Introduction
Normal-tension glaucoma (NTG) is a type of
progressive glaucomatous optic neuropathy (GON). Its main
difference to primary open-angle glaucoma (POAG) is that NTG
presents with normal intraocular pressure (IOP) (1). The epidemiology of NTG varies, being
more prevalent among Asian populations, including Japanese and
Korean populations, than among Caucasians. NTG is also considered a
‘middle-age’ disease with the mean age of patients being in the 6th
decade of life. Of note, although IOP is measured within a normal
range in NTG, optic disc excavation and visual field (VF) defects
are detected (2). This suggests an
IOP-independent multifactorial background of NTG etiology. The main
factors contributing to NTG-related GON can be distinguished into
two main categories, namely vascular and mechanical. These factors
can lead to retinal ganglion cell (RGC) loss and axonal damage via
several pathways (3). In addition,
various genes have been found to be involved in NTG, thus
suggesting a genetic predisposition for NTG development (4).
The aim of this review was to summarize and analyze
the various pathogenetic factors which contribute to the
development of NTG-related GON and to present the recent research
findings on the genetics of NTG.
Vascular factors and ocular blood flow
The keystone of the vascular pathogenesis of GON in
NTG seems to be a disruption in ocular blood flow (OBF) (5). Although the measurement of OBF remains
challenging, with a great variety of available methods and no gold
standard method available, studies on patients with glaucoma have
indicated a reduction in OBF in ocular tissues (optic nerve, retina
and choroid), being more prominent in cases of progressive NTG than
in POAG (5,6).
The reduction in OBF is a multifactorial phenomenon.
The role of ocular perfusion pressure (OPP) is known, which is
equal to arterial blood pressure (BP) minus IOP, with IOP being
equal to retinal venous pressure (RVP) (7). Since IOP is within a normal range in
NTG, alterations in BP or RVP can influence OPP. Low OPP,
particularly if fluctuating, contributes to the progression of GON
(8). Indeed, low systemic blood
pressure, particularly during the night, reduces OBF via a
reduction in OPP, thus being a major risk factor of NTG
progression. Additionally, RVP is increased in patients with
glaucoma, resulting also in a lower OPP (9,10). These
facts indicate a component related to OPP reduction. As a result,
structural damage and low OPP constitute a secondary element of OBF
reduction.
However, a primary component may be involved.
Nailfold capillaries also demonstrate a reduction in blood flow,
which cannot be connected to glaucomatous damage or IOP. Moreover,
a low BP does not enhance GON progression in all cases (11,12). OBF
and OPP demonstrate a complex interaction in combination with local
flow resistance, with a reduction in resistivity as a regulatory
reaction to OPP reduction (5). There
is evidence to suggest that in subjects with low BP, a reduction in
OPP reduces OBF due to altered autoregulation and defective
adaptation (13). OBF reduction in
retrobulbar arteries with a distortion of vascular properties,
concerning resistance and pulsatility, was recently reported in
patients with NTG (14). In
addition, OBF reduction often precedes GON, being a predictor of
its progression (15). All these
observations have led to the term ‘vascular dysregulation’ which,
in general, describes the lack of maintaining an adequate tissue
blood supply, despite alterations in perfusion pressure, embracing
irregular systemic or local vasospastic/vasodilating phenomena
(5). Vascular dysregulation is
considered to be the key primary component of GON pathogenesis in
patients with NTG (12,16).
Despite this evidence regarding OBF reduction and
GON, OBF reduction in animal models through the administration of
endothelin-1 (ET-1), and in patients with multiple sclerosis, has
led to only mild optic nerve head (ONH) atrophy and minor
excavation (17,18). Thus, it has been proposed that an OBF
fluctuation initiates and promotes glaucomatous damage more
effectively than baseline OBF, a fact that has been proven
(5). OBF instability may be due to
high IOP or low BP fluctuation exceeding normal autoregulation, or
normal IOP and/or BP fluctuation in the case of altered
autoregulation (19). IOP
fluctuations are discussed below as a part of mechanical factors in
NTG. As regards vascular factors in the case of normal
autoregulation, systemic hypotension, particularly nocturnal drops,
is responsible for low BP, resulting in an OPP deficit. On the
other hand, improper autoregulation implies vascular dysregulation
(10,16).
Hence, below, we analyze systemic hypotension with
reference to nocturnal drops, and vascular dysregulation, as the
major vascular factors of NTG-related GON.
Systemic hypotension
BP plays a crucial role in NTG. Various studies have
indicated systemic hypotension as a common risk factor in patients
with NTG with ONH excavation, VF progression, and optic disc
hemorrhage (10,20). Notably, Orgul et al reported
that the majority of patients with NTG with systemic hypotension
also present vasospasm (65%) (21),
indicating an association between low BP and vasospastic disorders,
possibly reflecting the additional role of vascular dysregulation
(21,22). As previously mentioned, hypotension
through OPP reduction creates OBF reduction/fluctuation,
particularly when low BP exceeds the capacity of vascular
autoregulation, resulting in perfusion deficits and ischemic
injuries in optic nerve fibers (10).
These alterations are more prominent in sustained
nocturnal BP drops, which are more frequent among NTG cases, almost
always being observed in patients with NTG with vasospasm (10,23). A
meta-analysis of the correlation between GON and the circadian
rhythm of BP, including NTG cases, reported that nocturnal BP drops
>10% of diurnal BP were a risk factor for VF defect progression
(24). A prospective study was
conducted including 85 patients with NTG, and BP was monitored over
a 48-h period in 30-min intervals and during follow-up at 6 and 12
months. VF defect progression was observed in 29% of patients, with
a significant predictor of it being sustained nocturnal BP drops 10
mmHg lower compared to diurnal BP (23). Consequently, systemic hypotension,
particularly nocturnal BP drops, may be an important risk factor
for GON and VF defect progression in NTG, although these findings
are still under debate (25).
Vascular dysregulation
It has been suggested that vascular dysregulation is
a primary step initiating glaucomatous damage in NTG via OBF
instability, affecting optic nerve and retina by the enhancement of
ischemic damage, as well as by promoting the apoptosis of RGCs and
their axons (19). The main cause is
a systemic form of vascular dysregulation, accompanied by symptoms
and signs, thus constituting a ‘vascular dysregulation syndrome’.
Considering its primary nature in NTG pathogenesis, the term
primary vascular dysregulation syndrome or Flammer syndrome (FS) is
used (26,27). Other components of vascular
dysregulation include migraines, systemic vascular diseases and
endothelial dysfunction (28,29). All
of these are considered risk factors for GON in NTG cases (4).
Primary vascular dysregulation or
FS
Josef Flammer, who first reported this condition,
described it as a combination of signs and symptoms together with
primary vascular dysregulation, occurring idiopathically in
individuals with no vascular disease and resulting in transiently
impaired adaptation of blood flow to tissue needs (12,35).
FS is more prevalent among females, Asians, slender
subjects and academics, with the initial symptoms becoming evident
during puberty (26,27). Although there is no gold standard
diagnostic method, diverse signs and symptoms are indicative of FS.
Subjects with FS usually have cold hands or/and feet, a reduced
feeling of thirst, low BP with pronounced nocturnal drops, an
enhanced drug sensitivity, are more susceptive to migraines, and
they often have a prolonged sleep onset time (11,30–33).
As regards circulation, subjects with FS display an
inborn tendency towards an altered response to various stimuli,
such as cold or stress (34). The
most overt pathological reactions are irregular vasoconstrictions
(vasospasms) (35); hence, FS was
firstly named ‘vasospastic syndrome’ (36). In these subjects, the peripheral
vessels, particularly in their skin and extremities are
constricted, in order to decrease heat loss. In addition, the
vascular properties of peripheral circulation in patients with FS
(e.g., in nailfold capillaries) are associated with their ocular
vessels and OBF (34,35).
Although the majority of subjects with FS are
healthy, they generally demonstrate several ocular signs (26). Their retinal vessels present less
flexibility, due to high pulse wave conduction, and higher spatial
variability (37). They also exhibit
larger choroidal vasoconstriction as a response to a hand-grip test
(38). Subjects with FS also have
altered responses to BP and IOP due to vessel dysregulation,
resulting in an instability of OPP and OBF (8,27). FS is
often observed in patients with NTG with progressive GON,
contributing to its prevalence and pathogenesis, being a crucial
risk factor (26,27,35). FS
in NTG is associated with an increased RVP and blood flow
resistance in retrobulbar vessels, diffuse VF defects and splinter
optic disc hemorrhages, possibly being the main cause of the latter
(26,39). Moreover, patients with FS with
glaucoma tend to have low diurnal BP, patients with nocturnal drops
present similar retrobulbar dynamics with patients with NTG with
FS, and NTG cases with systemic hypotension and/or nocturnal drops
are largely accompanied by vasospasm (21,31).
These facts indicate a possible interrelation between FS and
systemic hypotension in NTG.
Therefore, FS leads to GON in patients with NTG
mainly due to low BP, increased RVP and improper autoregulation,
acting either combined or independently, resulting in OBF
instability (31).
Migraines
A number of studies have suggested an association
between migraines and NTG, with migraines being a risk factor for
NTG progression (4,20,40).
Migraines have been described as a vasospastic disorder, frequently
observed in females and particularly in patients with NTG in
contrast to those with POAG or control subjects (28). Corbett et al, using various
neurological investigations including imaging techniques (CT and
ECG) in NTG cases, with 44% of them having a positive migraine
history, revealed that ischemia related to migraine possibly
conduces to NTG pathogenesis (40).
The ‘Low-Pressure Glaucoma Treatment Study’ reported migraines as a
predicting factor of optic disc hemorrhage in NTG cases (20).
All of the above suggest a common vascular basis of
migraines and NTG. Migraines are related to temporary vasospasm,
leading to the disruption of autoregulation in cerebral blood flow.
Moreover, rapid VF impairment in NTG cases with silent cerebral
infarct signifies an additional role of cerebral ischemic damage in
GON progression. Impotent vascular autoregulation in subjects with
GON can result in various ischemic phenomena, including
microinfarctions in ONH and optic disc hemorrhage (19,41).
Thus, migraines may be an important indicator of microvascular
dysregulation related to NTG (20).
Systemic vascular diseases
Various autoimmune and inflammatory vascular
diseases, such as multiple sclerosis, rheumatoid arthritis, lupus
erythematosus, giant cell arteritis, antiphospholipid syndrome,
Berger's disease and pre-eclampsia, among others, have been linked
to vascular dysregulation as a secondary effect of these conditions
(19,22). However, it has been suggested that
these diseases have a minor impact on autoregulation, hence leading
to OBF reduction via increased levels of ET-1 in circulation
(26). In addition, a common
characteristic of these conditions is the presence of vasospasm
which, although transiently occurs, it is well known to be involved
in ONH damage in NTG (22,35).
On the whole, considering these vascular diseases,
vasospasm and increased plasma ET-1 levels seem to be responsible
for OBF dysregulation in NTG.
Endothelial dysfunction
Microcirculation, including retrobulbar
microvessels, is mainly regulated through secretion of
vasoregulatory factors from endothelial cells, namely nitric oxide
(NO) and ET-1, affecting the vascular smooth muscle tone (42). Consequently, an impairment of these
factors, or endothelial dysfunction, alters vascular regulation. NO
primarily induces vasodilation. Oxidative stress is the major
mechanism which impairs NO signaling. NO has also been reported to
be involved in ocular autoregulation, presenting an endothelial and
neuronal protective role against pathologic alterations in glaucoma
(43).
By contrast, ET-1 significantly promotes
vasoconstriction interacting with its receptors ETA and
ETB. A number of studies have indicated an association
between plasma ET-1 levels and NTG pathogenesis (44–47).
Increased plasma ET-1 levels have been described in NTG cases
compared to controls, being higher at the primary stage of VF loss,
whereas normal levels have also been described (44,46).
Other studies have revealed abnormal ET-1 levels only following a
change in body position or cold stimulation, suggesting that
vascular dysregulation is probably involved in NTG-related GON
(45,48).
Su et al reported a generalized endothelial
dysfunction in patients with NTG, by utilizing the brachial artery
ultrasound assessment of endothelium-dependent flow-mediated
vasodilation (49). Henry et
al observed an altered peripheral vasodilation, caused by
abnormal vascular response to endothelial vasoregulatory factors,
due to impaired properties of ET receptors (50). Buckley et al, via cutaneous
artery biopsies in patients with NTG, identified a defect in the
release of vasodilating factors and enhanced sensitivity to ET-1,
contributing to vasospasm (29).
These findings demonstrate a systemic endothelium-derived vascular
dysfunction in NTG. The structural and functional affinity between
ocular and acral vasculature implies a similar dysfunction causing
OBF disturbance in NTG (34). Of
note, ET-1 largely exerts its vasoconstrictive effects on retinal
microcirculation, and vascular inadequacy in posterior ciliary
arteries supplying ONH has been indicated through color Doppler
imaging (46), suggesting that such
a local ET-1-dependent dysregulation could negatively affect OBF in
the ONH and adjacent retina (22).
FS is the most important cause of vascular
dysregulation in NTG (27). An
endothelial dysfunction presenting high ET-1 and low NO plasmatic
levels, may be the basis of FS, an imbalance that could disrupt
OBF, particularly in the ONH, through irregular retrobulbar
vasoconstriction (47). Moreover, an
endothelial dysfunction has been reported in migraines (51). Plasma ET-1 levels have been
positively associated with the resistivity index of retrobulbar
vessels, as well as with an improper neuro-endothelial function via
vasospasm in NTG cases (47).
To sum up, these findings indicate that an
endothelial dysfunction seems to be the connective factor between
systemic or local vascular dysregulation and irregular retrobulbar
hemodynamics including vasoconstriction, which can induce OBF
impairment occurring either primarily, promoting vasospasm and GON,
or secondarily as a result of these changes in NTG.
Mechanical factors
The mechanical pathogenesis of NTG includes IOP and
lamina cribrosa.
The role of IOP. Although an elevated IOP leads to
GON through a recognized pathophysiological pathway, the normal IOP
range in NTG implies that other mechanisms may coexist inducing
glaucomatous damage (5).
The ‘Collaborative Normal-Tension Glaucoma Study’,
which was conducted among 145 patients with NTG, reported that a
30% IOP reduction, even though favorable, did not prevent the
progression of VF defects, indicating that IOP was not solely
involved in GON (52). Agnifili
et al observed the presence of conjunctival epithelial
microcysts in NTG cases using in vivo confocal microscopy,
suggesting an alteration in aqueous humor hydrodynamics concerning
trans-scleral outflow in NTG (53).
Epidemiological studies have demonstrated that NTG is more
prevalent in Asian (52–92%) compared with Caucasian (30–39%)
glaucoma cases, possibly reflecting a demographically different
tolerance and/or genetic susceptibility to IOP (2,52). IOP
evaluation through contact lens sensor has revealed notable IOP
fluctuations, presenting nocturnal acrophase and prolonged IOP
peaks in 80% of NTG individuals (54). High IOP peaks (>90 mV) have been
linked to glaucomatous progression and IOP fluctuations can lead to
OBF instability in view of disturbed autoregulation (55,56).
Furthermore, an elevated nocturnal IOP in the supine position may
contribute to ONH and/or VF damage in NTG (57). FS, the major cause of vascular
dysregulation, also prevalent among Asian populations, may play an
important role in IOP-induced damage in NTG. Moreover, an altered
ONH morphology has been demonstrated in patients with NTG, with
deeper and larger optic disc cups than POAG cases (58). Various studies have reported a
thinner central corneal thickness in NTG than POAG and normal
individuals which is also associated with VF defects, although
these findings are considered controversial (59–61).
Thus, the above-mentioned findings create the
hypothesis of a lower threshold for IOP-related damage in some
patients with NTG as regards epidemiology, ocular morphology and
dysregulation, where normal IOP and its fluctuations can induce
mechanical stress and axonal damage in ONH.
The impact of lamina cribrosa. Lamina cribrosa (LC)
is a thin mesh-like collagenous tissue containing non-myelinated
RGC axons, forming a barrier between intraocular and orbital
subarachnoid spaces (SAS). It has been hypothesized that the
trans-laminar pressure gradient (TLPG), expressed as the difference
between IOP and intracranial pressure (ICP), is associated with
mechanical damage to the optic nerve fibers in NTG (62,63).
An elevated retrograde TLPG through a reduction in
ICP can induce optic nerve damage, either baro-traumatically or
through damage to the capillaries, resulting in a posteriorly
displaced LC (64). Ren et al
indicated a significantly reduced ICP using lumbar puncture in NTG
regarding POAG or normal individuals, in agreement with the
above-mentioned theory (65). A
meta-analysis also reported a significantly reduced ICP and
elevated TLPG in NTG and POAG than in normal subjects (66). Furthermore, multivariate analyses
have revealed that TLPG was positively associated with perimetric
VF defects and negatively with the neuroretinal rim area in both
glaucoma types (65,66). Nevertheless, other studies on
patients with NTG have argued that ICP is normal and that TLPG is
not significantly associated with VF defects (67,68). A
recent prospective study on 13 NTG cases demonstrated a normal TLPG
and ICP, presenting no association with VF defects (68). Therefore, the issue of TLPG in NTG
pathogenesis remains a matter of debate. Future methodological
improvements may provide a more accurate assessment of TLPG,
clarifying its role in NTG.
Jonas et al described a distinct difference
in the appearance of optic nerve head in highly myopic eyes
(refraction more than −8.00 diopters), with significantly larger
discs and of a more oval configuration, which may render highly
myopic eyes more susceptible to nerve fibre loss (69).
ET-1 and its receptors may also interact with LC
function. Rao et al described an altered function and
expression of ETA and ETB receptors in LC
cells following prolonged stimulation by ET-1 (70,71).
This implies that high ET-1 levels, a frequent observation in NTG
cases, may affect LC via the remodeling of the extracellular matrix
(ECM), thus causing LC deformation. An impaired LC structure and
function can influence local RGC axons, astrocytes and capillaries,
enhancing sensitivity to IOP-related stress in NTG (63).
Obstructive sleep apnea/hypopnea
syndrome
There is recent evidence to suggest the involvement
of obstructive sleep apnea/hypopnea syndrome (OSAHS) in NTG-related
GON (72). OSAHS is characterized by
upper airway obstruction during sleep. NTG is particularly
prevalent among patients with OSAHS. Elevated IOP, VF disruption,
glaucomatous disc alterations and thinning of the retinal nerve
fiber layer have been reported in OSAHS cases (72,73). GON
pathogenesis in OSAHS includes both vascular and mechanical factors
(72).
Repetitive sustained upper airway obstruction leads
to recurrent hypoxemia (and thus hypoxia) and hypercapnia, in
addition to increased vascular resistance which derives from
enhanced sympathetic stimulation. Transient hypoxia combined with
increased vascular resistance can harm the vascular endothelium,
thus producing endothelial dysfunction and impaired autoregulation,
possibly disturbing blood flow to the optic nerve and retina
(74). Such an unstable oxygen
concentration can directly cause insults to the optic nerve fibers,
or indirectly via reperfusion and subsequent oxidative stress and
inflammation (75). A diminished
cerebral perfusion pressure may also affect blood flow to the optic
nerve (76). Moreover, patients with
OSAHS present an IOP nocturnal acrophase, which is associated with
the supine position and immoderate orbital adipose tissue due to
obesity (73,77). Elastic fiber depletion in LC is also
observed in patients with OSAHS, increasing the glaucomatous risk
(72).
The neurovascular hypothesis of RGC damage
in NTG
RGC cells and their axons, endothelium and glial
cells, such as astrocytes, form a ‘neurovascular unit’, which
contributes to the homeostasis of the microenvironment and OBF in
ONH and retina, via vascular autoregulation, glial support, balance
between ET-1 and NO, trophic supply, proper blood-brain barrier
(BBB) and controlled immunity (19,78–80). Any
disruption in these factors, mostly due to IOP-independent
mechanisms, can damage the ONH and retina, resulting in NTG-related
GON.
A primary pathogenetic step seems to be the OBF
instability leading to RGC loss (78,81). As
demonstrated above, FS, migraines, vascular diseases, OSAHS,
nocturnal systemic hypotension, IOP fluctuations and LC deformation
induce OPP impairment and vascular dysregulation through complex
mechanisms, thus resulting in an unstable OBF, a prominent finding
of NTG (9,22,25). A
common characteristic of dysregulation in these conditions is an
interplay between vasospasm and an ET-1-dependent endothelial
dysfunction, occurring either primary or secondary to other
changes, which can harm autoregulation and increase ONH
susceptibility to vascular challenges (22). It should be noted that in particular,
the short posterior ciliary arteries supplying the ONH and choroid
are more sensitive to OPP alterations and vascular dysregulation
than the central retinal artery, possibly indicating ONH as the
initial location of damage (82).
Although an improper OBF can directly lead to optic nerve atrophy
and RGC loss through major sustained hypoxic insults, creating
tissue infarction, such events rarely occur in NTG. Therefore, it
is a mild, repetitive and reversible hypoxic pattern, namely an
unstable oxygen tissue supply, as a result of oxygen saturation
fluctuation due to impaired OBF and OSAHS, which contributes to
glaucomatous damage. This phenomenon is known as
ischemia-reperfusion injury (IRI) (35,36). The
main effect of IRI is the induction of chronic oxidative stress,
particularly in local mitochondria of RGCs and their axons
(36,83).
Oxidative stress is a major factor of NTG-related
GON. The reduction in the oxygen concentration impairs electron
flow in mitochondrial complexes, which results in the formation of
free radicals or reactive oxygen species (ROS), mainly superoxide
(O2−), increasing the hydrogen peroxide
(H2O2) concentration.
H2O2 further impairs electron flow and
increases ROS production, leading to mitochondrial dysfunction.
High or sustained ROS levels induce oxidative damage to the
mitochondria, as well as to cellular proteins and DNA, causing
insults to RGCs and their axons (84,85). The
upregulated expression of proteosome 20S α subunit and numerous DNA
breaks in circulating lymphocytes in glaucoma cases support the
hypothesis of oxidative stress involvement in GON (86). In addition, hypoxia and oxidative
stress upregulates hypoxia-inducible factor-1α (HIF-1α) in the
optic nerve and retina, which stimulates p53 and caspase, leading
to RGC apoptosis (87). RGC axons in
ONH, particularly in the prelaminar region, are mostly
non-myelinated and consequently present high density of
mitochondria due to high energy demands. Thus, ONH is the major
site of mitochondrial dysfunction as a result of oxidative stress
creating energy deficiency (35). LC
cells also demonstrate significant mitochondrial dysfunction and
enhanced ROS production with impaired antioxidant capacity in
glaucoma (88).
Concurrently, glial cells vigorously respond to
microenvironment alterations, leading to gliosis (35). Gliosis is defined as an activation of
glial cells presenting hypertrophy and proliferation. Mechanical
stress (IOP fluctuations, LC deformation and increased TLPG),
through the release of epidermal growth factor receptor (EGFR),
elevated ET-1, IRI, ROS and oxidative stress can result in gliosis
(35,85). Indeed, stimulated astrocytes and
microglia have been observed in glaucoma, promoting demyelination
and damage to the axons (89). Apart
from ONH, gliosis also occurs in the retina through chronic
stimulation of Müller cells, causing the apoptosis of RGC bodies
via degenerative and inflammatory processes. Activated glial cells
further disrupt the microenvironment in ONH and retina through the
release of various factors being involved in neurodegeneration
(90). The astrocytic release of
ET-1 further impairs OBF, mediates RGC apoptosis and disrupts
axonal transport (91). The glial
production of tumor necrosis factor-α (TNF-α) and interleukin-1β
(IL-1β) can induce the apoptosis of RGC and axons through binding
to TNF receptor and the activation of nerve factor (NF)-κB,
respectively, leading to an enhanced inflammatory response
(90).
Moreover, the cytokine-mediated upregulation of NO
synthase-2 (NOS-2) by glial cells has been shown to elevate local
NO levels (35). NO, although
harmless, diffuses into RGC axons in ONH where it reacts with high
levels of superoxide, due to intense local mitochondrial
dysfunction and oxidative stress, creating peroxynitrite
(ONOO−). Peroxynitrite, as well as superoxide, are able
to diffuse along the RGC axons into both the retina and lateral
geniculate nucleus, leading to neuronal apoptosis through oxidative
DNA damage (92).
Glutamate seems to play an important role in GON.
The extracellular glutamate concentration in retina is maintained
below neurotoxic levels by uptake via glutamate/aspartate
transporter (GLAST) into Müller glial cells. Oxidative stress could
increase extracellular glutamate due to ROS-induced reuptake
failure of glutamate, a fact which is evident in NTG animal
studies. An impaired glial GLAST function, possibly due to genetic
polymorphisms may be responsible for elevated glutamate levels, as
reported in transgenic mice, although in vivo evidence in
NTG is needed (84,92). Glutamate neurotoxicity is mediated
through the over-stimulation of N-methyl-D-asparate (NMDA)
membrane receptors by glutamate in RGCs enhancing intracellular
Ca2+ influx, which activates various apoptotic pathways,
damaging the mitochondria, endoplasmic reticulum and DNA (93). Additionally, an NMDA-related
activation of NF-κB has been detected in Müller glial cells,
leading to TNF-α formation and RGC apoptosis via TNFR signaling and
Ca2+ influx (94). This
glutamate-induced damage is known as ‘excitotoxicity’.
Oxidative stress, IRI and cell damage, upregulate
ET-1 release in NTG, as a result of activated astrocytes and
endothelial dysfunction (36,91).
There are diverse mechanisms of ET-1-related GON in NTG. ET-1
induces chronic ONH ischemia through irregular vasoconstriction and
enhances glutamate toxicity in retina through ETA
receptors. In addition, ET-1 promotes astrocyte activation and
amplifies neuroinflammation through ETB receptors
(18,44). Combined ETA and
ETB stimulation by increased ET-1 promotes superoxide
synthesis and NOS upregulation (95), disturbs retrograde and anterograde
axonal transport in optic nerve, contributes to ECM remodeling in
ONH, particularly in LC (70,71), and
causes RGC apoptosis (91,93,95).
Moreover, tissue remodeling triggered by activated
astrocytes and mechanical stress occurs concurrently to the damage
in RGC and axons, leading to the overproduction of
metalloproteinases (MMPs), which enzymatically alter ECM morphology
and composition (63). Upregulated
MMP-2 and MMP-9 levels have been detected both locally in ONH, as
well as in circulating lymphocytes in glaucoma (96). Therefore, tissue remodeling
contributes to ONH excavation, as well as LC bowing and deformation
(27).
BBB damage has been indicated in NTG (97). Capillaries of the retina and ONH
present a complete BBB, with endothelial tight-junctions between
adjacent endothelial cells. However, capillaries in prelaminar
region of ONH have an improper BBB, and choroid vessels are highly
fenestrated (98). Although elevated
plasma ET-1 levels exert a neutral effect in retinal and ONH
vessels when BBB is intact, ET-1 can diffuse from choroid
capillaries into the ONH and adjacent retina, resulting in
vasoconstriction, impaired ONH blood flow, increased RVP and
weakening of endothelial tight-junctions. The hypoxic retina
upregulates HIF-1α, stimulating the release of ET-1 in local
vessels, which also leads to vasoconstriction and damage to the BBB
(97). Simultaneous ET-1 and MMP-9
diffusion from the choroid into the ONH can disrupt endothelial
tight-junctions and basal membrane with the subsequent leak of
erythrocytes, clinically observed as optic disc hemorrhage in NTG
(97,99). BBB damage also includes the
extravasation of proteins, tissue deposition of toxins such as Aβ,
and the release of plasma ET-1 and MMP-9 promoting BBB damage
(96,100).
The aforementioned factors may be involved in RGC
loss and in the synthesis of inflammatory antigens or cytokines.
Moreover, interplay between oxidative stress and damage, ROS
secretion and gliosis can cause an aberrant immune response due to
the loss of normal immunosuppression, increased antigenity, and
increased antigen presentation or exposure. All of these combined
can result in immune dysregulation and possibly trigger autoimmune
neurodegenerative processes (87,90).
Indeed, the presence of elevated levels of anti-phosphatidylserine
and anti-rhodopsin antibodies in the serum of NTG patients has been
reported, pointing towards an autoimmune aspect of NTG pathogenesis
(27).
Finally, mechanical and oxidative stress,
astrocytes and microglia activation, ET-1, lateral geniculate
nucleus (LGN) degeneration and axonal damage disrupt the signaling
and bioactivity of neuronal growth factors, including brain-derived
neurotrophic factor (BDNF), vascular endothelial growth factor
(VEGF) and nerve growth factor (NGF), causing a diminished
neurotrophic support negatively impacting axonal myelination in ONH
and survival of RGC in the retina (101).
Fig. 1 summarizes
the interaction of the aforementioned mechanisms in NTG
pathogenesis.
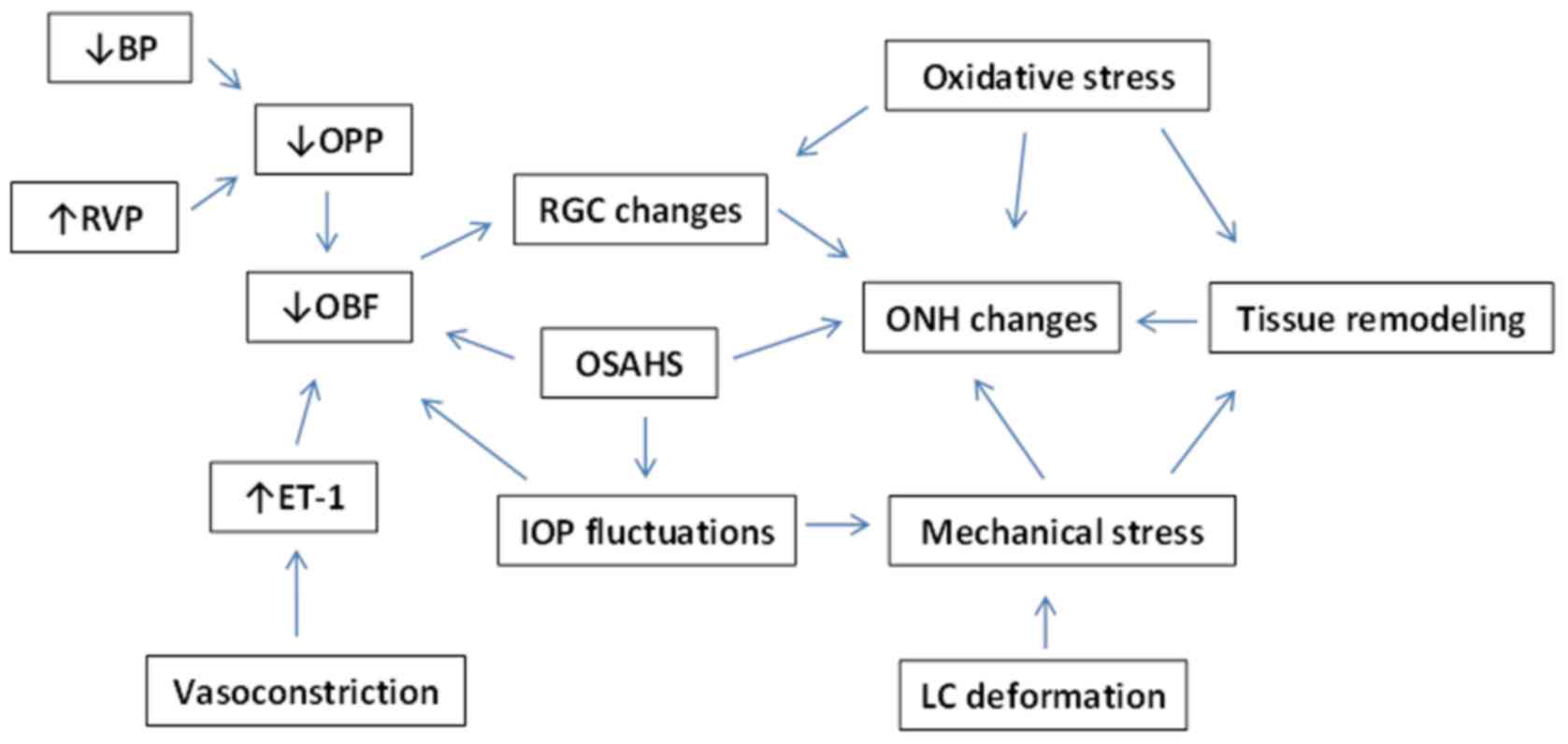 | Figure 1.Factors contributing to
normal-tension glaucoma (NTG). BP, blood pressure; RVP, retinal
venous pressure; OPP, ocular perfusion pressure; OBF, ocular blood
flow; ET-1, endothelin-1; RGC, retinal ganglion cell; OSAHS,
obstructive sleep apnea/hypopnea syndrome; IOP, intraocular
pressure; ONH, optic nerve head; LC, lamina cribrosa. |
Genetic associations in NTG
NTG is a heterogeneous disease, involving multiple
genes in its pathogenesis. The presence of a family history in NTG
cases suggests a possible genetic predisposition to NTG (102).
Variants in genes including WD repeat-containing
protein 36 (WDR36), myocilin (MYOC) and optineurin
(OPTN), are considered causative for NTG, although further
evidence is required (103,104). The association between OPTN
gene alterations in NTG cases and families has been reported
(105). OPTN plays a
neuroprotective, as well as an IOP-controlling role. Studies have
also revealed a possible link between OPTN, NTG and Alzheimer's
disease (102,106). Mutations in the optic atrophy type
1 (OPA1) gene may be related to hereditary NTG. OPA1 is
involved in mitochondrial function, protecting RGC from retinal
mechanical stress. OPA1 polymorphisms in NTG have been
confirmed, which can induce RGC apoptosis probably via
mitochondrial dysfunction (107,108).
The duplication of TANK binding kinase 1 (TBK1) gene in
glaucoma 1P (GLC1P) locus on 12q14 chromosome has also been
identified in familial NTG cases. TBK1 is expressed in RGC
and retinal microcirculation, and can thus contribute to apoptosis
and vascular impairment in NTG through a dysregulated expression
(109). Polymorphisms of the
endothelin receptor type A (EDNRA) gene of ETA
receptor, such as C+70G, have been related to more severe VF
defects in NTG, also being revealed in a Korean NTG population
study (110). Various single
nucleotide polymorphisms (SNPs) in the toll-like receptor 4
(TLR4) gene have been indicated in NTG cases in Japanese
contrary to Korean populations. Since TLR4 mediates immune
responses, such alterations may result in aberrant immunity and
autoimmune phenomena in patients with NTG (111). Human leukocyte antigen (HLA) class
II variants have also been associated with NTG, contributing to
autoimmunity. RAR-related orphan receptor C (RORC) gene
upregulation in blood lymphocytes has been detected in patients
with NTG, which can promote apoptosis and improper immunity
(112). As regards oxidative stress
and mitochondrial dysfunction, a variety of mitochondrial
irregularities and mitochondrial DNA alterations have been
determined in patients with NTG, including a Korean population,
pointing towards a genetic profile of this condition (113). Concerning tissue remodeling, the
upregulated gene expression of MMP-9 and MMP-14 has
been detected in patients with NTG, as well as in circulating
lymphocytes (96). Numerous other
candidate gene variants have been reported in NTG, such as glaucoma
1B (GLC1B) (114), glaucoma
1F (GLC1F) (115),
mitofusin-1 (MFN1) (116),
mitofusin-2 (MFN2) (116),
S1 RNA-binding domain (SRBD1) (117), presenilin-associated rhomboid-like
(PARL) (116) and fatty acid
elongase 5 (ELOVL5) (117).
Studies on NTG-related models of transgenic mice
have helped to further elucidate NTG pathogenesis. E50K mutant
transgenic mice overexpressing OPTN have demonstrated RGC loss,
also reflecting the role of oxidative stress in NTG (118). WDR36-mutant mice exhibit axonal
growth impairment, resulting in progressive RGC degeneration
(119). Mice with the OPA1 variant
exhibit RGC death and ONH atrophy (120). The endothelial overexpression of
ET-1 in TET-1 mice reveals the degeneration of RGC and their axons,
BBB damage and retinal gliosis, implying a vascular-related
component of NTG (121). Damage to
the RGCs and their axons has been reported in P301S and amyloid
precursor protein/presenilin 1 (APP/PS1) mice, indicating an
association between NTG and Alzheimer's disease (122,123).
GLAST and excitatory carrier-1 (EAAC1) deficient mice present
impulsive RGC loss and optic nerve degeneration with normal IOP.
Furthermore, mice deficient with apoptosis signal-regulating
kinase-1 (Ask-1) are more tolerant to ischemia, exhibit diminished
RGC and axonal loss, and mild VF defects, suggesting Ask-1 as a
potential target for NTG therapy (124). Table
I summarizes the genes reported to be associated with NTG, to
the best of the our knowledge.
 | Table I.Glaucoma-associated candidate
genes. |
Table I.
Glaucoma-associated candidate
genes.
|
|
| Glaucoma
subtype |
|
|---|
|
|
|
|
|
|---|
| Gene | Chromosomal
location | NTG | POAG | Author (Ref.),
year |
|---|
| WDR36 (WD
repeat-containing protein 36) | 5q22.1 |
| + | Monemi et al
(132), 2005 |
|
|
|
|
| Rangachari et
al (135), 2011 |
|
|
|
|
| Kumar et al
(129), 2016 |
| MYOC
(myocilin) | 1q24.3 | + | + | Stone et al
(139), 1997 |
| OPTN
(optineurin) | 10p13 | + | + | Rezaie et al
(136), 2002 |
|
|
|
|
| Kumar et al
(129), 2016 |
| TBK1
(TANK-binding kinase 1) | 12q14 |
| + | Fingert et
al (127), 2011 |
| GLC1B
(glaucoma 1, open angle, B) | 2cen-q13 |
| APOAG | Stoilova et
al (138), 1996 |
|
|
|
| + |
|
| ASB10
(GLC1F, open-angle glaucoma-1F) | 7q36.1 |
| + | Wirtz et al
(140),1999 |
|
|
|
|
| Pasutto et
al (134), 2012 |
| EDNRA
(endothelin receptor, type A) | 4q31.22-q31.23 |
| + | Janssen et
al (128), 2013 |
| TLR4
(Toll-like receptor 4) | 9q33.1 | + | + | Janssen et
al (128), 2013 |
| OPA1 (optic
atrophy 1) | 3q29 | + |
| Lascaratos et
al (130), 2012 |
|
|
|
|
| Yu-Wai-Man et
al (142), 2010 |
| MFN1
(mitofusin 1) | 3q26.33 |
| + | Lascaratos et
al (130), 2012 |
| MFN2
(mitofusin 2) | 1p36.22 |
| + | Lascaratos et
al (130), 2012 |
| PARL
(presenilin-associated rhomboid-like protein) | 3q27.1 |
| + | Lascaratos et
al (130), 2012 |
| RORC
(RAR-related orphan receptor C gene) | 1q21.3 | + |
| Fraenkl et
al (112), 2013 |
| MMP-9
(matrix metallopeptidase 9) | 20q13.12 | + | + | Golubnitschaja
et al (96), 2004 |
|
|
|
|
| Sahay et al
(137), 2017 |
| MMP-14
(matrix metallopeptidase 14) | 14q11.2 | + | + | Golubnitschaja
et al (96), 2004 |
| SRBD1 (S1
RNA binding domain 1) | 2p21 | + |
| Writing Committee
for the |
|
|
|
|
| Normal-Tension
Glaucoma Genetic |
|
|
|
|
| Study Group of
Japan Glaucoma |
|
|
|
|
| Society et
al (117), 2010 |
| ELOVL5
(ELOVL fatty acid elongase 5) | 6p12.1 | + |
| Writing Committee
for the |
|
|
|
|
| Normal-Tension
Glaucoma Genetic |
|
|
|
|
| Study Group of
Japan Glaucoma |
|
|
|
|
| Society et
al (117), 2010 |
| CDKN2B
(cyclin-dependent kinase inhibitor 2B) | 9p213 | + |
| Mabuchi et
al (131), 2012 |
| ATOH7
(atonal, Drosophila, homolog of, 7) | 10q21.3 | + |
| Mabuchi et
al (131), 2012 |
| DCLK1
(doublecortin-like kinase 1) | 13q13.3 | + | HTG | Mabuchi et
al (131), 2012 |
|
|
|
| + |
|
| RERE (RE
repeats-encoding gene) | 1p36.23 | + | HTG | Mabuchi et
al (131), 2012 |
|
|
|
| + |
|
| RPGRIP1
(retinitis pigmentosa GTPase regulator-interacting protein 1) | 14q11.2 | + | JOAG | Fernández-Martínez
et al (126), 2011 |
|
|
|
| + |
|
| APOE
(apolipoprotein E) | 19q13.32 | + |
| Janssen et
al (128), 2013 |
|
|
|
|
| Nowak et al
(133), 2015 |
| HSP70-1A
(heat shock 70-kDa protein 1A) | 6p21.33 | + | PACG | Ayub et al
(125), 2010 |
|
|
|
| + |
|
|
|
|
|
| Nowak et al
(133), 2015 |
| MTHFR
(5,10-methylenetetrahydrofolate reductase) | 1p36.22 | + | + | Woo et al
(141), 2009 |
Conclusions
All things considered, NTG is a multifactorial
disease, leading to progressive GON without an elevated IOP.
Vascular dysregulation may be the key of vascular pathogenesis
resulting in OBF instability in ONH, retina and choroid through an
interplay between retrobulbar vasospasm and endothelial
dysfunction. Mechanical factors, such as increased sensitivity to
IOP fluctuations, LC dysfunction and TLPG also play a significant
role. OSAHS and may also be a mixed risk factor for NTG. All these
factors, through oxidative stress, gliosis, excitotoxicity, ET-1
upregulation, BBB damage, abnormal immunity and deficient trophic
support can promote axonal damage and RGC apoptosis via several
extrinsic and intrinsic pathways. In addition, current literature
indicates a strong genetic component in NTG, with genes
significantly contributing to its pathogenesis. Nevertheless, NTG
remains a complex disease. Further studies are warranted to focus
on clarifying the mechanisms responsible for NTG-related GON and
its genetic basis, aiding to the development of diagnostic and
therapeutic strategies.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
AT conceived and designed the study. AT, IK, CT and
GD researched the literature, performed analysis of data and
drafted the manuscript. GNG, ETD, DAS and CSS researched the
literature, performed the analysis of the data and critically
revised the article for important intellectual content. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
DAS is the Editor-in-Chief for the journal, but had
no personal involvement in the reviewing process, or any influence
in terms of adjudicating on the final decision, for this article.
The other authors declare that they have no competing
interests.
References
|
1
|
Lee BL, Bathija R and Weinreb RN: The
definition of normal-tension glaucoma. J Glaucoma. 7:366–371. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cho HK and Kee C: Population-based
glaucoma prevalence studies in Asians. Surv Ophthalmol. 59:434–447.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Budak Y and Akdogan M: Retinal ganglion
cell death. Glaucoma-Basic and Clinical Concepts. Rumelt S: InTech;
Rijeka: pp. 33–56. 2011, https://www.intechopen.com/books/glaucoma-basic-and-clinical-concepts/retinal-ganglion-cell-death
|
|
4
|
Drance S, Anderson DR and Schulzer M:
Collaborative Normal-Tension Glaucoma Study Group: Risk factors for
progression of visual field abnormalities in normal-tension
glaucoma. Am J Ophthalmol. 131:699–708. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Flammer J, Orgül S, Costa VP, Orzalesi N,
Krieglstein GK, Serra LM, Renard JP and Stefánsson E: The impact of
ocular blood flow in glaucoma. Prog Retin Eye Res. 21:359–393.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Grunwald JE, Piltz J, Hariprasad SM and
DuPont J: Optic nerve and choroidal circulation in glaucoma. Invest
Ophthalmol Vis Sci. 39:2329–2336. 1998.PubMed/NCBI
|
|
7
|
Quaranta L and Floriani I: The rate of
progression and ocular perfusion pressure in the Low-pressure
Glaucoma Treatment Study. Am J Ophthalmol. 152:880–881; author
reply 880-881. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sung KR, Cho JW, Lee S, Yun SC, Choi J, Na
JH, Lee Y and Kook MS: Characteristics of visual field progression
in medically treated normal-tension glaucoma patients with unstable
ocular perfusion pressure. Invest Ophthalmol Vis Sci. 52:737–743.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Choi J, Kim KH, Jeong J, Cho HS, Lee CH
and Kook MS: Circadian fluctuation of mean ocular perfusion
pressure is a consistent risk factor for normal-tension glaucoma.
Invest Ophthalmol Vis Sci. 48:104–111. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Okumura Y, Yuki K and Tsubota K: Low
diastolic blood pressure is associated with the progression of
normal-tension glaucoma. Ophthalmologica. 228:36–41. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gasser P and Flammer J: Blood-cell
velocity in the nailfold capillaries of patients with
normal-tension and high-tension glaucoma. Am J Ophthalmol.
111:585–588. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Flammer J: The vascular concept of
glaucoma. Surv Ophthalmol. 38 Suppl:S3–S6. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Flammer J and Mozaffarieh M:
Autoregulation, a balancing act between supply and demand. Can J
Ophthalmol. 43:317–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Abegão Pinto L, Vandewalle E and Stalmans
I: Disturbed correlation between arterial resistance and
pulsatility in glaucoma patients. Acta Ophthalmol. 90:e214–e220.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Galassi F, Sodi A, Ucci F, Renieri G,
Pieri B and Baccini M: Ocular hemodynamics and glaucoma prognosis:
A color Doppler imaging study. Arch Ophthalmol. 121:1711–1715.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Emre M, Orgül S, Gugleta K and Flammer J:
Ocular blood flow alteration in glaucoma is related to systemic
vascular dysregulation. Br J Ophthalmol. 88:662–666. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pache M, Kaiser HJ, Akhalbedashvili N,
Lienert C, Dubler B, Kappos L and Flammer J: Extraocular blood flow
and endothelin-1 plasma levels in patients with multiple sclerosis.
Eur Neurol. 49:164–168. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chauhan BC, LeVatte TL, Jollimore CA, Yu
PK, Reitsamer HA, Kelly ME, Yu DY, Tremblay F and Archibald ML:
Model of endothelin-1-induced chronic optic neuropathy in rat.
Invest Ophthalmol Vis Sci. 45:144–152. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Flammer J and Mozaffarieh M: What is the
present pathogenetic concept of glaucomatous optic neuropathy? Surv
Ophthalmol. 52 (Suppl 2):S162–S173. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Furlanetto RL, De Moraes CG, Teng CC,
Liebmann JM, Greenfield DS, Gardiner SK, Ritch R and Krupin T:
Low-Pressure Glaucoma Treatment Study Group: Risk factors for optic
disc hemorrhage in the Low-Pressure Glaucoma Treatment Study. Am J
Ophthalmol. 157:945–952. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Orgül S, Kaiser HJ, Flammer J and Gasser
P: Systemic blood pressure and capillary blood-cell velocity in
glaucoma patients: A preliminary study. Eur J Ophthalmol. 5:88–91.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Moore D, Harris A, Wudunn D, Kheradiya N
and Siesky B: Dysfunctional regulation of ocular blood flow: A risk
factor for glaucoma? Clin Ophthalmol. 2:849–861. 2008.PubMed/NCBI
|
|
23
|
Charlson ME, de Moraes CG, Link A, Wells
MT, Harmon G, Peterson JC, Ritch R and Liebmann JM: Nocturnal
systemic hypotension increases the risk of glaucoma progression.
Ophthalmology. 121:2004–2012. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bowe A, Grünig M, Schubert J, Demir M,
Hoffmann V, Kütting F, Pelc A and Steffen HM: Circadian variation
in arterial blood pressure and glaucomatous optic neuropathy - a
systematic review and meta-analysis. Am J Hypertens. 28:1077–1082.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee J, Choi J, Jeong D, Kim S and Kook MS:
Relationship between daytime variability of blood pressure or
ocular perfusion pressure and glaucomatous visual field
progression. Am J Ophthalmol. 160:522–537.e1. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Flammer J, Konieczka K and Flammer AJ: The
primary vascular dysregulation syndrome: Implications for eye
diseases. EPMA J. 4:142013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Konieczka K, Ritch R, Traverso CE, Kim DM,
Kook MS, Gallino A, Golubnitschaja O, Erb C, Reitsamer HA, Kida T,
et al: Flammer syndrome. EPMA J. 5:112014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cursiefen C, Wisse M, Cursiefen S,
Jünemann A, Martus P and Korth M: Migraine and tension headache in
high-pressure and normal-pressure glaucoma. Am J Ophthalmol.
129:102–104. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Buckley C, Hadoke PW, Henry E and O'Brien
C: Systemic vascular endothelial cell dysfunction in normal
pressure glaucoma. Br J Ophthalmol. 86:227–232. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Teuchner B, Orgül S, Ulmer H, Haufschild T
and Flammer J: Reduced thirst in patients with a vasospastic
syndrome. Acta Ophthalmol Scand. 82:738–740. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gherghel D, Orgül S, Gugleta K and Flammer
J: Retrobulbar blood flow in glaucoma patients with nocturnal
over-dipping in systemic blood pressure. Am J Ophthalmol.
132:641–647. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wunderlich K, Zimmerman C, Gutmann H,
Teuchner B, Flammer J and Drewe J: Vasospastic persons exhibit
differential expression of ABC-transport proteins. Mol Vis.
9:756–761. 2003.PubMed/NCBI
|
|
33
|
Pache M, Kräuchi K, Cajochen C,
Wirz-Justice A, Dubler B, Flammer J and Kaiser HJ: Cold feet and
prolonged sleep-onset latency in vasospastic syndrome. Lancet.
358:125–126. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mozaffarieh M, Osusky R, Schotzau A and
Flammer J: Relationship between optic nerve head and finger blood
flow. Eur J Ophthalmol. 20:136–141. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mozaffarieh M and Flammer J: New insights
in the pathogenesis and treatment of normal tension glaucoma. Curr
Opin Pharmacol. 13:43–49. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Flammer J: Glaucomatous optic neuropathy:
A reperfusion injury. Klin Monbl Augenheilkd. 218:290–291. 2001.(In
German). View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gugleta K, Zawinka C, Rickenbacher I,
Kochkorov A, Katamay R, Flammer J and Orgul S: Analysis of retinal
vasodilation after flicker light stimulation in relation to
vasospastic propensity. Invest Ophthalmol Vis Sci. 47:4034–4041.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gugleta K, Orgül S, Hasler PW, Picornell
T, Gherghel D and Flammer J: Choroidal vascular reaction to
hand-grip stress in subjects with vasospasm and its relevance in
glaucoma. Invest Ophthalmol Vis Sci. 44:1573–1580. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nitta K: Disc hemorrhage is a sign of
progression in normal-tension glaucoma. J Glaucoma.
21:2762012.PubMed/NCBI
|
|
40
|
Corbett JJ, Phelps CD, Eslinger P and
Montague PR: The neurologic evaluation of patients with low-tension
glaucoma. Invest Ophthalmol Vis Sci. 26:1101–1104. 1985.PubMed/NCBI
|
|
41
|
Kruit MC, Launer LJ, Ferrari MD and van
Buchem MA: Infarcts in the posterior circulation territory in
migraine. The population-based MRI CAMERA study. Brain.
128:2068–2077. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Haefliger IO, Flammer J, Bény JL and
Lüscher TF: Endothelium-dependent vasoactive modulation in the
ophthalmic circulation. Prog Retin Eye Res. 20:209–225. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Toda N and Nakanishi-Toda M: Nitric oxide:
Ocular blood flow, glaucoma, and diabetic retinopathy. Prog Retin
Eye Res. 26:205–238. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sugiyama T, Moriya S, Oku H and Azuma I:
Association of endothelin-1 with normal tension glaucoma: Clinical
and fundamental studies. Surv Ophthalmol. 39 Suppl 1:S49–S56. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kaiser HJ, Flammer J, Wenk M and Lüscher
T: Endothelin-1 plasma levels in normal-tension glaucoma: Abnormal
response to postural changes. Graefes Arch Clin Exp Ophthalmol.
233:484–488. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cellini M, Possati GL, Profazio V, Sbrocca
M, Caramazza N and Caramazza R: Color Doppler imaging and plasma
levels of endothelin-1 in low-tension glaucoma. Acta Ophthalmol
Scand Suppl. 224:S22411–13. 1997.
|
|
47
|
Galassi F, Giambene B and Varriale R:
Systemic vascular dysregulation and retrobulbar hemodynamics in
normal-tension glaucoma. Invest Ophthalmol Vis Sci. 52:4467–4471.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nicolela MT, Ferrier SN, Morrison CA,
Archibald ML, LeVatte TL, Wallace K, Chauhan BC and LeBlanc RP:
Effects of cold-induced vasospasm in glaucoma: The role of
endothelin-1. Invest Ophthalmol Vis Sci. 44:2565–2572. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Su WW, Cheng ST, Hsu TS and Ho WJ:
Abnormal flow-mediated vasodilation in normal-tension glaucoma
using a noninvasive determination for peripheral endothelial
dysfunction. Invest Ophthalmol Vis Sci. 47:3390–3394. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Henry E, Newby DE, Webb DJ, Hadoke PWF and
O'Brien CJ: Altered endothelin-1 vasoreactivity in patients with
untreated normal-pressure glaucoma. Invest Ophthalmol Vis Sci.
47:2528–2532. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hamed SA, Hamed EA, Ezz Eldin AM and
Mahmoud NM: Vascular risk factors, endothelial function, and
carotid thickness in patients with migraine: Relationship to
atherosclerosis. J Stroke Cerebrovasc Dis. 19:92–103. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Collaborative Normal-Tension Glaucoma
Study Group, . The effectiveness of intraocular pressure reduction
in the treatment of normal-tension glaucoma. Am J Ophthalmol.
126:498–505. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Agnifili L, Carpineto P, Fasanella V,
Mastropasqua R, Zappacosta A, Di Staso S, Costagliola C and
Mastropasqua L: Conjunctival findings in hyperbaric and low-tension
glaucoma: An in vivo confocal microscopy study. Acta Ophthalmol.
90:e132–e137. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Agnifili L, Mastropasqua R, Frezzotti P,
Fasanella V, Motolese I, Pedrotti E, Di Iorio A, Mattei PA,
Motolese E and Mastropasqua L: Circadian intraocular pressure
patterns in healthy subjects, primary open angle and normal tension
glaucoma patients with a contact lens sensor. Acta Ophthalmol.
93:e14–e21. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
De Moraes CG, Jasien JV, Simon-Zoula S,
Liebmann JM and Ritch R: Visual field change and 24-hour
IOP-related profile with a contact lens sensor in treated glaucoma
patients. Ophthalmology. 123:744–753. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Choi J and Kook MS: Systemic and ocular
hemodynamic risk factors in glaucoma. Biomed Res Int.
2015:1419052015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sakata R, Aihara M, Murata H, Saito H,
Iwase A, Yasuda N and Araie M: Intraocular pressure change over a
habitual 24-hour period after changing posture or drinking water
and related factors in normal tension glaucoma. Invest Ophthalmol
Vis Sci. 54:5313–5320. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Adlina AR, Alisa-Victoria K, Shatriah I,
Liza-Sharmini AT and Ahmad MS: Optic disc topography in Malay
patients with normal-tension glaucoma and primary open-angle
glaucoma. Clin Ophthalmol. 8:2533–2539. 2014.PubMed/NCBI
|
|
59
|
Copt RP, Thomas R and Mermoud A: Corneal
thickness in ocular hypertension, primary open-angle glaucoma, and
normal tension glaucoma. Arch Ophthalmol. 117:14–16. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lee JW, Wong RL, Chan JC, Wong IY and Lai
JS: Differences in corneal parameters between normal tension
glaucoma and primary open-angle glaucoma. Int Ophthalmol. 35:67–72.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Cao KY, Kapasi M, Betchkal JA and Birt CM:
Relationship between central corneal thickness and progression of
visual field loss in patients with open-angle glaucoma. Can J
Ophthalmol. 47:155–158. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jonas JB, Wang N and Yang D: Translamina
cribrosa pressure difference as potential element in the
pathogenesis of glaucomatous optic neuropathy. Asia Pac J
Ophthalmol (Phila). 5:5–10. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Burgoyne CF: A biomechanical paradigm for
axonal insult within the optic nerve head in aging and glaucoma.
Exp Eye Res. 93:120–132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wostyn P, De Groot V, Van Dam D, Audenaert
K and De Deyn PP: Senescent changes in cerebrospinal fluid
circulatory physiology and their role in the pathogenesis of
normal-tension glaucoma. Am J Ophthalmol. 156:5–14.e2. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ren R, Jonas JB, Tian G, Zhen Y, Ma K, Li
S, Wang H, Li B, Zhang X and Wang N: Cerebrospinal fluid pressure
in glaucoma: A prospective study. Ophthalmology. 117:259–266. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Siaudvytyte L, Januleviciene I, Daveckaite
A, Ragauskas A, Bartusis L, Kucinoviene J, Siesky B and Harris A:
Literature review and meta-analysis of translaminar pressure
difference in open-angle glaucoma. Eye (Lond). 29:1242–1250. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Pircher A, Remonda L, Weinreb RN and
Killer HE: Translaminar pressure in Caucasian normal tension
glaucoma patients. Acta Ophthalmol. 95:e524–e531. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lindén C, Qvarlander S, Jóhannesson G,
Johansson E, Östlund F, Malm J and Eklund A: Normal-tension
glaucoma has normal intracranial pressure: A prospective study
ofintracranial pressure and intraocular pressure in different body
positions. Ophthalmology. 125:361–368. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Jonas JB, Gusek GC and Naumann GO: Optic
disk morphometry in high myopia. Graefes Arch Clin Exp Ophthalmol.
226:587–590. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Rao VR, Krishnamoorthy RR and Yorio T:
Endothelin-1, endothelin A and B receptor expression and their
pharmacological properties in GFAP negative human lamina cribrosa
cells. Exp Eye Res. 84:1115–1124. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Rao VR, Krishnamoorthy RR and Yorio T:
Endothelin-1 mediated regulation of extracellular matrix collagens
in cells of human lamina cribrosa. Exp Eye Res. 86:886–894. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Pérez-Rico C, Gutiérrez-Díaz E,
Mencía-Gutiérrez E, Díaz-de-Atauri MJ and Blanco R: Obstructive
sleep apnea-hypopnea syndrome (OSAHS) and glaucomatous optic
neuropathy. Graefes Arch Clin Exp Ophthalmol. 252:1345–1357. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Lin PW, Friedman M, Lin HC, Chang HW,
Wilson M and Lin MC: Normal tension glaucoma in patients with
obstructive sleep apnea/hypopnea syndrome. J Glaucoma. 20:553–558.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Karakucuk S, Goktas S, Aksu M, Erdogan N,
Demirci S, Oner A, Arda H and Gumus K: Ocular blood flow in
patients with obstructive sleep apnea syndrome (OSAS). Graefes Arch
Clin Exp Ophthalmol. 246:129–134. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Nadeem R, Molnar J, Madbouly EM, Nida M,
Aggarwal S, Sajid H, Naseem J and Loomba R: Serum inflammatory
markers in obstructive sleep apnea: A meta-analysis. J Clin Sleep
Med. 9:1003–1012. 2013.PubMed/NCBI
|
|
76
|
Thurtell MJ, Bruce BB, Newman NJ and
Biousse V: An update on idiopathic intracranial hypertension. Rev
Neurol Dis. 7:e56–e68. 2010.PubMed/NCBI
|
|
77
|
Hara T, Hara T and Tsuru T: Increase of
peak intraocular pressure during sleep in reproduced diurnal
changes by posture. Arch Ophthalmol. 124:165–168. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Carvey PM, Hendey B and Monahan AJ: The
blood-brain barrier in neurodegenerative disease: A rhetorical
perspective. J Neurochem. 111:291–314. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Pournaras CJ, Rungger-Brändle E, Riva CE,
Hardarson SH and Stefansson E: Regulation of retinal blood flow in
health and disease. Prog Retin Eye Res. 27:284–330. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Iadecola C and Nedergaard M: Glial
regulation of the cerebral microvasculature. Nat Neurosci.
10:1369–1376. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
81
|
Cioffi GA and Sullivan P: The effect of
chronic ischemia on the primate optic nerve. Eur J Ophthalmol. 9
Suppl 1:S34–S36. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Harris A, Siesky B and Wirostko B:
Cerebral blood flow in glaucoma patients. J Glaucoma. 22 Suppl
5:S46–S48. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Mozaffarieh M, Grieshaber MC and Flammer
J: Oxygen and blood flow: Players in the pathogenesis of glaucoma.
Mol Vis. 14:224–233. 2008.PubMed/NCBI
|
|
84
|
Bunting H, Still R, Williams DR, Gravenor
M and Austin MW: Evaluation of plasma glutamate levels in normal
tension glaucoma. Ophthalmic Res. 43:197–200. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Chrysostomou V, Rezania F, Trounce IA and
Crowston JG: Oxidative stress and mitochondrial dysfunction in
glaucoma. Curr Opin Pharmacol. 13:12–15. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Mozaffarieh M, Schoetzau A, Sauter M,
Grieshaber M, Orgül S, Golubnitschaja O and Flammer J: Comet assay
analysis of single-stranded DNA breaks in circulating leukocytes of
glaucoma patients. Mol Vis. 14:1584–1588. 2008.PubMed/NCBI
|
|
87
|
Tezel G and Wax MB: Hypoxia-inducible
factor 1alpha in the glaucomatous retina and optic nerve head. Arch
Ophthalmol. 122:1348–1356. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
McElnea EM, Quill B, Docherty NG, Irnaten
M, Siah WF, Clark AF, O'Brien CJ and Wallace DM: Oxidative stress,
mitochondrial dysfunction and calcium overload in human lamina
cribrosa cells from glaucoma donors. Mol Vis. 17:1182–1191.
2011.PubMed/NCBI
|
|
89
|
Yuan L and Neufeld AH: Activated microglia
in the human glaucomatous optic nerve head. J Neurosci Res.
64:523–532. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Tezel G and Wax MB: The immune system and
glaucoma. Curr Opin Ophthalmol. 15:80–84. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Prasanna G, Krishnamoorthy R and Yorio T:
Endothelin, astrocytes and glaucoma. Exp Eye Res. 93:170–177. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Harada T, Harada C, Nakamura K, Quah HM,
Okumura A, Namekata K, Saeki T, Aihara M, Yoshida H, Mitani A, et
al: The potential role of glutamate transporters in the
pathogenesis of normal tension glaucoma. J Clin Invest.
117:1763–1770. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Munemasa Y and Kitaoka Y: Molecular
mechanisms of retinal ganglion cell degeneration in glaucoma and
future prospects for cell body and axonal protection. Front Cell
Neurosci. 6:602013. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Lebrun-Julien F, Duplan L, Pernet V,
Osswald I, Sapieha P, Bourgeois P, Dickson K, Bowie D, Barker PA
and Di Polo A: Excitotoxic death of retinal neurons in vivo occurs
via a non-cell-autonomous mechanism. J Neurosci. 29:5536–5545.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Oku H, Fukuhara M, Komori A, Okuno T,
Sugiyama T and Ikeda T: Endothelin-1 (ET-1) causes death of retinal
neurons through activation of nitric oxide synthase (NOS) and
production of superoxide anion. Exp Eye Res. 86:118–130. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Golubnitschaja O, Yeghiazaryan K, Liu R,
Mönkemann H, Leppert D, Schild H, Haefliger IO and Flammer J:
Increased expression of matrix metalloproteinases in mononuclear
blood cells of normal-tension glaucoma patients. J Glaucoma.
13:66–72. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Grieshaber MC and Flammer J: Does the
blood-brain barrier play a role in Glaucoma? Surv Ophthalmol. 52
Suppl 2:S115–S121. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Hofman P, Hoyng P, vanderWerf F, Vrensen
GF and Schlingemann RO: Lack of blood-brain barrier properties in
microvessels of the prelaminar optic nerve head. Invest Ophthalmol
Vis Sci. 42:895–901. 2001.PubMed/NCBI
|
|
99
|
Grieshaber MC, Terhorst T and Flammer J:
The pathogenesis of optic disc splinter haemorrhages: A new
hypothesis. Acta Ophthalmol Scand. 84:62–68. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Farrall AJ and Wardlaw JM: Blood-brain
barrier: Ageing and microvascular disease - systematic review and
meta-analysis. Neurobiol Aging. 30:337–352. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Tong L, Balazs R, Soiampornkul R,
Thangnipon W and Cotman CW: Interleukin-1 beta impairs brain
derived neurotrophic factor-induced signal transduction. Neurobiol
Aging. 29:1380–1393. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Allingham RR, Liu Y and Rhee DJ: The
genetics of primary open-angle glaucoma: A review. Exp Eye Res.
88:837–844. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Weisschuh N, Neumann D, Wolf C, Wissinger
B and Gramer E: Prevalence of myocilin and optineurin sequence
variants in German normal tension glaucoma patients. Mol Vis.
11:284–287. 2005.PubMed/NCBI
|
|
104
|
Weisschuh N, Wolf C, Wissinger B and
Gramer E: Variations in the WDR36 gene in German patients with
normal tension glaucoma. Mol Vis. 13:724–729. 2007.PubMed/NCBI
|
|
105
|
Tang S, Toda Y, Kashiwagi K, Mabuchi F,
Iijima H, Tsukahara S and Yamagata Z: The association between
Japanese primary open-angle glaucoma and normal tension glaucoma
patients and the optineurin gene. Hum Genet. 113:276–279. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Liu YH and Tian T: Hypothesis of
optineurin as a new common risk factor in normal-tension glaucoma
and Alzheimer's disease. Med Hypotheses. 77:591–592. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Mi XS, Yuan TF and So KF: The current
research status of normal tension glaucoma. Clin Interv Aging.
9:1563–1571. 2014.PubMed/NCBI
|
|
108
|
Guo Y, Chen X, Zhang H, Li N, Yang X,
Cheng W and Zhao K: Association of OPA1 polymorphisms with NTG and
HTG: A meta-analysis. PLoS One. 7:e423872012. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Kawase K, Allingham RR, Meguro A, Mizuki
N, Roos B, Solivan-Timpe FM, Robin AL, Ritch R and Fingert JH:
Confirmation of TBK1 duplication in normal tension glaucoma. Exp
Eye Res. 96:178–180. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Kim SH, Kim JY, Kim DM, Ko HS, Kim SY, Yoo
T, Hwang SS and Park SS: Investigations on the association between
normal tension glaucoma and single nucleotide polymorphisms of the
endothelin-1 and endothelin receptor genes. Mol Vis. 12:1016–1021.
2006.PubMed/NCBI
|
|
111
|
Shibuya E, Meguro A, Ota M, Kashiwagi K,
Mabuchi F, Iijima H, Kawase K, Yamamoto T, Nakamura M, Negi A, et
al: Association of Toll-like receptor 4 gene polymorphisms with
normal tension glaucoma. Invest Ophthalmol Vis Sci. 49:4453–4457.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Fraenkl SA, Golubnitschaja O, Yeghiazaryan
K, Orgül S and Flammer J: Differences in gene expression in
lymphocytes of patients with high-tension, PEX, and normal-tension
glaucoma and in healthy subjects. Eur J Ophthalmol. 23:841–849.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Jeoung JW, Seong MW, Park SS, Kim DM, Kim
SH and Park KH: Mitochondrial DNA variant discovery in
normal-tension glaucoma patients by next-generation sequencing.
Invest Ophthalmol Vis Sci. 55:986–992. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Akiyama M, Yatsu K, Ota M, Katsuyama Y,
Kashiwagi K, Mabuchi F, Iijima H, Kawase K, Yamamoto T, Nakamura M,
et al: Microsatellite analysis of the GLC1B locus on chromosome 2
points to NCK2 as a new candidate gene for normal tension glaucoma.
Br J Ophthalmol. 92:1293–1296. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Murakami K, Meguro A, Ota M, Shiota T,
Nomura N, Kashiwagi K, Mabuchi F, Iijima H, Kawase K, Yamamoto T,
et al: Analysis of microsatellite polymorphisms within the GLC1F
locus in Japanese patients with normal tension glaucoma. Mol Vis.
16:462–466. 2010.PubMed/NCBI
|
|
116
|
Wolf C, Gramer E, Müller-Myhsok B, Pasutto
F, Reinthal E, Wissinger B and Weisschuh N: Evaluation of nine
candidate genes in patients with normal tension glaucoma: A case
control study. BMC Med Genet. 10:912009. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Writing Committee for the Normal Tension
Glaucoma Genetic Study Group of Japan Glaucoma Society, . Meguro A,
Inoko H, Ota M, Mizuki N and Bahram S: Genome-wide association
study of normal tension glaucoma: Common variants in SRBD1 and
ELOVL5 contribute to disease susceptibility. Ophthalmology.
117:1331.e5–1338.e5. 2010.
|
|
118
|
Chi ZL, Akahori M, Obazawa M, Minami M,
Noda T, Nakaya N, Tomarev S, Kawase K, Yamamoto T, Noda S, et al:
Overexpression of optineurin E50K disrupts Rab8 interaction and
leads to a progressive retinal degeneration in mice. Hum Mol Genet.
19:2606–2615. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Chi ZL, Yasumoto F, Sergeev Y, Minami M,
Obazawa M, Kimura I, Takada Y and Iwata T: Mutant WDR36 directly
affects axon growth of retinal ganglion cells leading to
progressive retinal degeneration in mice. Hum Mol Genet.
19:3806–3815. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Heiduschka P, Schnichels S, Fuhrmann N,
Hofmeister S, Schraermeyer U, Wissinger B and Alavi MV:
Electrophysiological and histologic assessment of retinal ganglion
cell fate in a mouse model for OPA1-associated autosomal dominant
optic atrophy. Invest Ophthalmol Vis Sci. 51:1424–1431. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Mi XS, Zhang X, Feng Q, Lo AC, Chung SK
and So KF: Progressive retinal degeneration in transgenic mice with
overexpression of endothelin-1 in vascular endothelial cells.
Invest Ophthalmol Vis Sci. 53:4842–4851. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Gasparini L, Crowther RA, Martin KR, Berg
N, Coleman M, Goedert M and Spillantini MG: Tau inclusions in
retinal ganglion cells of human P301S tau transgenic mice: Effects
on axonal viability. Neurobiol Aging. 32:419–433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Ning A, Cui J, To E, Ashe KH and Matsubara
J: Amyloid-beta deposits lead to retinal degeneration in a mouse
model of Alzheimer disease. Invest Ophthalmol Vis Sci.
49:5136–5143. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Harada C, Namekata K, Guo X, Yoshida H,
Mitamura Y, Matsumoto Y, Tanaka K, Ichijo H and Harada T: ASK1
deficiency attenuates neural cell death in GLAST-deficient mice, a
model of normal tension glaucoma. Cell Death Differ. 17:1751–1759.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Ayub H, Khan MI, Micheal S, Akhtar F,
Ajmal M, Shafique S, Ali SH, den Hollander AI, Ahmed A and Qamar R:
Association of eNOS and HSP70 gene polymorphisms with glaucoma in
Pakistani cohorts. Mol Vis. 16:18–25. 2010.PubMed/NCBI
|
|
126
|
Fernández-Martínez L, Letteboer S, Mardin
CY, Weisschuh N, Gramer E, Weber BH, Rautenstrauss B, Ferreira PA,
Kruse FE, Reis A, et al: Evidence for RPGRIP1 gene as risk factor
for primary open angle glaucoma. Eur J Hum Genet. 19:445–451. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Fingert JH, Robin AL, Stone JL, Roos BR,
Davis LK, Scheetz TE, Bennett SR, Wassink TH, Kwon YH, Alward WL,
et al: Copy number variations on chromosome 12q14 in patients with
normal tension glaucoma. Hum Mol Genet. 20:2482–2494. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Janssen SF, Gorgels TG, van der Spek PJ,
Jansonius NM and Bergen AA: In silico analysis of the molecular
machinery underlying aqueous humor production: Potential
implications for glaucoma. J Clin Bioinforma. 3:212013. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Kumar S, Malik MA, Goswami S, Sihota R and
Kaur J: Candidate genes involved in the susceptibility of primary
open angle glaucoma. Gene. 577:119–131. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Lascaratos G, Garway-Heath DF, Willoughby
CE, Chau KY and Schapira AH: Mitochondrial dysfunction in glaucoma:
Understanding genetic influences. Mitochondrion. 12:202–212. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Mabuchi F, Sakurada Y, Kashiwagi K,
Yamagata Z, Iijima H and Tsukahara S: Association between genetic
variants associated with vertical cup-to-disc ratio and phenotypic
features of primary open-angle glaucoma. Ophthalmology.
119:1819–1825. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Monemi S, Spaeth G, DaSilva A, Popinchalk
S, Ilitchev E, Liebmann J, Ritch R, Héon E, Crick RP, Child A, et
al: Identification of a novel adult-onset primary open-angle
glaucoma (POAG) gene on 5q22.1. Hum Mol Genet. 14:725–733. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Nowak A, Majsterek I, Przybyłowska-Sygut
K, Pytel D, Szymanek K, Szaflik J and Szaflik JP: Analysis of the
expression and polymorphism of APOE, HSP, BDNF, and GRIN2B genes
associated with the neurodegeneration process in the pathogenesis
of primary open angle glaucoma. Biomed Res Int. 2015:2582812015.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Pasutto F, Keller KE, Weisschuh N, Sticht
H, Samples JR, Yang YF, Zenkel M, Schlötzer-Schrehardt U, Mardin
CY, Frezzotti P, et al: Variants in ASB10 are associated with
open-angle glaucoma. Hum Mol Genet. 21:1336–1349. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Rangachari K, Dhivya M, Eswari
Pandaranayaka PJ, Prasanthi N, Sundaresan P, Krishnadas SR and
Krishnaswamy S: Glaucoma database. Bioinformation. 5:398–399. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Rezaie T, Child A, Hitchings R, Brice G,
Miller L, Coca-Prados M, Héon E, Krupin T, Ritch R, Kreutzer D, et
al: Adult-onset primary open-angle glaucoma caused by mutations in
optineurin. Science. 295:1077–1079. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Sahay P, Rao A, Padhy D, Sarangi S, Das G,
Reddy MM and Modak R: Functional activity of matrix
metalloproteinases 2 and 9 in tears of patients with glaucoma.
Invest Ophthalmol Vis Sci. 58:BIO106–BIO113. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Stoilova D, Child A, Trifan OC, Crick RP,
Coakes RL and Sarfarazi M: Localization of a locus (GLC1B) for
adult-onset primary open angle glaucoma to the 2cen-q13 region.
Genomics. 36:142–150. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Stone EM, Fingert JH, Alward WL, Nguyen
TD, Polansky JR, Sunden SL, Nishimura D, Clark AF, Nystuen A,
Nichols BE, et al: Identification of a gene that causes primary
open angle glaucoma. Science. 275:668–670. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Wirtz MK, Samples JR, Rust K, Lie J,
Nordling L, Schilling K, Acott TS and Kramer PL: GLC1F, a new
primary open-angle glaucoma locus, maps to 7q35-q36. Arch
Ophthalmol. 117:237–241. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Woo SJ, Kim JY, Kim DM, Park SS, Ko HS and
Yoo T: Investigation of the association between 677C>T and
1298A>C 5,10-methylenetetra- hydrofolate reductase gene
polymorphisms and normal-tension glaucoma. Eye (Lond). 23:17–24.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Yu-Wai-Man P, Stewart JD, Hudson G,
Andrews RM, Griffiths PG, Birch MK and Chinnery PF: OPA1 increases
the risk of normal but not high tension glaucoma. J Med Genet.
47:120–125. 2010. View Article : Google Scholar : PubMed/NCBI
|














