Introduction
Renal fibrosis is a common outcome of virtually all
progressive kidney diseases. It includes renal interstitial
fibrosis and glomerular sclerosis. Renal interstitial fibrosis is
the major cause of renal dysfunction. It is characterized by
excessive accumulation of extracellular matrix (ECM) in renal
interstitium and overproliferation of fibroblasts. Due to the high
solute transport activity of renal tubular epithelial cells,
sufficient energy supply by mitochondria is extremely important to
maintain normal renal function. Therefore, renal tubular epithelial
cells are sensitive to oxidative stress, which makes oxidative
stress a key factor of renal fibrosis. In the injured kidney,
levels of reactive oxygen species (ROS) may increase dramatically
and cause damage to the cell structure. Multiple signaling pathways
are involved in ROS transduction. Nuclear factor
κ-light-chain-enhancer of activated B cells (NF-κB) is a known
transcriptional factor that is redox-regulated by ROS (1). Activation of NF-κB triggers a series of
cellular processes, including inflammation, immunity, cell
proliferation and apoptosis.
Surfactant protein A (SP-A) is a newly identified
factor that has been associated with pulmonary fibrosis (2,3). SP-A is
encoded by two homologous genes: SFTPA1 encodes SP-A1 and SFTPA2
encodes SP-A2 (4). SP-A acts in a
dual manner depending on the binding orientation. When SP-A binds
signal regulatory protein α, the NF-κB pathway is inhibited to
prevent damage from excessive expression of inflammation factors;
when SP-A binds calreticulin (CRT), the production of
proinflammatory mediators is stimulated (5). CRT, also known as calregulin, CRP55 and
calsequestrin-like protein, is a multifunctional protein that binds
Ca2+ ions in endoplasmic and sarcoplasmic reticulum
(6). Overexpression of CRT as a
response to oxidative stress has been reported in previous studies
(7–9).
A recent study reported that SP-A was also involved
in renal fibrosis induced by unilateral ureter obstruction. SP-A
expression was induced in kidney epithelium due to increased
inflammation (10). Based on these
previous results, the present study hypothesized that SP-A2 may
contribute to the pathogenesis of renal fibrosis through binding to
CRT and influence the oxidative stress response.
Materials and methods
Cell culture and treatment
The human HK-2 cell line was obtained from the
American Type Culture Collection (Manassas, VA, USA) and used as a
model of proximal tubular cells. The cells were cultured in
Dulbecco's modified Eagle's medium/F12 (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum at 37°C in a humidified atmosphere containing 5%
CO2.
Cells were seeded into 6-well plates
(1.0×105 cells/well) and allowed to attach by incubation
for 24 h. Subsequently, 10 µg/ml SP-A was added, followed by
incubation for 30, 60 or 120 min. SP-A2 was isolated from whole
lung lavage fluid taken from patients with pulmonary alveolar
proteinosis as previously described (11). The lung lavage fluid was used with
the informed consent of 20 patients treated at Shanxi Dayi Hospital
(Taiyuan, China) between January 2015 and October 2016. This study
was approved by the ethics committee of Shanxi Dayi Hospital
(Taiyuan, China). Cells without SP-A2 treatment were used as a
negative control. To assess the role of SP-A in renal fibrosis
through binding to CRT, the cells were also pre-treated with
anti-CRT (10 µg/well; cat. no. sc-101436; 1:1,000; Santa Cruz
Biotechnology Inc., Dallas, TX, USA) for 1 h and then treated with
10 µg/ml of SP-A2 for 1 h. After the treatment, the cells were
harvested for further assays.
Measurement of ROS production
HK-2 cells were incubated in 96-well plates
(1×104 cells/well) and treated with SP-A2 with/without
anti-CRT as described above. The production of ROS (mostly
peroxide) was measured using a Reactive Oxygen Species Assay kit
(Beyotime Institute of Biotechnology, Haimen, China). Fluorescence
intensity was measured at an excitation wavelength of 488 nm and
emission at 525 nm using a fluorescence microplate reader.
Measurement of type I collagen
The culture media were collected for detection of
type I collagen. An ELISA kit for human type I collagen (Kamai Shu
Biotechnology, China) was used according to the manufacturer's
instructions.
Annexin V assay
The apoptotic rate was detected using an Annexin V
assay. Different cell groups were harvested with 0.25% trypsin and
washed with PBS. After centrifugation for 5 min, cells were
re-suspended and labeled using an Annexin V-FITC Apoptosis
Detection kit (Beyotime Institute of Biotechnology) according to
the manufacturer's instructions, followed by flow cytometric
analysis.
Western blot analysis
The cells were treated and harvested as described
above, then homogenized in radioimmunoprecipitation assay lysis
buffer (Beyotime Institute of Biotechnology) and centrifuged at
12,000 × g at 4°C for 30 min. The protein concentration was assayed
via the bicinchoninic acid method. Protein samples (30 µg of each
sample) were subjected to 10% SDS-PAGE and then transferred onto
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA), which were blocked with 5% skimmed milk at room temperature
for 1 h. The membranes were incubated overnight at 4°C with the
following primary antibodies: Mouse monoclonal antibody to matrix
metalloproteinase (MMP)2 (1:500 dilution; cat. no. sc-13594), MMP9
(1:500 dilution; cat. no. sc-21733), tissue inhibitor of matrix
metalloproteinases (TIMP)-1 (1:500 dilution; cat. no. sc-21734),
p38 mitogen-activated protein kinase (p38 MAPK; 1:500 dilution;
cat. no. sc-136210), phosphorylated (p)-p38 MAPK (1:1,000 dilution;
cat. no. sc-7973), p65 NF-κB (1:1,000 dilution; cat. no. sc-56735),
p-p65 NF-κB (1:1,000 dilution; cat. no. sc-136548), NADPH oxidase 2
(NOX2; 1:500 dilution; cat. no. sc-130543) and GAPDH (1:1,000
dilution; cat. no. sc-47724). Subsequently, the membranes were
incubated at room temperature for 1 h with horseradish
peroxidase-conjugated secondary antibody to mouse immunoglobulin G
(1:2,000 dilution; cat. no. sc-516102). All antibodies were
purchased from Santa Cruz Biotechnology, Inc. Blots were detected
using enhanced chemiluminescence (Beyotime Institute of
Biotechnology, Haimen, China).
Statistical analysis
Each experiment was performed in triplicate and
results were expressed as mean ± standard deviation. Statistical
analysis was performed using SPSS 20.0 software (IBM Corp., Armonk,
NY, USA). The significance of differences between groups was
determined using one-way analysis of variance followed by a
post-hoc Dunnett's test. P<0.05 was considered to indicate a
statistically significant difference.
Results
SP-A induces oxidative stress and
fibrosis in HK-2 cells
To determine whether SP-A induces oxidative stress
in HK-2 cells, the effect of SP-A on ROS production was first
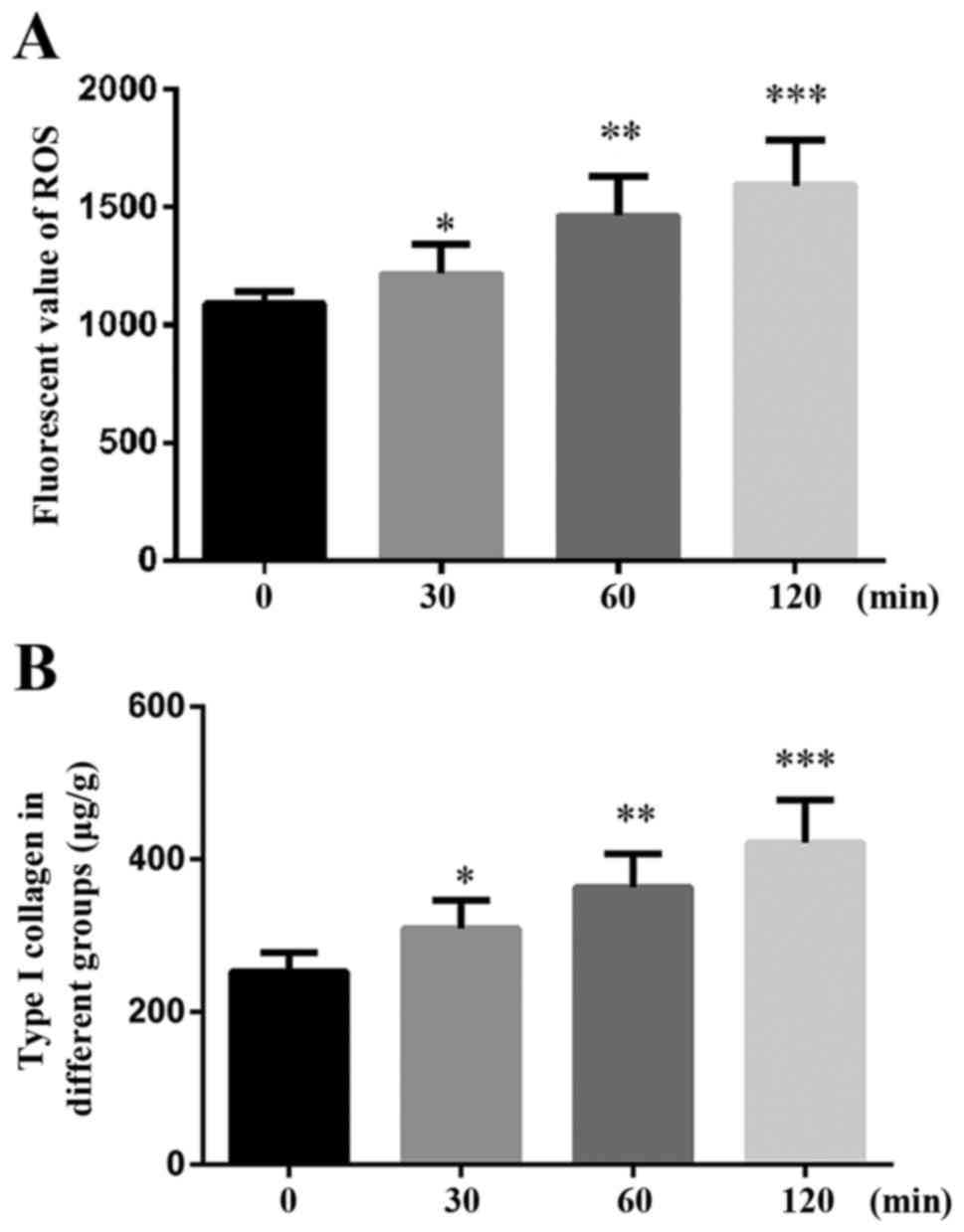
examined. As presented in Fig. 1A,
ROS production was increased in a time-dependent manner (30, 60 and
120 min).
Furthermore, the amount of Type I collagen was also
increased in a time-dependent manner, as demonstrated by the ELISA
(Fig. 1B).
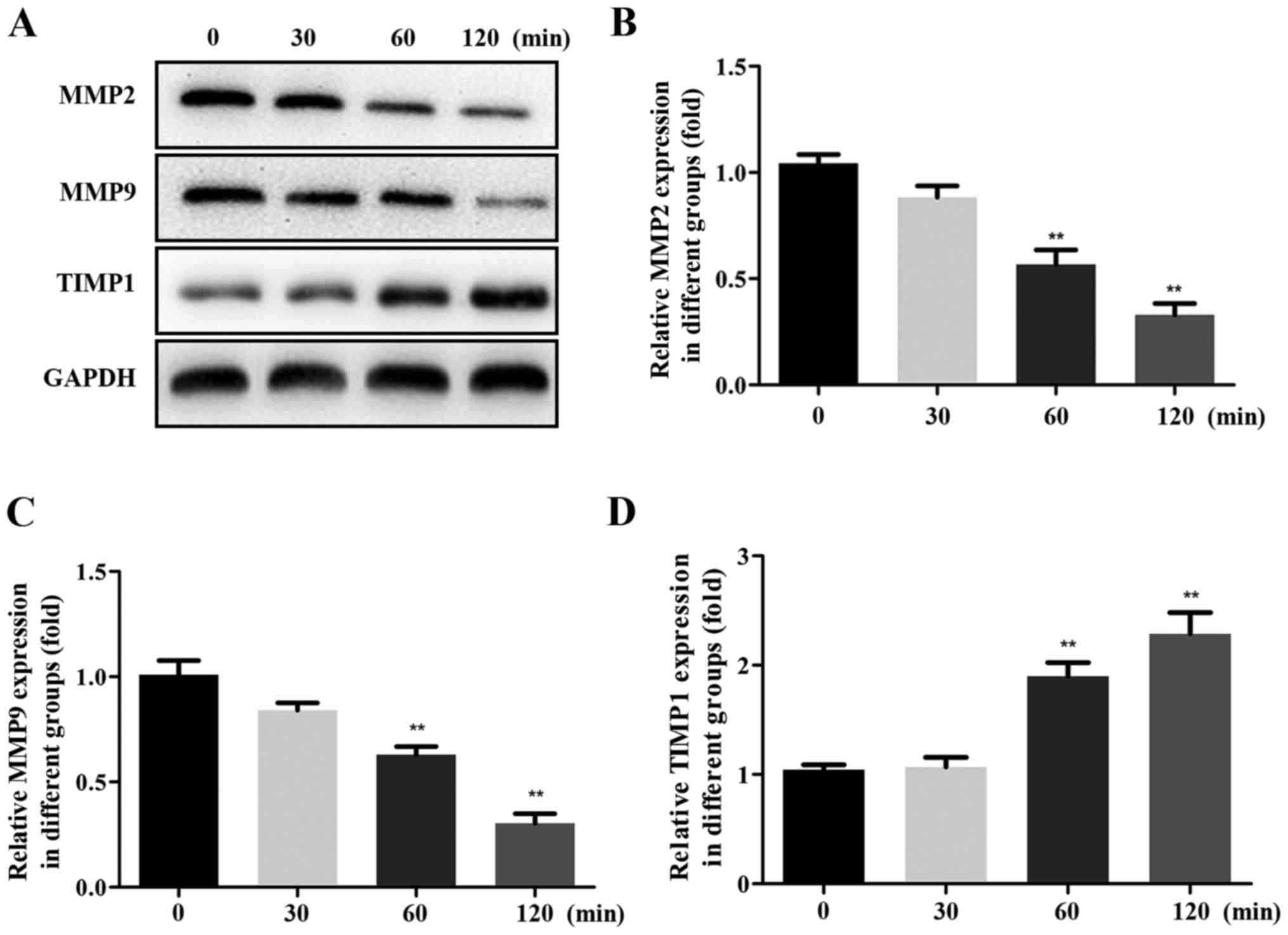
To characterize the effects of SP-A in inducing
renal fibrosis by MMPs, western blot analysis was performed to
measure the protein levels of MMP2, MMP9 and TIMP1. The expression
of MMP2 and MMP9 protein was decreased, while that of TIMP1 protein
was increased in a time-dependent manner (Fig. 2).
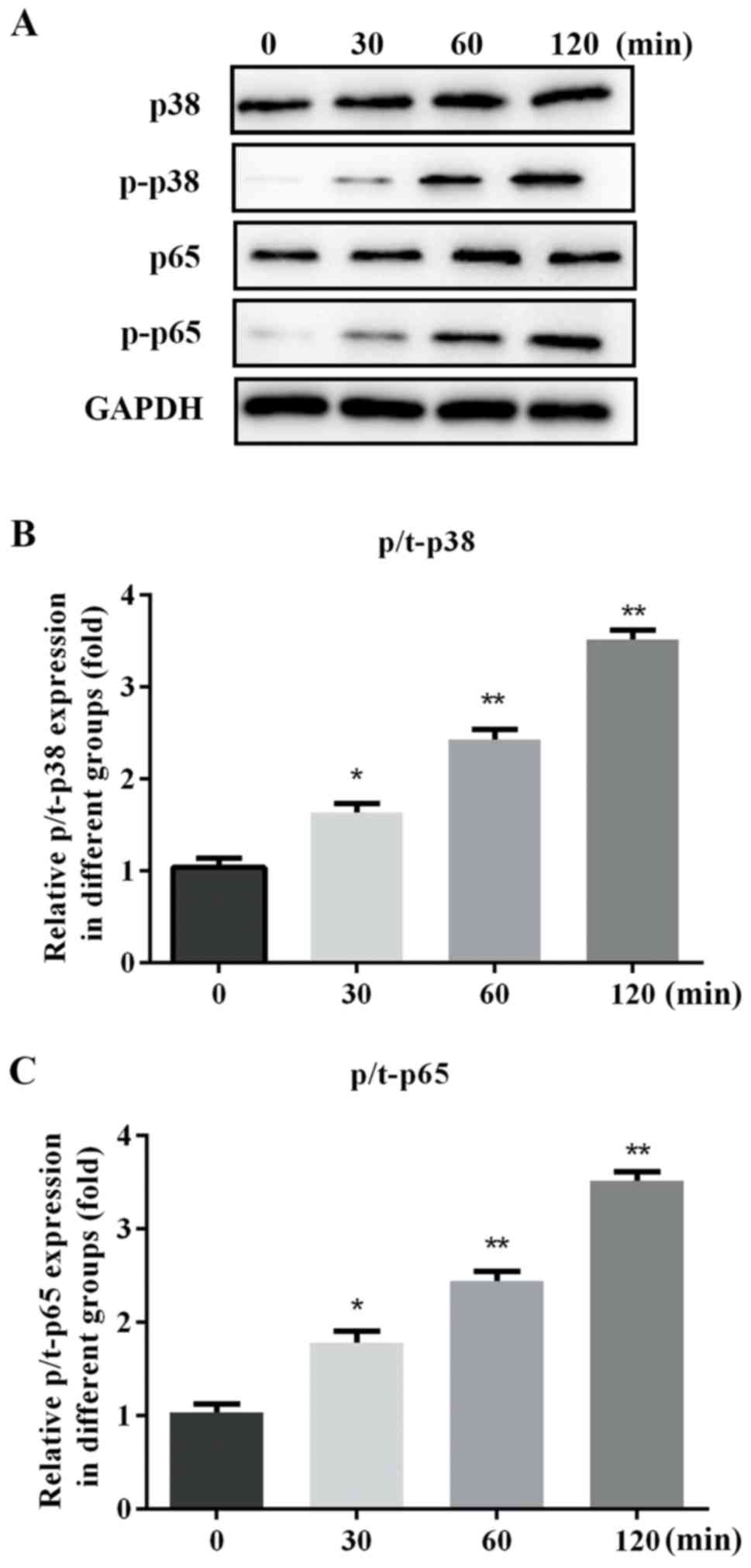
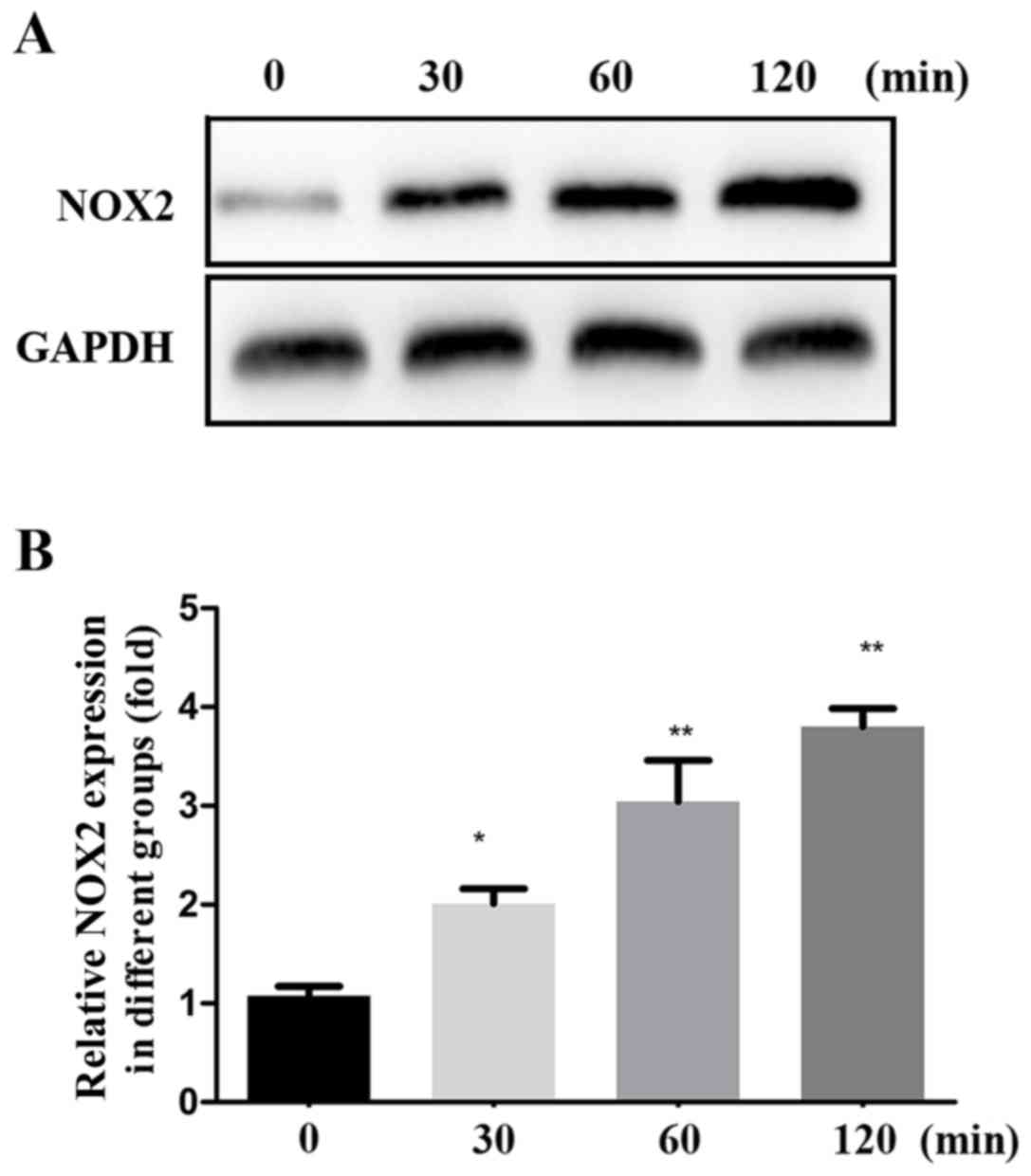
The mechanism of the renal fibrosis induction effect
of SP-A via the MAPK/NF-κB signaling pathways was further verified
by assessing the levels of MAPK p38, MAPK p-p38, NF-κB p65, NF-κB
p-p65 and NOX2 protein within total cell lysate. The expression of
p/t-p38, p/t-p65 and NOX2 proteins was gradually increased by SP-A
in a time-dependent manner (Figs. 3
and 4).
These results indicated that SP-A induces oxidative
stress and renal fibrosis in HK-2 cells.
Anti-CRT alleviates oxidative stress
and fibrosis in HK-2 cells induced by SP-A
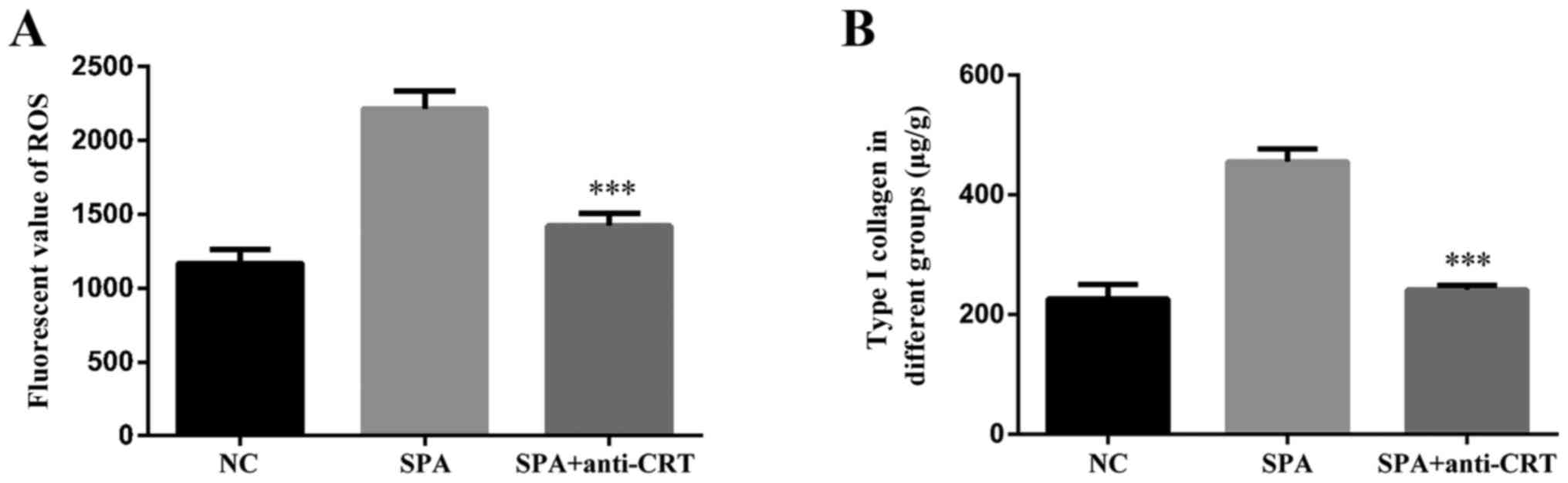
To determine whether SP-A induces oxidative stress
through binding to CRT, HK-2 cells were pre-treated with anti-CRT
for 1 h, and the production of ROS and the content of type I
collagen were assessed using a Reactive Oxygen Species Assay Kit
and ELISA kit. Addition of anti-CRT obviously reduced the
SP-A-induced production of ROS (Fig.
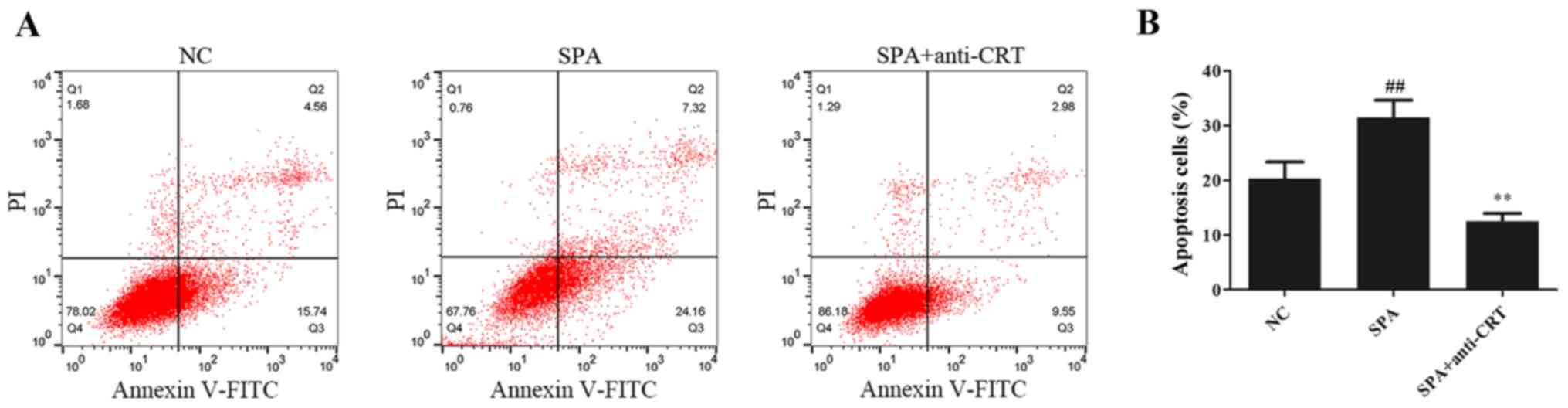
5A) and secretion of type I collagen by HK-2 cells (Fig. 5B). As presented in Fig. 6, compared with the control group,
SP-A treatment significantly promoted cell apoptosis and anti-CRT
partially alleviated this increase.
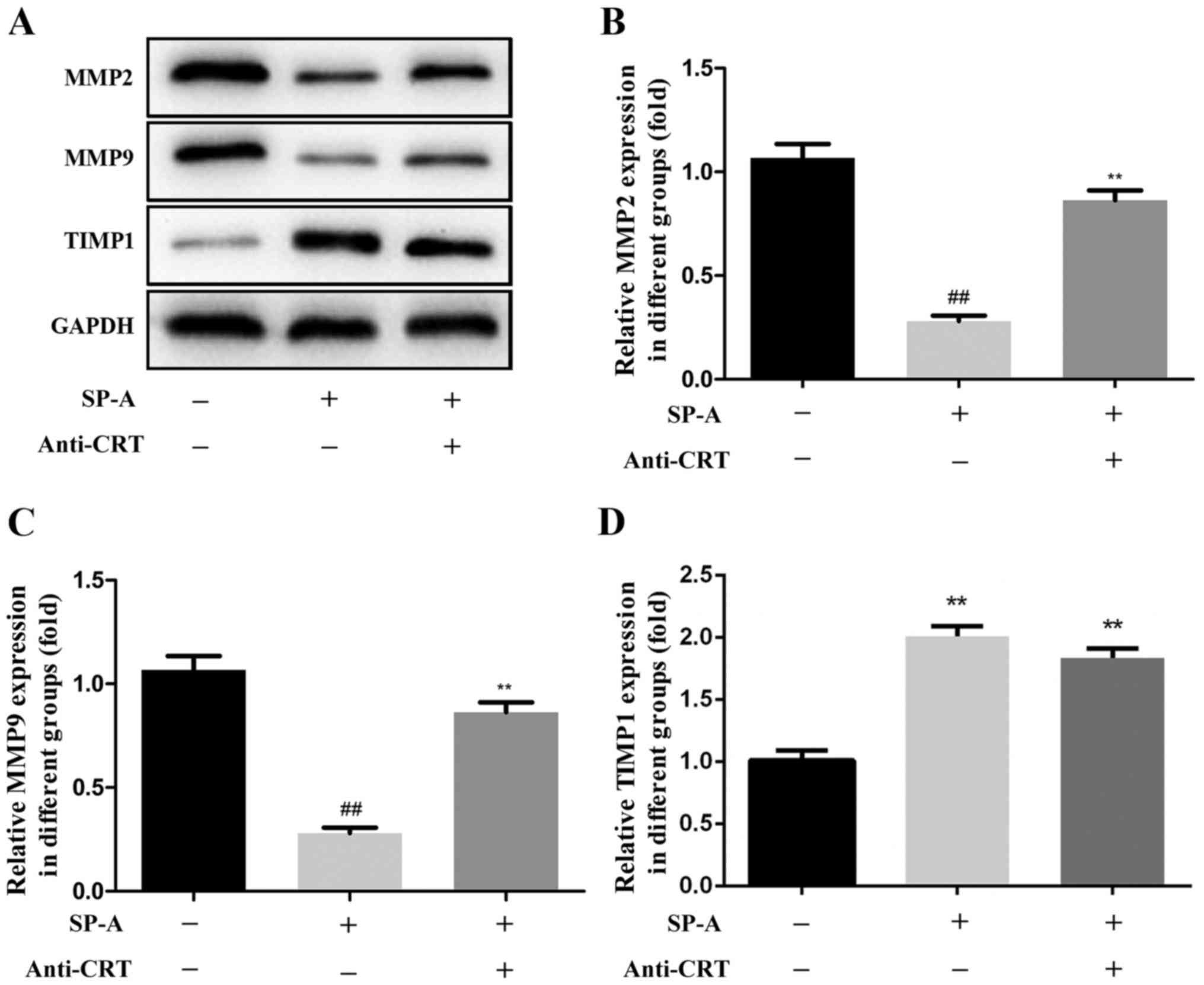
To characterize the effects of SP-A on MMPs through
binding to CRT, the levels of MMP2, MMP9 and TIMP1 protein were
assessed by western blot analysis. The addition of anti-CRT
inhibited the SP-A-induced decreases in the protein expression of
MMP2 and MMP9, and the increase in the expression of TIMP1 protein
(Fig. 7).
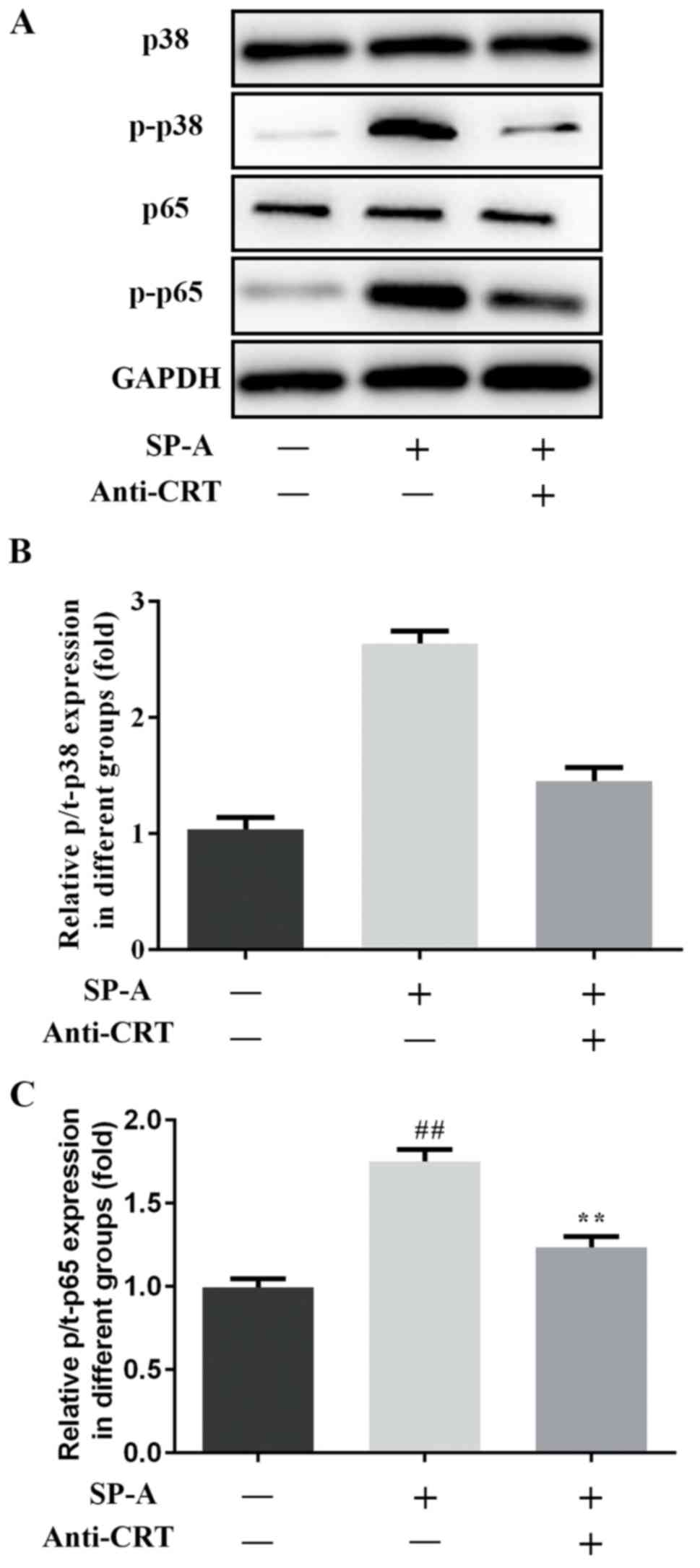
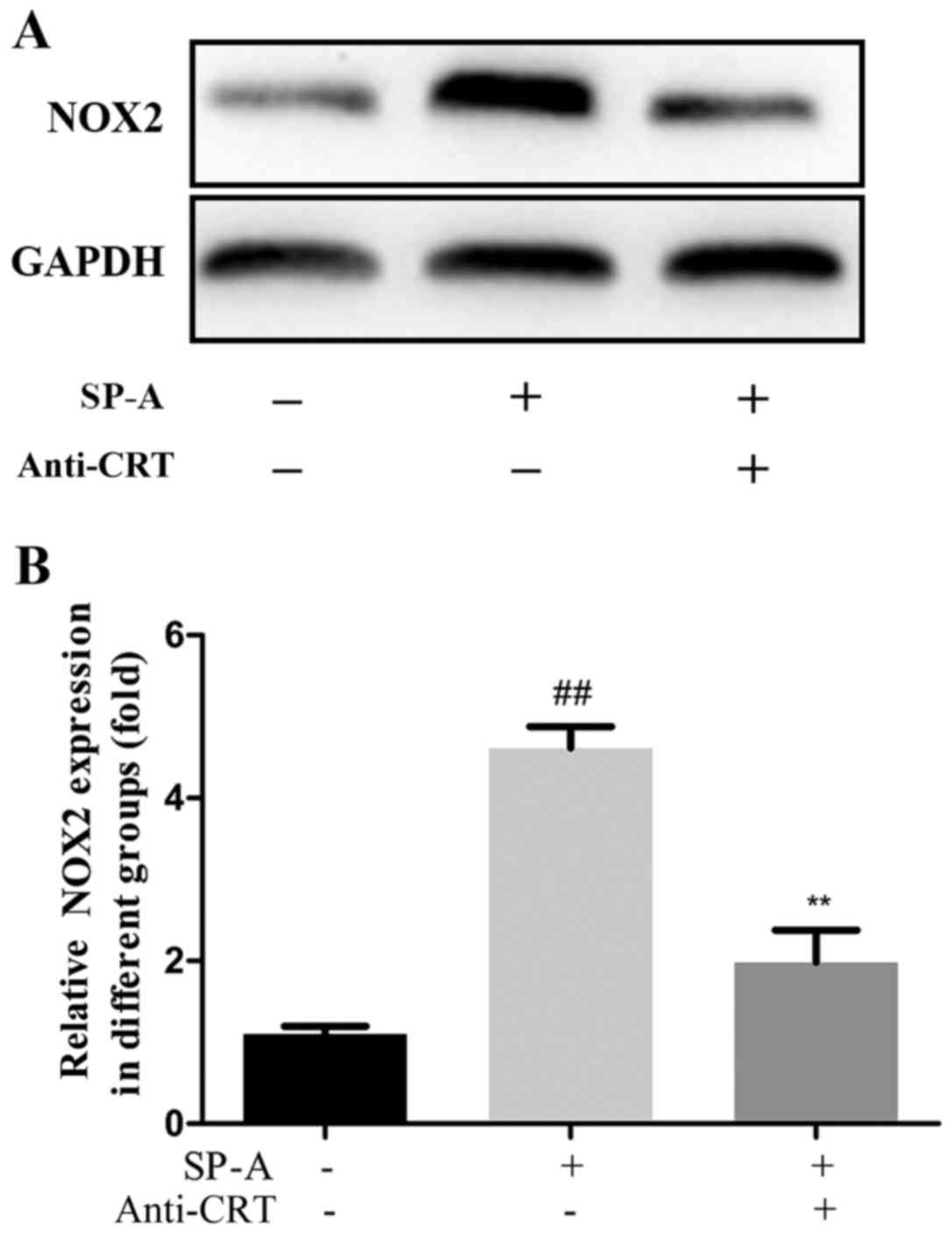
To further characterize whether SP-A has a
significant role in renal fibrosis through binding to CRT, the
levels of MAPK p38, MAPK p-p38, NF-κB p65, NF-κB p-p65 and NOX2
protein were assessed by western blot analysis. The expression of
these proteins was increased by SP-A, and this increase was
reversed by anti-CRT (Figs. 8 and
9).
These results suggested that anti-CRT alleviates
oxidative stress and fibrosis in HK-2 cells induced by SP-A.
Overall, SP-A has an important role in renal fibrosis through
binding to CRT.
Discussion
The main features of chronic kidney disease are
excessive deposition and fibrosis of ECM, leading to decreased
renal function. Renal interstitial fibrosis is a common pathway and
main pathological characteristic of chronic kidney disease
progression to end-stage renal disease (12–14).
SP-A is a novel factor that has been associated with pulmonary
fibrosis (2,3).
MMPs have a key role in the degradation of ECM and
promote the degradation of ECM (particularly Type I and Type III
collagen), while TIMPs have the effect of inhibiting the ECM, which
reduces the degradation of the ECM and leads to fibrosis.
MMPs/TIMPs have been confirmed to have a pivotal role in liver,
myocardial and lung fibrosis (15–17). In
the present study, western blot analysis was performed to determine
the protein expression of MMP2, MMP9 and TIMP1. The results
indicated that SP-A inhibits the expression of MMP2 and MMP9 and
promotes the expression of TIMP1, while the addition of anti-CRT
reduced these effects. In addition, ELISA indicated that SP-A
promotes the secretion of Type I collagen, which was attenuated by
the addition of anti-CRT. These results indicated that SP-A
regulates renal fibrosis through binding to CRT.
In the initial stage of renal fibrosis, numerous
factors affect the redox state of renal cells and macrophages to
stimulate the production of ROS. In the development stage of renal
fibrosis, various stimuli lead to multiple fibrogenetic signaling.
ROS are an important intracellular secondary messenger, activating
the MAPK/NF-κB signaling pathway (1,18).
Gardai et al (5) demonstrated
that SP-A regulates NF-κB p65 and stimulates the production of
proinflammatory mediators by binding to CRT. In the present study,
SP-A treatment increased the production of ROS, and anti-CRT
partially alleviated this increase. The western blot results
illustrated that the protein expression of MAPK p38, MAPK p-p38,
NF-κB p65, NF-κB p-p65 and NOX2 was increased by SP-A, while this
increase was inhibited by anti-CRT. These results confirmed that
SP-A upregulates the expression of p65 and stimulates the
production of proinflammatory mediators by binding to CRT.
In conclusion, the present results demonstrated that
SP-A may participate in the pathogenesis of kidney fibrosis and
activate the MAPK/NF-κB oxidative stress signaling pathway by
binding to CRT.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The analyzed data sets generated during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ wrote the manuscript and interpreted the data.
JH, XZ and WY analyzed the data and revised the manuscript. XH and
YH searched the literature and collected the data. JH designed the
study. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the ethics committee of
Shanxi Dayi Hospital (Taiyuan, China).
Patient consent for publication
The lung lavage fluid was used with the informed
consent of 20 patients treated at Shanxi Dayi Hospital (Taiyuan,
China) between January 2015 and October 2016.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gloire G, Legrand-Poels S and Piette J:
NF-kappaB activation by reactive oxygen species: Fifteen years
later. Biochem Pharmacol. 72:1493–1505. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Samukawa T, Hamada T, Uto H, Yanagi M,
Tsukuya G, Nosaki T, Maeda M, Hirano T, Tsubouchi H and Inoue H:
The elevation of serum napsin A in idiopathic pulmonary fibrosis,
compared with KL-6, surfactant protein-A and surfactant protein-D.
BMC Pulm Med. 12:552012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Takahashi H, Fujishima T, Koba H, Murakami
S, Kurokawa K, Shibuya Y, Shiratori M, Kuroki Y and Abe S: Serum
surfactant proteins A and D as prognostic factors in idiopathic
pulmonary fibrosis and their relationship to disease extent. Am J
Respir Crit Care Med. 162:1109–1114. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mccormack FX: Structure, processing and
properties of surfactant protein A. Biochim Biophys Acta.
1408:109–131. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gardai SJ, Xiao Y, Dickinson M, Nick JA,
Voelker DR, Greene KE and Henson PM: By binding SIRPalpha or
calreticulin/CD91, lung collectins act as dual function
surveillance molecules to suppress or enhance inflammation. Cell.
115:13–23. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang L, Wu G, Tate CG, Lookene A and
Olivecrona G: Calreticulin promotes folding/dimerization of human
lipoprotein lipase expressed in insect cells (Sf21). J Biol Chem.
278:29344–29351. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang Y, Liu L, Jin L, Yi X, Dang E, Yang
Y, Li C and Gao T: Oxidative stress-induced calreticulin expression
and translocation: New insights into the destruction of
melanocytes. J Invest Dermatol. 134:183–191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ihara Y, Urata Y, Goto S and Kondo T: Role
of calreticulin in the sensitivity of myocardiac H9c2 cells to
oxidative stress caused by hydrogen peroxide. Am J Physiol Cell
Physiol. 290:C208–C221. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ihara Y, Kageyama K and Kondo T:
Overexpression of calreticulin sensitizes SERCA2a to oxidative
stress. Biochem Biophys Res Commun. 329:1343–1349. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tian S, Li C, Ran R and Chen SY:
Surfactant protein A deficiency exacerbates renal interstitial
fibrosis following obstructive injury in mice. Biochim Biophys
Acta. 1863:509–517. 2017. View Article : Google Scholar
|
|
11
|
Allen MJ, Voelker DR and Mason RJ:
Interactions of surfactant proteins A and D with Saccharomyces
cerevisiae and Aspergillus fumigatus. Infect Immun. 69:2037–2044.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Boor P, Ostendorf T and Floege J: Renal
fibrosis: Novel insights into mechanisms and therapeutic targets.
Nat Rev Nephrol. 6:643–656. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Farris AB and Colvin RB: Renal
interstitial fibrosis: Mechanisms and evaluation. Curr Opin Nephrol
Hypertens. 21:289–300. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lawson J, Elliott J, Wheeler-Jones C, Syme
H and Jepson R: Renal fibrosis in feline chronic kidney disease:
Known mediators and mechanisms of injury. Vet J. 203:18–26. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hemmann S, Graf J, Roderfeld M and Roeb E:
Expression of MMPs and TIMPs in liver fibrosis-a systematic review
with special emphasis on anti-fibrotic strategies. J Hepatol.
46:955–975. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pei Z, Meng R, Li G, Yan G, Xu C, Zhuang
Z, Ren J and Wu Z: Angiotensin-(1–7) ameliorates myocardial
remodeling and interstitial fibrosis in spontaneous hypertension:
Role of MMPs/TIMPs. Toxicol Lett. 199:173–181. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Manoury B, Nénan S, Guénon I, Lagente V
and Boichot E: Influence of early neutrophil depletion on
MMPs/TIMP-1 balance in bleomycin-induced lung fibrosis. Int
Immunopharmacol. 7:900–911. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jian KL, Zhang C, Shang ZC, Yang L and
Kong LY: Eucalrobusone C suppresses cell proliferation and induces
ROS-dependent mitochondrial apoptosis via the p38 MAPK pathway in
hepatocellular carcinoma cells. Phytomedicine. 25:71–82. 2017.
View Article : Google Scholar : PubMed/NCBI
|