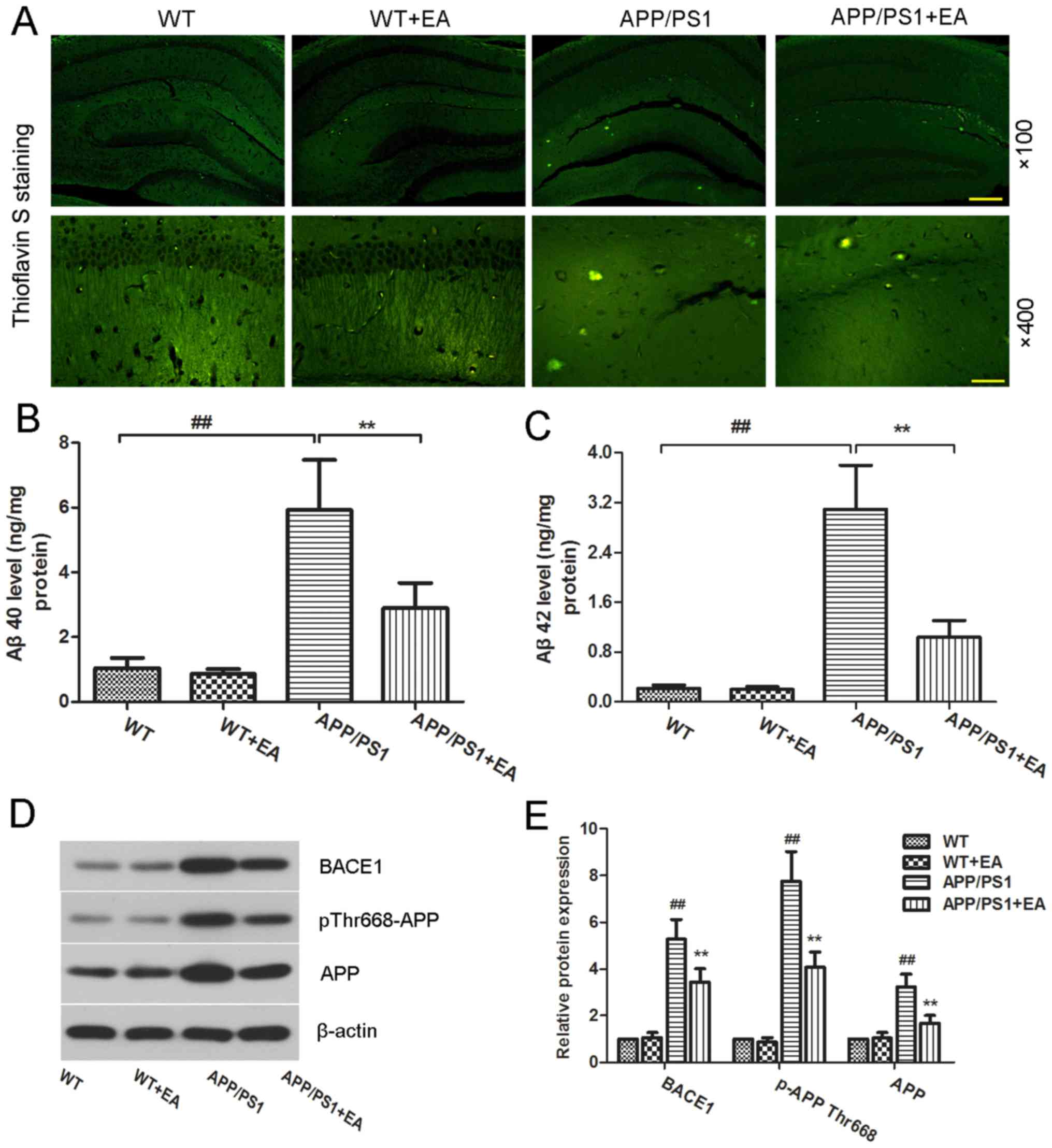 | Figure 3.Effect of EA on β-amyloid plaque
deposition in the hippocampus of APP/PS1 mice. Following completion
of the behavioral tests, the brain tissues from each group were
subjected to analysis. (A) Thioflavin S-stained Aβ plaques in the
hippocampus. Magnification, ×100; scale bar, 200 µm (upper panels)
and magnification, ×400; scale bar, 50 µm (lower panels).
Concentration of (B) Aβ40 and (C) Aβ42 in the hippocampal tissues
was determined using ELISA kits. (D) Western blotting and (E)
quantitative analysis of APP and pThr668-APP protein expression
levels in the hippocampus. β-actin served as an internal control
for grayscale analysis. Data are presented as the mean ± standard
deviation. ##P<0.01 vs. WT mice; **P<0.01 vs.
APP/PS1 transgenic mice. EA, ellagic acid WT, wild-type; APP,
amyloid precursor protein; Aβ, β-amyloid; BACE1, β-secretase 1. |
Aβ peptides are produced from the APP;
phosphorylation of APP and BACE1 kinase are responsible for Aβ
generation (25,26). To evaluate whether EA affects Aβ
production, the protein expression levels of pThr668-APP and BACE1
were determined. The western blot analysis results revealed that
pThr668-APP and BACE1 protein expression levels in the APP/PS1
group were increased compared with the WT group, whereas EA
treatment reduced these expression levels (Fig. 3D). Notably, these differences were
indicated to be statistically significant (P<0.01; Fig. 3E). These results suggested that EA
reduced Aβ deposition by suppressing Aβ production.
EA inhibits tau hyperphosphorylation
in the hippocampus of APP/PS1 transgenic mice
In addition to Aβ deposition, tau
hyperphosphorylation is another characteristic feature of the brain
in APP/PS1 mice (27). In the
present study, the extent of Tau hyperphosphorylation in the
hippocampus was determined by immunohistochemical staining and
western blot analysis. Immunohistochemical results revealed high
expression of pSer396-tau in the APP/PS1 group compared with the WT
group. Notably, EA treatment reduced the expression of pSer396-tau
in the hippocampus (Fig. 4A).
Furthermore, western blot analysis also determined that EA
downregulated the protein expression levels of pSer199-tau and
pSer396-tau in the APP/PS1 group (Fig.
4B). Furthermore, the difference between the protein expression
levels of pSer199-tau and pSer396-tau in the APP/PS1 group compared
with the APP/PS1+EA group was indicated to be statistically
significant (P<0.01; Fig. 4C).
Taken together, these results indicated that the beneficial role of
EA in AD may be associated with the downregulation of tau
phosphorylation.
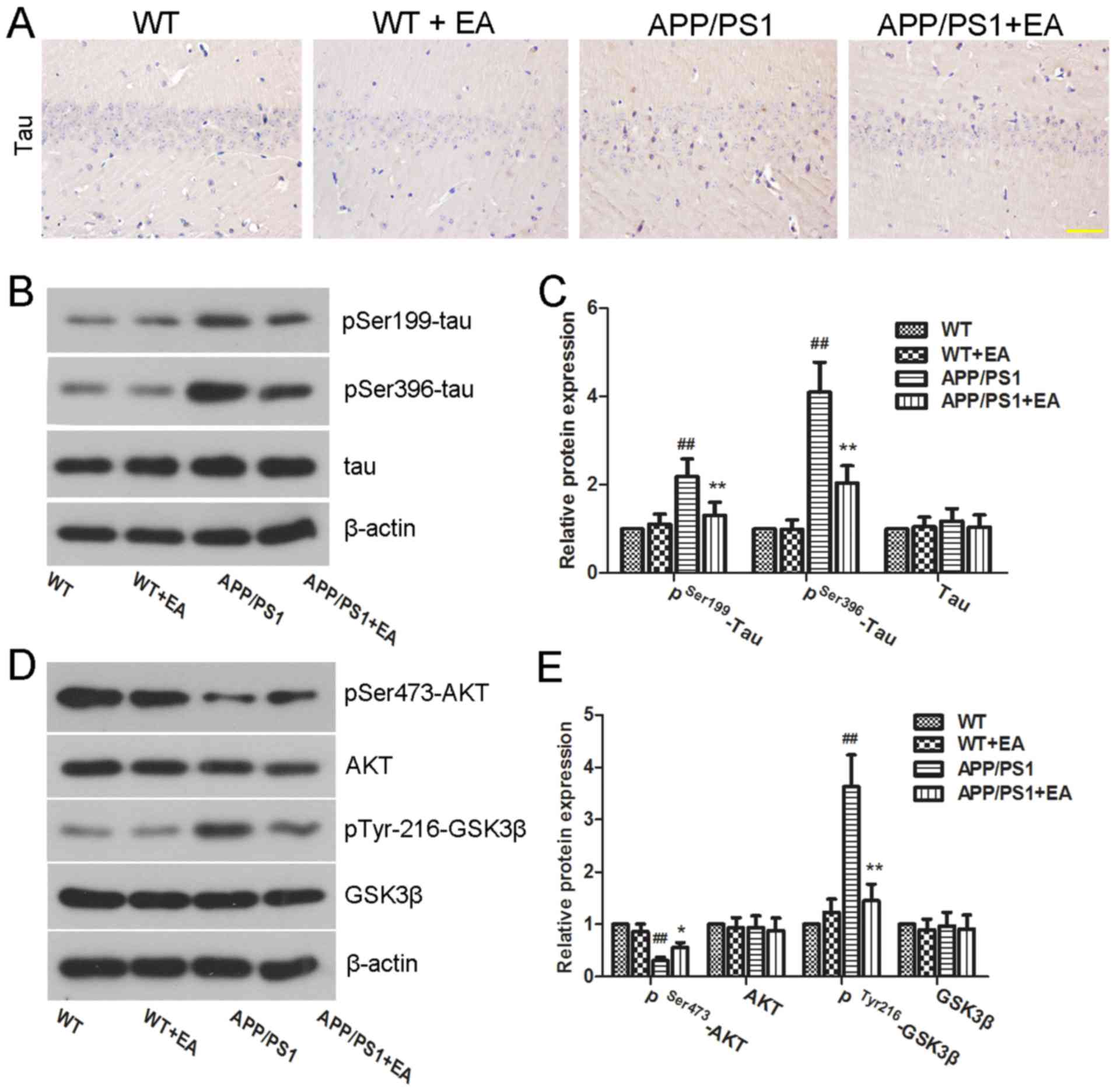 | Figure 4.Effect of EA on tau
hyperphosphorylation in APP/PS1 transgenic mice. (A) Protein
expression of pSer396-tau in the hippocampus was detected by
immunohistochemical staining (magnification, ×400; scale bar, 50
µm). (B) Western blotting and (C) quantitative analysis of
pSer199-tau, pSer396-tau and tau protein expression levels in the
hippocampus. β-actin served as an internal control for grayscale
analysis. (D) Western blotting and (E) quantitative analysis of
pSer473-AKT, pTyr216-GSK3β, AKT, and GSK3β protein expression
levels in the hippocampus. β-actin served as an internal control
for grayscale analysis. Data are presented as the mean ± standard
deviation. ##P<0.01 vs. WT mice; *P<0.05 or
**P<0.01 vs. APP/PS1 transgenic mice. EA, ellagic acid; WT,
wild-type; GSK3β, glycogen synthase kinase 3β; AKT, RAC-α
serine/threonine-protein kinase. |
The AKT/GSK3β signaling pathway has been
demonstrated to have an important role in tau hyperphosphorylation.
Activated GSK3β has been demonstrated to facilitate tau
phosphorylation and NFT formation (28). To further investigate the underlying
mechanism of action of EA, the effect of EA on the regulation of
the AKT/GSK3β signaling pathway was examined by western blot
analysis. As presented in Fig. 4D,
mice in the APP/PS1 group exhibited low protein expression levels
of pSer473-AKT and high expression levels of pTyr216-GSK3β compared
with those in the WT group. Notably, these differences were
indicated to be statistically significant (P<0.01; Fig. 4E). These results suggested GSK3β
activation. Conversely, EA treatment significantly upregulated the
protein expression levels of pSer473-AKT and decreased the protein
expression levels of pTyr216-GSK3β in APP/PS1 mice (P<0.05 and
P<0.01, respectively; Fig. 4E),
which indicated activation of AKT and deactivation of GSK3β
following EA treatment. These results suggested that EA inhibited
tau phosphorylation in the hippocampus of APP/PS1 mice and that
this inhibitory effect may be partially mediated by the AKT/GSK3β
signaling pathway.
Discussion
AD, which is the most common form of dementia,
severely affects the life quality of elderly patients, and there is
currently no effective treatment (29). To further understand the underlying
mechanisms and develop novel pharmacological agents for AD, animal
models of AD have been employed. A growing body of evidence has
demonstrated that APP/PS1 transgenic mice exhibit similar
pathological alterations to those of patients with AD (30). In APP/PS1 transgenic mice, the memory
impairment and amyloid deposition appear at 4–6 months of age and
progress in an age-dependent manner (31,32). In
the present study, APP/PS1 mice were selected to investigate the
therapeutic efficacy of EA in AD. The results demonstrated that
administration of EA (50 mg/kg/day for 60 days) ameliorated the
learning and memory deficits in APP/PS1 mice, significantly reduced
neuronal apoptosis and amyloid deposition, and also significantly
inhibited tau hyperphosphorylation in the hippocampus, which
suggested a beneficial role of EA in AD. A previous study also
reported the beneficial role of EA in an Aβ-induced AD mouse model
(16). Notably, the present findings
further demonstrated the therapeutic efficacy of EA in APP/PS1 mice
and the beneficial effects were indicated to be associated with the
inhibition of Aβ production and tau hyperphosphorylation.
Aβ aggregates exhibit crucial roles in AD-associated
cognitive impairment and neuronal loss (24,33).
Targeted inhibition of Aβ production has been indicated to exert
beneficial effects on APP/PS1 mice (34). In the present study, APP/PS1 mice
exhibited significant deficits in their learning and memory
abilities alongside increased neuronal apoptosis and Aβ deposition.
Learning and memory impairments of APP/PS1 mice in the present
study were in line with those reported by other studies (35,36).
Notably, EA treatment improved the learning and memory abilities,
and significantly decreased Aβ deposition and Aβ expression levels
in the hippocampus. Furthermore, EA treatment significantly reduced
the expression levels of pThr668APP and BACE1. As previously
reported, the phosphorylation of APP at the Thr668 site promotes Aβ
generation (25) and BACE1 is the
key enzyme for Aβ generation through cleavage of APP (26,37). The
present results revealed that EA treatment alleviated cognitive
impairment and decreased Aβ production. These results were
consistent with a previous study reporting the anti-amyloidogenic
effect of EA on potassium sorbate and glucose-induced human serum
albumin fibril formation (38).
Hyperphosphorylated tau is the primary component of
intracellular NFTs, which have an important role in AD pathology
(2,39). It has been demonstrated that
hyperphosphorylated tau may also result in neuronal death and
memory impairment (5,40,41). A
number of serine phosphorylation sites of tau are elevated in AD
mice, including Ser199 and Ser396 (42). Targeting inhibition of tau
phosphorylation may therefore provide an effective therapeutic
approach to AD (43). In the present
study, phosphorylation of tau at Ser199 and Ser396 was
significantly increased in the hippocampus of APP/PS1 mice, whereas
EA treatment efficiently inhibited tau phosphorylation. Since the
phosphorylation of tau is regulated by multiple signals, including
AKT/GSK3β signaling (35,44), the present study further investigated
the effect of EA on the AKT/GSK3β signaling pathway. The results
demonstrated that EA significantly increased the phosphorylation of
AKT at the Ser473 site and significantly decreased the
phosphorylation of GSK3β at the Tyr216 site, indicating
de-activation of GSK3β (45). These
results suggested that EA inhibited tau phosphorylation in APP/PS1
mice and that the inhibitory effect of EA may be partially mediated
by the AKT/GSK3β signaling pathway. However, several other kinases
are involved in the regulation of tau phosphorylation and further
investigation is required to identify the precise mechanisms of
action of EA in AD.
In conclusion, the present study demonstrated the
beneficial effects of EA on spatial learning and memory in APP/PS1
mice, which included inhibition of neuronal death, Aβ production
and tau hyperphosphorylation. Furthermore, the beneficial effects
of EA were indicated to be partially mediated by the AKT/GSK3β
signaling pathway. These findings suggest that EA may be a
promising therapeutic agent for the treatment of AD.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Natural Science Foundation of Heilongjiang Province (grant no.
QC2015101 and QC2015102), the Postdoctoral Foundation of
Heilongjiang Province Government (grant no. LBH-Z14196 and
LBH-Z15207), the Postdoctoral Science Foundation of China (grant
nos. 2015M581496 and 2016M591565) and the National Natural Science
Foundation of China (grant no. 81403288).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LZ, HL and ZS contributed to the conception and
design of the study, wrote and revised the manuscript. LZ, HL and
WZ conducted the studies and analyzed and interpreted the data. XL,
BJ and HF analyzed the data and interpreted the data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The animal experiments were performed in accordance
with the guidelines for the Care and Use of Laboratory Animals and
were approved by the Institutional Animal Care and Use Committee of
Heilongjiang University of Chinese Medicine (Harbin, China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ballard C, Gauthier S, Corbett A, Brayne
C, Aarsland D and Jones E: Alzheimer's disease. Lancet.
377:1019–1031. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Selkoe DJ: Alzheimer's disease results
from the cerebral accumulation and cytotoxicity of amyloid
beta-protein. J Alzheimers Dis. 3:75–80. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nelson PT, Braak H and Markesbery WR:
Neuropathology and cognitive impairment in Alzheimer disease: A
complex but coherent relationship. J Neuropathol Exp Neurol.
68:1–14. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bloom GS: Amyloid-β and tau: The trigger
and bullet in Alzheimer disease pathogenesis. JAMA Neurol.
71:505–508. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ruan YY, Zhai W, Shi XM, Zhang L and Hu
YL: Safflower yellow ameliorates cognition deficits and reduces tau
phosphorylation in APP/PS1 transgenic mice. Metab Brain Dis.
31:1133–1142. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sobów T, Flirski M and Liberski PP:
Amyloid-beta and tau proteins as biochemical markers of Alzheimer's
disease. Acta Neurobiol Exp (Wars). 64:53–70. 2004.PubMed/NCBI
|
|
7
|
Sengupta U, Nilson AN and Kayed R: The
role of Amyloid-β oligomers in toxicity, propagation, and
immunotherapy. EBioMedicine. 6:42–49. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang B, Li Q, Chu X, Sun S and Chen S:
Salidroside reduces tau hyperphosphorylation via up-regulating
GSK-3β phosphorylation in a tau transgenic Drosophila model of
Alzheimer's disease. Transl Neurodegener. 5:212016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Amakura Y, Okada M, Tsuji S and Tonogai Y:
High-performance liquid chromatographic determination with
photodiode array detection of ellagic acid in fresh and processed
fruits. J Chromatogr A. 896:87–93. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Han DH, Lee MJ and Kim JH: Antioxidant and
apoptosis-inducing activities of ellagic acid. Anticancer Res.
26:3601–3606. 2006.PubMed/NCBI
|
|
11
|
Mansouri MT, Hemmati AA, Naghizadeh B,
Mard SA, Rezaie A and Ghorbanzadeh B: A study of the mechanisms
underlying the anti-inflammatory effect of ellagic acid in
carrageenan-induced paw edema in rats. Indian J Pharmacol.
47:292–298. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ahmed T, Setzer WN, Nabavi SF, Orhan IE,
Braidy N, Sobarzo-Sanchez E and Nabavi SM: Insights into effects of
ellagic acid on the nervous system: A mini review. Curr Pharm Des.
22:1350–1360. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Farbood Y, Sarkaki A, Dianat M, Khodadadi
A, Haddad MK and Mashhadizadeh S: Ellagic acid prevents cognitive
and hippocampal long-term potentiation deficits and brain
inflammation in rat with traumatic brain injury. Life Sci.
124:120–127. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sarkaki A, Farbood Y, Dolatshahi M,
Mansouri SM and Khodadadi A: Neuroprotective effects of ellagic
acid in a rat model of Parkinson's disease. Acta Med Iran.
54:494–502. 2016.PubMed/NCBI
|
|
15
|
Feng Y, Yang SG, Du XT, Zhang X, Sun XX,
Zhao M, Sun GY and Liu RT: Ellagic acid promotes Abeta42
fibrillization and inhibits Abeta42-induced neurotoxicity. Biochem
Biophys Res Commun. 390:1250–1254. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kiasalari Z, Heydarifard R, Khalili M,
Afshin-Majd S, Baluchnejadmojarad T, Zahedi E, Sanaierad A and
Roghani M: Ellagic acid ameliorates learning and memory deficits in
a rat model of Alzheimer's disease: An exploration of underlying
mechanisms. Psychopharmacology (Berl). 234:1841–1852. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baluchnejadmojarad T, Rabiee N, Zabihnejad
S and Roghani M: Ellagic acid exerts protective effect in
intrastriatal 6-hydroxydopamine rat model of Parkinson's disease:
Possible involvement of ERbeta/Nrf2/HO-1 signaling. Brain Res.
1662:23–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang ZX, Zhao RP, Wang DS and Wang AN:
Fuzhisan ameliorates Aβ production and tau phosphorylation in
hippocampal of 11 month old APP/PS1 transgenic mice: A Western blot
study. Exp Gerontol. 84:88–95. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yoshiyama Y, Higuchi M, Zhang B, Huang SM,
Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ and Lee VM:
Synapse loss and microglial activation precede tangles in a P301S
tauopathy mouse model. Neuron. 53:337–351. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ma J, Gao Y, Jiang L, Chao FL, Huang W,
Zhou CN, Tang W, Zhang L, Huang CX, Zhang Y, et al: Fluoxetine
attenuates the impairment of spatial learning ability and prevents
neuron loss in middle-aged APPswe/PSEN1dE9 double transgenic
Alzheimer's disease mice. Oncotarget. 8:27676–27692.
2017.PubMed/NCBI
|
|
21
|
Garcia-Alloza M, Robbins EM, Zhang-Nunes
SX, Purcell SM, Betensky RA, Raju S, Prada C, Greenberg SM, Bacskai
BJ and Frosch MP: Characterization of amyloid deposition in the
APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis.
24:516–524. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Niikura T, Tajima H and Kita Y: Neuronal
cell death in Alzheimer's disease and a neuroprotective factor,
humanin. Curr Neuropharmacol. 4:139–147. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mucke L: Neuroscience: Alzheimer's
disease. Nature. 461:895–897. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jahn H: Memory loss in Alzheimer's
disease. Dialogues Clin Neurosci. 15:445–454. 2013.PubMed/NCBI
|
|
25
|
Lee MS, Kao SC, Lemere CA, Xia W, Tseng
HC, Zhou Y, Neve R, Ahlijanian MK and Tsai LH: APP processing is
regulated by cytoplasmic phosphorylation. J Cell Biol. 163:83–95.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gandhi S, Refolo LM and Sambamurti K:
Amyloid precursor protein compartmentalization restricts
beta-amyloid production: Therapeutic targets based on BACE
compartmentalization. J Mol Neurosci. 24:137–143. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kurt MA, Davies DC, Kidd M, Duff K and
Howlett DR: Hyperphosphorylated tau and paired helical
filament-like structures in the brains of mice carrying mutant
amyloid precursor protein and mutant presenilin-1 transgenes.
Neurobiol Dis. 14:89–97. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cohen P and Frame S: The renaissance of
GSK3. Nat Rev Mol Cell Biol. 2:769–776. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sabbagh M and Cummings J: Progressive
cholinergic decline in Alzheimer's disease: Consideration for
treatment with donepezil 23 mg in patients with moderate to severe
symptomatology. BMC Neurol. 11:212011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Serrano-Pozo A, Frosch MP, Masliah E and
Hyman BT: Neuropathological alterations in Alzheimer disease. Cold
Spring Harbor Persp Med. 1:a0061892011.
|
|
31
|
Parthsarathy V, McClean PL, Hölscher C,
Taylor M, Tinker C, Jones G, Kolosov O, Salvati E, Gregori M,
Masserini M and Allsop D: A novel retro-inverso peptide inhibitor
reduces amyloid deposition, oxidation and inflammation and
stimulates neurogenesis in the APPswe/PS1DeltaE9 mouse model of
Alzheimer's disease. PLoS One. 8:e547692013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Herran E, Perez-Gonzalez R, Igartua M,
Pedraz JL, Carro E and Hernandez RM: Enhanced Hippocampal
Neurogenesis in APP/Ps1 Mouse Model of Alzheimer's Disease After
Implantation of VEGF-loaded PLGA Nanospheres. Curr Alzheimer Res.
12:932–940. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Reiserer RS, Harrison FE, Syverud DC and
McDonald MP: Impaired spatial learning in the APPSwe + PSEN1DeltaE9
bigenic mouse model of Alzheimer's disease. Genes Brain Behav.
6:54–65. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dionísio PA, Amaral JD, Ribeiro MF, Lo AC,
D'Hooge R and Rodrigues CM: Amyloid-β pathology is attenuated by
tauroursodeoxycholic acid treatment in APP/PS1 mice after disease
onset. Neurobiol Aging. 36:228–240. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bao XQ, Li N, Wang T, Kong XC, Tai WJ, Sun
H and Zhang D: FLZ alleviates the memory deficits in transgenic
mouse model of Alzheimer's disease via decreasing beta-amyloid
production and tau hyperphosphorylation. PloS One. 8:e780332013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li C, Guo XD, Lei M, Wu JY, Jin JZ, Shi
XF, Zhu ZY, Rukachaisirikul V, Hu LH, Wen TQ and Shen X: Thamnolia
vermicularis extract improves learning ability in APP/PS1
transgenic mice by ameliorating both Aβ and Tau pathologies. Acta
Pharmacol Sin. 38:9–28. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chang KA, Kim HS, Ha TY, Ha JW, Shin KY,
Jeong YH, Lee JP, Park CH, Kim S, Baik TK and Suh YH:
Phosphorylation of amyloid precursor protein (APP) at Thr668
regulates the nuclear translocation of the APP intracellular domain
and induces neurodegeneration. Mol Cell Biol. 26:4327–4338. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Taghavi F, Habibi-Rezaei M, Bohlooli M,
Farhadi M, Goodarzi M, Movaghati S, Maghami P, Taghibiglou C,
Amanlou M, Haertlé T and Moosavi-Movahedi AA: Antiamyloidogenic
effects of ellagic acid on human serum albumin fibril formation
induced by potassium sorbate and glucose. J Mol Recognit.
29:611–618. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Roberson ED, Scearce-Levie K, Palop JJ,
Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ and Mucke L: Reducing
endogenous tau ameliorates amyloid beta-induced deficits in an
Alzheimer's disease mouse model. Science. 316:750–754. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Iqbal K, Alonso Adel C, Chen S, Chohan MO,
El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, et al: Tau
pathology in Alzheimer disease and other tauopathies. Biochim
Biophys Acta. 1739:198–210. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ma Q, Ruan YY, Xu H, Shi XM, Wang ZX and
Hu YL: Safflower yellow reduces lipid peroxidation, neuropathology,
tau phosphorylation and ameliorates amyloid beta-induced impairment
of learning and memory in rats. Biomed Pharmacother. 76:153–164.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Maas T, Eidenmüller J and Brandt R:
Interaction of tau with the neural membrane cortex is regulated by
phosphorylation at sites that are modified in paired helical
filaments. J Biol Chem. 275:15733–15740. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Matsuoka Y, Gray AJ, Hirata-Fukae C,
Minami SS, Waterhouse EG, Mattson MP, LaFerla FM, Gozes I and Aisen
PS: Intranasal NAP administration reduces accumulation of amyloid
peptide and tau hyperphosphorylation in a transgenic mouse model of
Alzheimer's disease at early pathological stage. J Mol Neurosci.
31:165–170. 2007.PubMed/NCBI
|
|
44
|
Ryder J, Su Y and Ni B: Akt/GSK3beta
serine/threonine kinases: Evidence for a signalling pathway
mediated by familial Alzheimer's disease mutations. Cell Signal.
16:187–200. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gassowska M, Czapski GA, Pajak B, Cieslik
M, Lenkiewicz AM and Adamczyk A: Extracellular α-synuclein leads to
microtubule destabilization via GSK-3β-dependent Tau
phosphorylation in PC12 cells. PloS One. 9:e942592014. View Article : Google Scholar : PubMed/NCBI
|