Introduction
Burkholderia gladioli pv.Alliicola
(Bga), one of the four pathovars known in B. gladioli, was
originally described by Severini in 1913 (1) as a phytopathogen causing rot of
Gladiolus corms. Initially, it was considered as a synonym
of Pseudomonas marginata (2),
and was then given the present name B. gladioli (3). As a gram-negative bacillus, B.
gladioli had been primarily considered to be a plant pathogen.
However, it was also reported to be associated with the onset of
pulmonary infections in humans, including cystic fibrosis (CF) and
chronic granulomatous disease (CGD) (4–7).
B. cepacia complex (Bcc) is a group of
closely related gram-negative bacteria with similar phenotypes and
genotypes (8). To date, a total of
17 genomovars have been identified from the environment and are
widely used as a reagent for biodegradation and biocontrol, acting
as a plant growth-promoting rhizobacterium (9–11).
Similar to Bga, Bcc may also induce fatal infections in vulnerable
individuals, for example those with CF and CGD (12–15), as
well as induce rot of onions (16).
These pathogenic similarities suggested a close association between
Bga and Bcc.
Currently, extensive studies have been conducted on
the taxonomy of Bcc; however, a high incidence of misidentification
for Bcc is reported as ~50% of isolated Bcc actually belongs to
B. gladioli (17). To date,
polymerase chain reaction (PCR)-based methods have been commonly
used to isolate unknown family members using 16S and 23S ribosomal
RNA, and recA or gyrB (18–24).
Among these markers, the gyrB gene encoding subunit B of DNA
gyrase was demonstrated to be effective in the discrimination of
species within Bcc and Bga due to its sufficiently variable rate
(25). Although such strategies have
been indicated to be successful in isolating closely related
sequences, failure is usually encountered when sequences are more
distantly related or are in low copy numbers.
In the present study, a novel strategy named after
COnsensus-DEgenerate Hybrid Oligonucleotide Primer (CODEHOP)
(26) was adopted to test its
applicability for PCR amplification of the gyrB gene of Bga
by CODEHOP primers designed upon related protein sequences of the
gyrB gene of Bcc. A total of 1,644 base pairs of the
gyrB gene of Bga were acquired and two probes, designed
based on the gyrB gene sequences of Bga and Bcc, were used
to distinguish between Bga and five genomovars of Bcc. A
phylogenetic tree was constructed based on the gyrB gene of
Bga and nine genomovars of Bcc that demonstrated a divergence
between Bga and Bcc. The present study demonstrated the utility of
CODEHOP in the study of homologous genes between Bga and Bcc.
Furthermore, the results indicated that the gyrB gene was a
suitable marker in distinguishing Bga and Bcc; however, it was less
effective for the genotyping of different genomovars of Bcc. In the
future, more effective markers should be developed or a combination
of different marker genes may be more effective and robust in the
genotyping of Bcc.
Materials and methods
Strains and reagents
Bga strain 20157 was kindly provided by Professor
Hasan Bolkan from the Campbell's Agricultural Research
Centre (Davis, CA, USA). The DNA of Bcc was provided by Dr
Guan-Ning Xie of Zhejiang University (Hangzhou, Zhejiang, China).
Taq DNA polymerase, deoxynucleotide (dNTP) mixture for the
PCR reaction, DNA marker DL2000, Escherichia coli competent
cells (DH5α) for transformation and a MiniBEST plasmid purification
kit were purchased from Takara Biotechnology Co., Ltd., (Dalian,
China). E.Z.N.A.® Bacterial DNA Kit and E.Z.N.A Gel
Extraction Kit was purchased from Omega Bio-Tek, Inc., (Norcross,
GA, USA). A T-clone kit (pGEM®-T Vector System)
including the pGEM®-T Vector and T4 DNA Ligase was
purchased from Promega Corp., (Madison, WI, USA). Ampicillin,
isopropyl β-D-1-thiogalactopyranoside and X-gal used for screening
of clones were provided by Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany). Primer synthesis and DNA sequencing were performed by
Takara Biotechnology Co., Ltd. The reagents used in the experiments
were all of analytical purity.
Design of CODEHOP primers
Clustalx software (http://www.clustal.org/) was utilized to investigate
the sequence homology of the gyrB gene with Bcc and Bga. The
gyrB protein sequence of Bcc was retrieved and downloaded
from the National Center for Biotechnology Information (NCBI) Gen
Bank database (https://www.ncbi.nlm.nih.gov/genbank/). The sequence
length was limited between 630 and 655 amino acids, which was close
to its full length. Furthermore, alignment of the protein sequence
was conducted by the online Block Maker program (http://blocks.fhcrc.org/blockmkr/make_blocks.html) to
identify the conservative sequences. The sequences obtained were
input into the CODEHOP primers designing tool (https://virology.uvic.ca/virology-ca-tools/j-codehop/)
to search for the appropriate primers, and the codon preference was
set as ‘Burkholderiaglumae’ as it was the only species of
Burkholderia in the provided codon preference list and was
homologous to Bga.
CODEHOP amplification
The CODEHOP amplification reaction was performed in
a total volume of 25 µl containing 10X PCR buffer, 25 mmol/ldNTPs,
20 µmol/l of each primer, 5 U/µlTaq DNA polymerase and 30 ng
Bga genomic DNA. Genomic DNA extraction was performed using the
E.Z.N.A.® Bacterial DNA kit(Omega Bio-tek, Inc.). The
amplification conditions were as follows: 94°C for 4 min followed
by 45 cycles of 94°C for 30 sec, 47–49.5°C for 60 sec and 72°C for
90 sec, as well as a final extension at 72°C for 7 min. Following
this, the products were electrophoresed on a 1.2% agarose gel and
dyed with ethidium bromide.
Verification
To verify the amplified fragment obtained using
CODEHOP primers, the verification primers were designed according
to the gyrB gene of B. gladioli downloaded from the
NCBI database using Primer Premier 6.0 software (Premier Biosoft
International, Palo Alto, California, USA). The primers were then
used for amplification of genomic DNA of Bga to test their
validity. The PCR reaction was performed in a total volume of 25 µl
containing 2.5 µl 10X PCR buffer, 2 µl dNTPs (25 mmol/l), 0.8 µl of
each primer (20 µmol/l), 0.2 µl Taq DNA polymerase (5 U/µl)
and 30 ng Bga genomic DNA template. The amplification conditions
were as follows: 94°C for 4 min followed by 35 cycles of 94°C for
30 sec, 50°C for 30 sec, 72°C for 45 sec and a final extension at
72°C for 7 min.
Cloning of CODEHOP amplification
products
Purification of CODEHOP amplification products was
performed with an E.Z.N.A gel extraction kit, according to the
manufacturer's instructions. Following purification, the fragments
were linked with a T-vector using pGEM®-T Vector Systems
kit purchased from Promega Corp., at 4°C overnight. Subsequently,
the products were transformed into E.coli competent cells
(DH5α) by performing heat-shock, followed by cultivation on a
shaker at a speed of 150 RPM for 1 h at 37°C.Subsequently, the
plasmid was extracted using a MiniBEST plasmid purification kit
(Takara Biotechnology Co., Ltd.) according to the manufacturer's
protocol. Sanger sequencing was then performed to verify the
inserted sequence. Sequencing was completed by Sangon Biotech Co.,
Ltd. (Shanghai, China).
Phylogenetic analysis
The gyrB gene sequences were compared with
known sequences deposited in the NCBI database using the BlastN
program (https://blast.ncbi.nlm.nih.gov/Blast.cgi). For each
genomovar of Bcc, the strain with the most similar sequence of the
gyrB gene of Bga was selected. Phylogenetic analysis was
performed using the free tool, MEGA 5(https://www.megasoftware.net/) with the
Neighbor-Joining algorithm, and the topological accuracy of the
trees was evaluated with 1,000 bootstrap replicates.
Microarray analysis
A universal primer of the gyrB gene was
designed according to the gyrB gene sequence obtained from
Bga and sequences of Bcc downloaded from GenBank. However, two
probes identifying Bga and several probes identifying Bcc and its
genomovar were designed using AlleleID software 7.60(PremierBiosoft
International, Palo Alto, CA, USA). Probes, with a 15-bp poly (dT)
and an amino group at the 5′ terminal were conjugated to the glass
substrate modified by an aldehyde group. Fluorescent labeling was
incorporated in the amplification products by cy3-dCTP within the
PCR procedure. PCR reaction was performed in a total volume of 10
µl containing 10X PCR buffer, 25 mmol/ldNTPs, 250 nmol Cy™3-dCTP,
20 µmol/l of each primer, 5 U/µl Taq DNA polymerase and 0.5
µl Bga/Bcc genomic DNA. The amplification instructions were as
follows: 94°C for 4 min followed by 35 cycles of 94°C for 30 sec,
56°C for 1 min, 72°C for 45 sec and 72°C for 7 min. Amplification
products were initially mixed with the hybridization buffer (6X
Saline Sodium Citrate, 0.5% SDS, 100 µg/ml, Salmon DNA
(Sigma-Aldrich; Merck KGaA) preheated to 50°C, followed by heating
at 95°C for 5 min. Subsequently, the mixture was incubated on ice
and water for 5 min. After a 2-h hybridization, the chip was washed
twice with washing buffer (20 mM Tris-HCl, 150 mM NaCl, 0.05%
Tween-20) and air-dried. Finally, a GenePix 4200A (Molecular
Devices, LLC, Sunnyvale, CA, USA) scanner was used for the
results.
Results
CODEHOP primers and verification
primers
Two sets of primer pairs were eventually selected
(Table I), in which Y, R, N in the
sequence represented the degenerate bases. The bases in the capital
form represented the consensus clamp and those in the lowercase
form represented the degenerate core according to the principal of
CODEHOP primers. Two pairs of verification primers were designed
based on the gyrB gene sequence of B. gladioli
(Table II).
 | Table I.Consensus-degenerate hybrid
oligonucleotide primers. |
Table I.
Consensus-degenerate hybrid
oligonucleotide primers.
| Name | Sequence
(5′-3′) | Degeneracy | Tm value, °C |
|---|
| BgyrbF |
GACGGCAAGAAGCGCttyatggartt | 4 | 60.2 |
| BgyrbR |
CACGGACACGCGCacrttnccrtg | 16 | 60.6 |
| BgyrbR' |
TGGTAGTCGGCGGTGtgytgraaytc | 8 | 60.3 |
| Table II.Verification primers. |
Table II.
Verification primers.
| Name | Sequence
(5′-3′) | Length of amplicon,
bp |
|---|
| bgrybseq1F |
CGAGTATCACTACGACATCC | 442 |
| bgrybseq1R |
CACCTTCACCGACAACAC |
|
| bgrybseq2F |
TCCGACGATCTTCCACAT | 271 |
| bgrybseq2R |
CACCTTCACCGACAACAC |
|
CODEHOP PCR amplification and
verification
CODEHOP PCR was performed in a gradient pattern with
a decrease in the annealing temperature of 0.5°C (from 49.5–47°C)
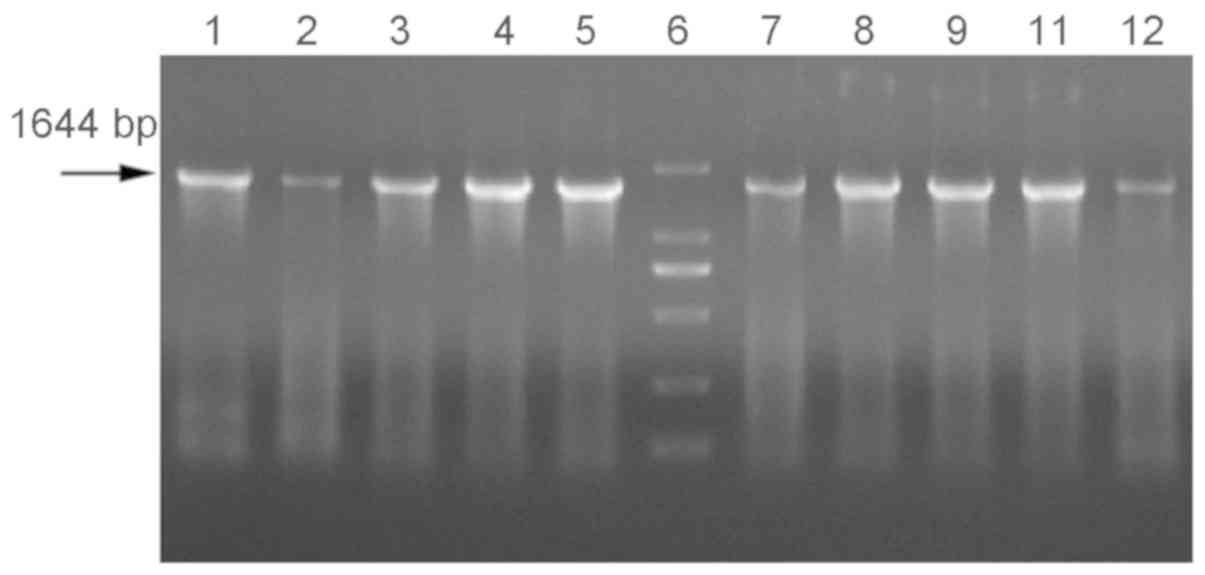
using the BgyrbF/BgyrbR primers. As demonstrated in Fig. 1, positive bands were obtained using
such primers at a temperature between 47.5 and 48.5°C. On this
basis, CODEHOP PCR was repeated using an annealing temperature of
48°C. The repeatability of the amplification results by
BgyrbF/BgyrbR are indicated in Fig.
2.
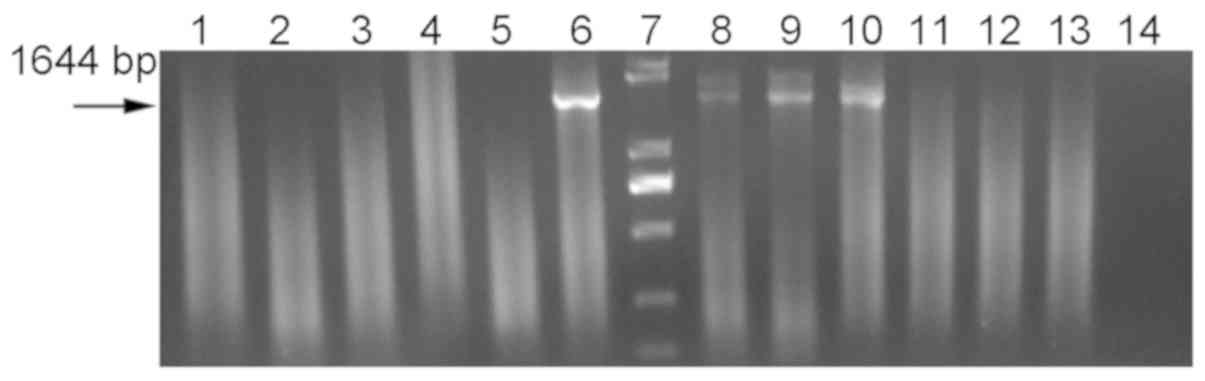 | Figure 1.COnsensus-DEgenerate Hybrid
Oligonucleotide Primer polymerase chain reaction results at
different annealing temperatures. Annealing temperatures: Lanes 1
and 2, 49.5°C; lanes 3 and 4, 49°C; lanes 5 and 6, 48.5°C; lane 7,
marker (100, 250, 500, 750, 1,000 and 2,000 bp); lanes 8 and 9,
48°C; lanes 10 and 11, 47.5°C; lanes 12 and 13, 47°C; lane 14,
blank control. The template used was Burkholderia gladioli
pv.alliicola20157 DNA. |
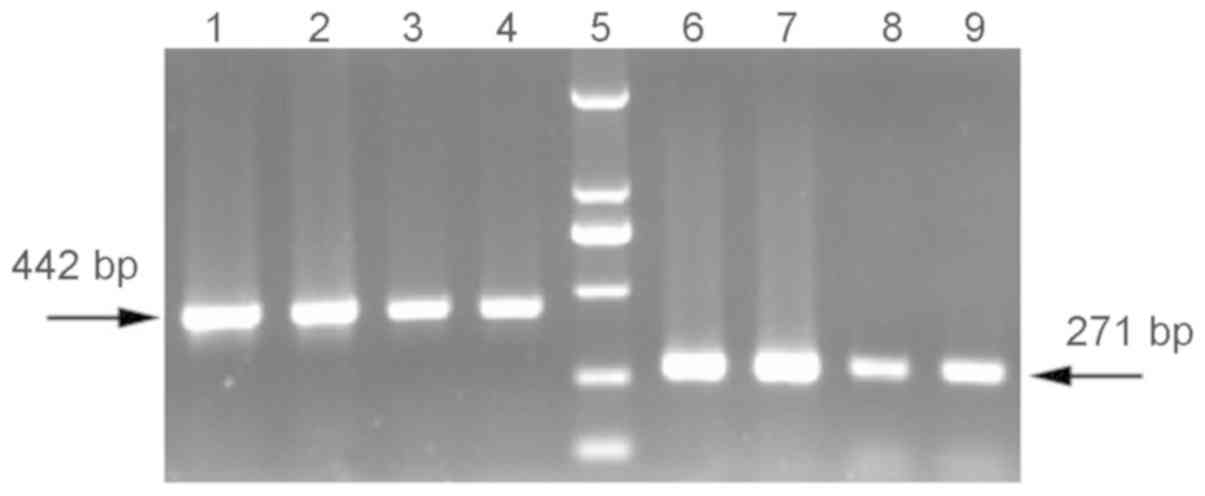
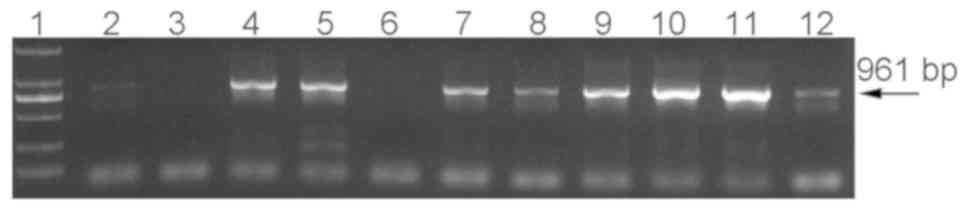
Two verification primers were used to verify the
amplified products of CODEHOP PCR. As demonstrated in Fig. 3, two pairs of primers
(bgrybseq1F/bgrybseq1R and bgrybseq2F/bgrybseq2R) were valid for
amplification of the CODEHOP product, and two amplification
fragments with lengths of 442 and 271 bp were obtained,
respectively.
Once the verification process was complete, DNA
sequencing was performed on the CODEHOP products. The full length
of the sequence was 1,644 bp, which was close to the full length of
the deposited gyrB gene of B. gladioli. Sequence
comparison was performed by the BlastN program, which revealed a
maximum identity of 87% between the gyrB gene of Bga and
that of the B. multivorans strain, FCF11. However, according
to the sequencing results, the non-degenerate sequences of CODEHOP
primers were BgyrbF (5′-GACGGCAAGAAGCGCTTTATGGAATT-3′) and BgyrbR
(5′-CACGGACACGCGCACATTTCCATG-3′).
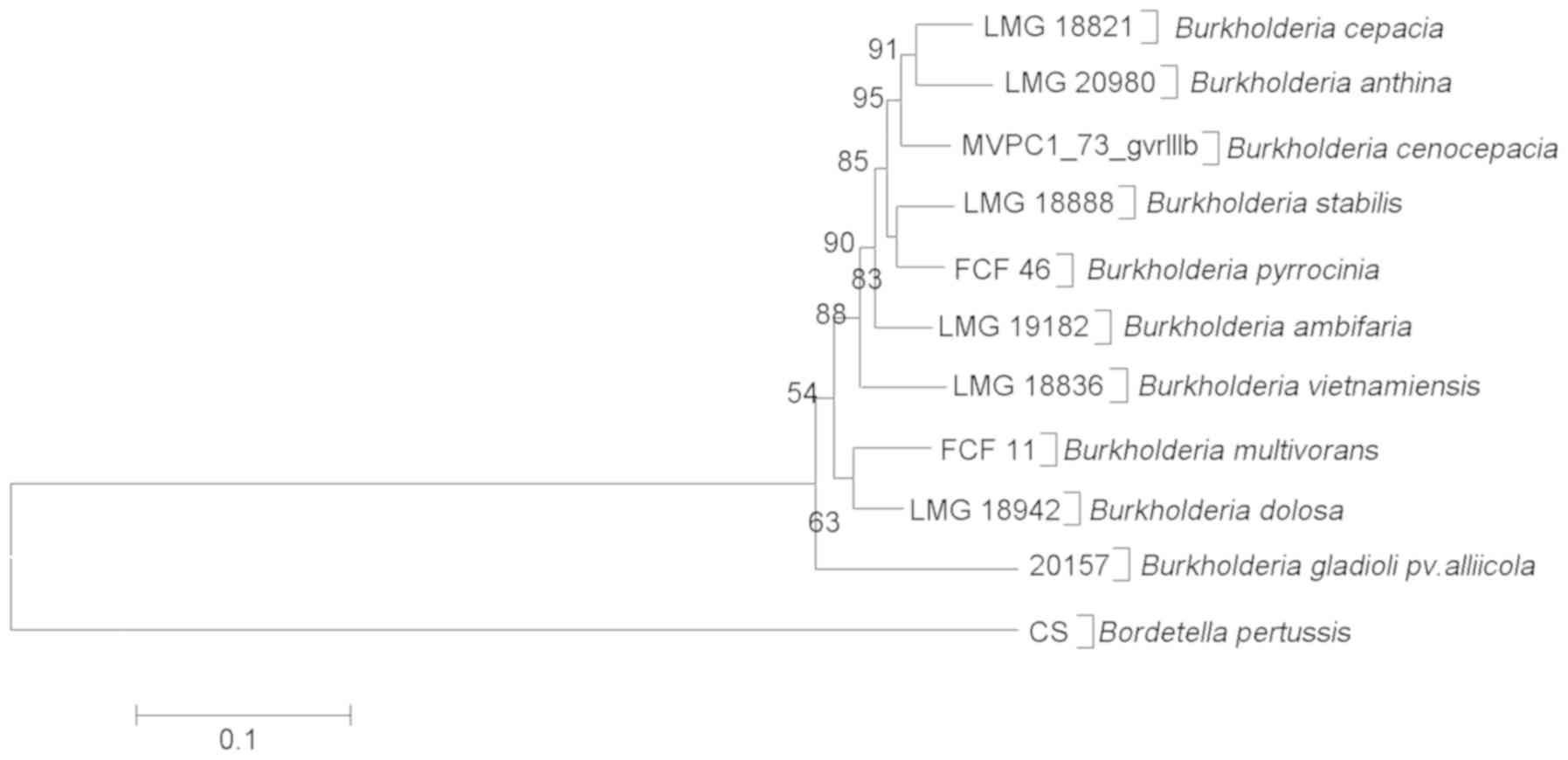
Phylogenetic analysis
Strains of Bcc selected for the phylogenetic
analysis are listed in Table III.
Multi-alignments including nine genomovar gyrB sequences of
Bcc and the gyrB sequence of Bordetella pertussis CS
as outgroup deposited in GenBank were used to construct the
phylogenetic tree. As demonstrated in Fig. 4, Bga formed a single cluster
separated from Bcc.
| Table III.Strains used for phylogenetic
analysis. |
Table III.
Strains used for phylogenetic
analysis.
| Strain | Accession no. | Coverage, % | Similarity
(nucleotide, %) | Similarity
(protein, %) |
|---|
|
Burkholderiacepacia strain LMG
18821 | EU240562 | 99 | 86 | 89 |
|
Burkholderiamultivorans strain
FCF11 | AY996876 | 100 | 87 | 87 |
|
Burkholderiacenocepacia strain
MVPC1_73_gvrIIIb | AY996886 | 99 | 86 | 90 |
|
Burkholderiastabilisstrain LMG
18888 | EU240566 | 99 | 86 | 89 |
|
Burkholderiavietnamiensis strain
LMG 18836 | EU240564 | 98 | 86 | 89 |
|
Burkholderiadolosa strain LMG
18942 | AY987924 | 100 | 87 | 88 |
|
Burkholderiaambifaria strain LMG
19182 | EU240568 | 99 | 86 | 90 |
|
Burkholderiaanthina strain LMG
20980 | AY987928 | 98 | 85 | 89 |
|
Burkholderiapyrrocinia strain
FCF46 | AY987932 | 100 | 86 | 89 |
| Bordetella
pertussis CS | CP002695 | 71 | 80 | – |
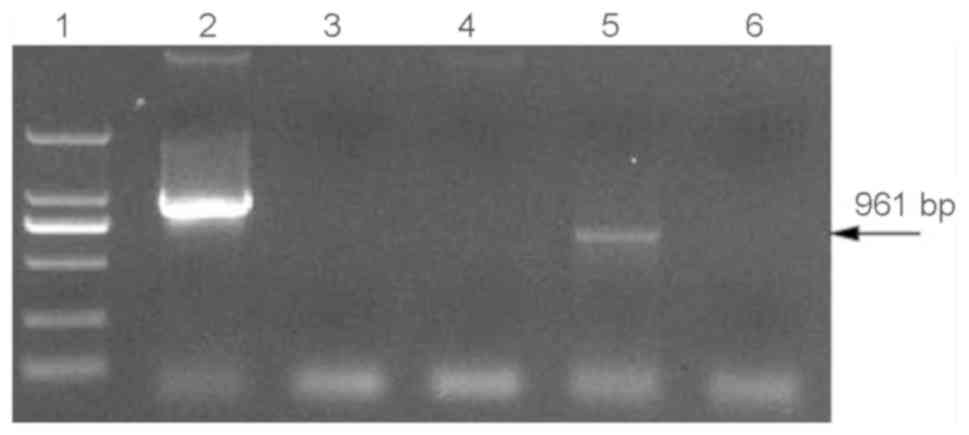
Results of the microarray assay
The primers and probes designed for the
discrimination of Bga and Bcc are summarized in Table IV. Universal primer pairs designated
as 20157F/20157R for the amplification of the gyrB gene of
Bga and Bcc were designed. A weak, non-specific band was observed
in B. cenocepacia, B. stabilis, B. anthina and B.
pyrrocinia (Figs. 5 and 6). After raising the annealing temperature
to 60°C, the non-specific bands in B. cenocepacia and B.
anthina were eliminated (data not shown). For B.
vietnamiensis, amplification was effective by decreasing the
annealing temperature to 50°C, as well as for B. cepacia. No
positive bands were obtained in the amplification performed using
the template obtained from B. andropogonis and B.
caryohpy, respectively. On the contrary, a weak, non-specific
band was obtained in B. glumae; however, the non-specific
positive bands were not observed after raising the annealing
temperature. A total of five genomovars and Bga amplified by
20157F/20157R were tested on the microchip assay, as demonstrated
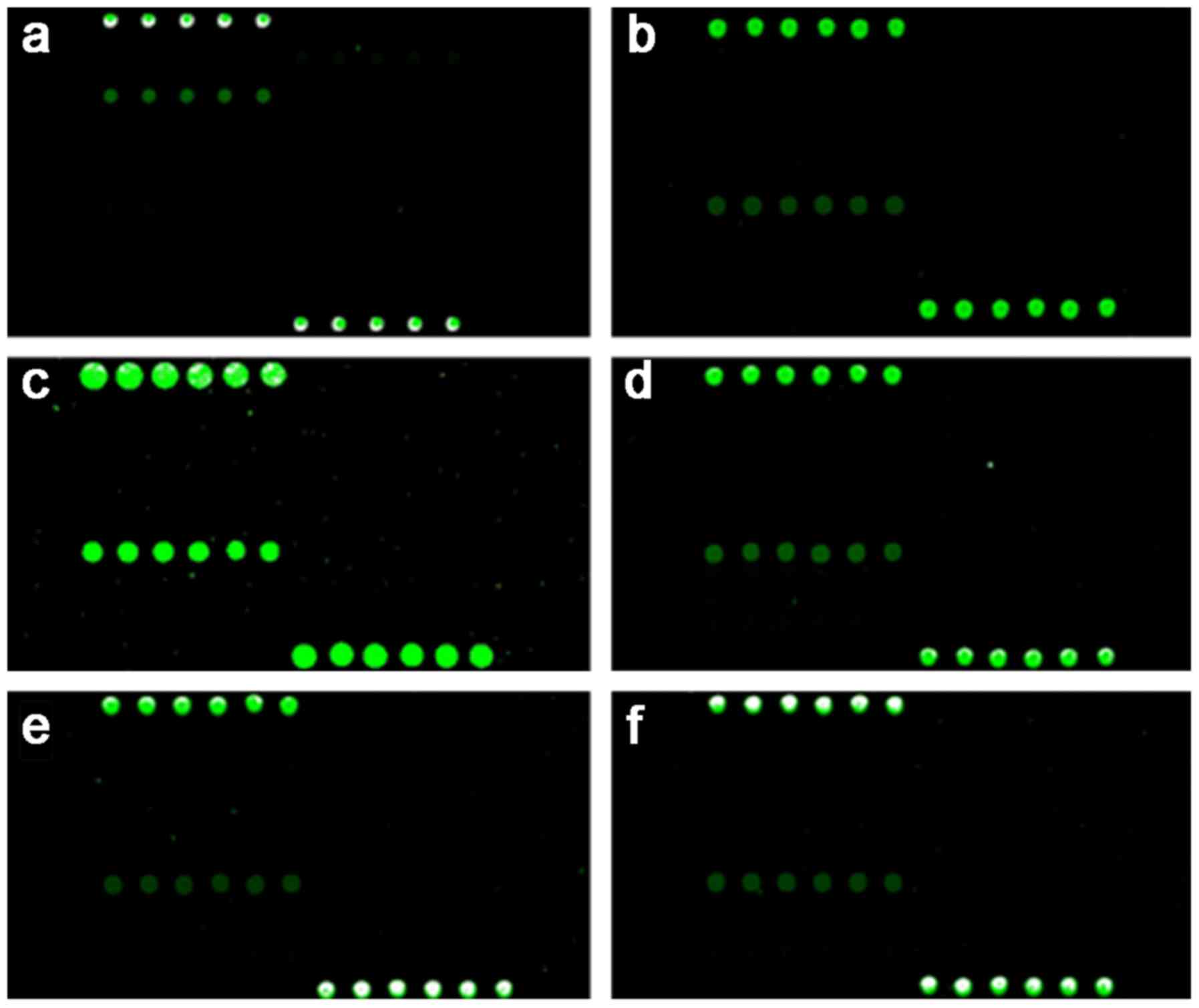
in Fig. 7. Between the two probes,
primer Bga-P1 (Fig. 7A) was more
effective for the recognition of Bga. Among the probes designed for
identification of Bcc and its genomovars, the probe Geno5-P2
(Fig. 7B-F) designed based on the
sequence of gyrB gene of B. vietnamiensis
demonstrated the best recognition rate over all test genomovars,
although it was originally designed for the recognition of B.
vietnamiensis. Geno5-P2 revealed a weak, non-specific
recognition rate with that of the amplification product of Bga,
which could be easily eliminated following an optimized
hybridization procedure. However, all the specific probes failed to
recognize the target genomovar, which was speculated to be
associated with the high identity between the gyrB gene of
different genomovars of Bcc.
 | Figure 5.Polymerase chain reaction results of
20157F/20157R. Lane 1, marker DL2000; lane 2,
Burkholderiacepacia Y3; lane 3, B. multivorans PW99;
lane 4, B. cenocepacia Y10; lane 5, B. cenocepacia
317; lane 6, B. stabilis J100; lane 7, B.
vietnamiensis 419; lane 8, B. anthina YP46; lane 9,
B. pyrrocinia 301; lane 10, B. arboris HT1; lane 11,
B. seminalis R45; lane 12, B. contaminans Y4. |
| Table IV.Sequence of primers and probes. |
Table IV.
Sequence of primers and probes.
| Name | Sequence
(5′-3′) |
|---|
| 20157F |
TCCTCCTTGCCGATCCCGCA |
| 20157R |
AGAACCGCGGCACCGAAGTG |
| Bga-P1 |
NH2-d(T)15-CTTCACCGACAACACGCAG |
| Geno5-P2 |
NH2-d(T)15-AAGGTGCTCAACGTCGA |
Discussion
CODEHOP strategy, a modified method of the ordinary
degenerate PCR, is more effective than the ordinary degenerate, PCR
particularly between distantly related species (26). A CODEHOP primer with a 5′
non-degenerate consensus sequence and a 3′ degenerate core
guarantees the stringency and degeneracy in the amplification
process.
In the present study, CODEHOP strategy was
successfully performed to amplify the sequence of the gyrB
gene of Bga based on the protein sequences of Bcc. One forward
primer and two reverse primers were designed (one of the reverse
primers failed to work). The amplification of the primer set
BgyrbF/BgyrbR was not as stable as expected during the
amplification performed on different strains of Bga. Thus, the most
stable annealing temperature was 48°C. Furthermore, the strategy
was more effective than the ordinary degenerate PCR, which failed
to work for the amplification of the gryB gene (data not
shown).
To obtain a magnified replication of CODEHOP
amplification products, the products were purified and linked with
a T-vector, and were transformed into E. coli competent
cells (DH5α). Sequencing results revealed that the gene obtained
showed an average similarity of 86% with the gyrB gene of
nine genomovars of Bcc and a maximum identity of 87% with that of
B. multivorans. In order to investigate the sequence
homology of the gyrB gene between Bcc and Bga, a comparison
was performed using Clustalx software, which revealed no evident
variable region in these sequences. Subsequently, the nucleotide
sequence was translated into a protein sequence, followed by
querying using the BlastX program in GenBank. The results revealed
an averaged similarity of 89% with the protein sequences of the
nine genomovars of Bcc. Additionally, the revolutionary rate of the
gyrB gene was faster at the nucleic acid level than at the
protein level in related species, thus the nucleic acid sequence
was employed to construct the phylogenetic tree.
A phylogenetic tree was constructed based on the
single gyrB gene sequence of Bga and Bcc, which revealed a
revolution divergence on the gyrB gene between these two
categories. The discrimination utility of the gyrB gene
between the two categories was then tested using microarray
methods. To the best of our knowledge, it is difficult to find a
well-performed universal primer for the gyrB gene of Bga and
Bcc due to sequence variation. Mass sequence data was analyzed to
find appropriate probes. In the present study, two probes for
recognition of gyrB of Bga, three universal probes for
recognition of that in Bcc, together with six probes specific for
recognition of five different genomovars were initially designed.
All the specific probes revealed cross-hybridization between
different genomovars on the chip. Two probes, Bga-P1 and
Geno5-P2, for recognition of Bgaand Bcc, respectively, demonstrated
the best hybridization result. Furthermore, sequence analysis
revealed that a difference was widely observed in the gyrB
gene within the strains of the same genomovar. A noteworthy
discovery was identified in B. multivoran; two obvious
groups existed in the gyrBgene of B. multivoranbased
on the similarity of gene sequences. This may explain the failure
in the amplification using a universal primer, although it
demonstrated a close similarity with the gyrB gene of
Bga.
It remains a challenge to discriminate Bga from Bcc
in clinical practice, and several molecular methods have been
developed for this based on the 16S and 23S rRNA genes (17,18). In
addition, a multiplex-PCR protocol has also been developed for the
specific detection of B. plantarii, B. glumae and, B.
gladioli in rice seeds based on the gyrB sequences
(23). In the present study, CODEHOP
strategy was used for amplification of the gyrB gene of Bga
based on the protein sequence of the gyrB gene of Bcc. As
demonstrated in the results, the strategy was practical, which
makes it a utility tool for homologous gene study or marker gene
selection between Bga and Bcc even within Bcc. In conclusion, the
gyrB gene was a useful marker gene in discriminating Bga
from Bcc based on the phylogenetic tree. Due to the high similarity
of the gyrB gene sequence of Bga and Bcc, a single probe was
not adequate to distinguish Bga from Bcc as revealed by the
microarray results. Therefore, more specific probes based on
discriminative genes that recognize different species would be
required.
Acknowledgements
The authors would like to thank Dr Guan-Ning Xie of
Zhejiang University for providing the DNA of Bcc.
Funding
The present study was supported by the fund of
General Administration of Quality Supervision, Inspection and
Quarantine of the People's Republic of China (grant no.
2013IK227).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LZ designed the experimental protocol, performed the
experiments, collected and analyzed raw data and wrote the
manuscript. WG modified the hybridization protocol of the microchip
array. YY modified the protocol of CODEHOP PCR amplification. ZW
provided the concept of the current study, reviewed all methods and
results, revised the manuscript and approved publication of the
final version of the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Severini G: Una batteriosidell
ixiamaculata e del gladiolus colvilli. Annali di Botanica.
11:413–424. 1913.
|
|
2
|
Hildebrand DC, Palleroni NJ and Doudoroff
M: Synonymy of pseudomonas gladioli severini 1913 and
pseudomonas marginata (McCulloch 1921) stapp 1928. Int J
Syst Evol Microbiol. 23:433–437. 1973.
|
|
3
|
Yabuuchi E, Kosako Y, Oyaizu H, Yano I,
Hotta H, Hashimoto Y, Ezaki T and Arakawa M: Proposal of
burkholderia gen. nov. and transfer of seven species of the
genus pseudomonas homology group II to the new genus, with
the type species burkholderia cepacia (Palleroni and Holmes
1981) comb. nov. Microbiol Immunol. 36:1251–1275. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mortensen JE, Schidlow DV and Stahl EM:
Pseudomonas gladioli (marginata) isolated from a patient
with cystic fibrosis. Clin Microbiol Newsletter. 10:29–30. 1988.
View Article : Google Scholar
|
|
5
|
Christenson JC, Welch DF, Mukwaya G,
Muszynski MJ, Weaver RE and Brenner DJ: Recovery of Pseudomonas
gladioli from respiratory tract specimens of patients with
cystic fibrosis. J Clin Microbiol. 27:270–273. 1989.PubMed/NCBI
|
|
6
|
Ross JP, Holland SM, Gill VJ, DeCarlo ES
and Gallin JI: Severe Burkholderia (Pseudomonas) gladioli
infection in chronic granulomatous disease: Report of two
successfully treated cases. Clin Infectr Dis. 21:1291–1293. 1995.
View Article : Google Scholar
|
|
7
|
Khan SU, Gordon SM, Stillwell PC, Kirby TJ
and Arroliga AC: Empyema and bloodstream infection caused by
Burkholderia gladioli in a patient with cystic fibrosis
after lung transplantation. Pediatr Infect Dis J. 15:637–639. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ursing JB, Rossell RA, Garcfa-Valdes E and
Lalucat J: Taxonomic note: A pragmatic approach to the nomenclature
of phenotypically similar genomic groups. Society for General
Microbiology, Reading, ROYAUME-Uni. 1995.
|
|
9
|
Yoshihisa H, Zenji S, Fukushi H, Katsuhiro
K, Haruhisa S and Takahito S: Production of antibiotics by
Pseudomonas cepacia as an agent for biological control of
soilborne plant pathogens. Soil Biology Biochem. 21:723–728. 1989.
View Article : Google Scholar
|
|
10
|
McLoughlin TJ, Quinn JP, Bettermann A and
Bookland R: Pseudomonas cepacia suppression of sunflower
wilt fungus and role of antifungal compounds in controlling the
disease. Appl Environmental Microbiol. 58:1760–1763. 1992.
|
|
11
|
King EB and Parke JL: Population density
of the biocontrol agent Burkholderia cepacia AMMDR1 on four
pea cultivars. Soil Biol Biochem. 28:307–312. 1996. View Article : Google Scholar
|
|
12
|
Isles A, Maclusky I, Corey M, Gold R,
Prober C, Fleming P and Levison H: Pseudomonas cepacia
infection in cystic fibrosis: An emerging problem. J Pediatr.
104:206–210. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thomassen MJ, Demko CA, Klinger JD and
Stern RC: Pseudomonas cepacia colonization among patients
with cystic fibrosis. A new opportunist. Am Rev Respir Dis.
131:791–796. 1985.PubMed/NCBI
|
|
14
|
Lacy DE, Spencer DA, Goldstein A, Weller
PH and Darbyshire P: Chronic granulomatous disease presenting in
childhood with Pseudomonas cepacia septicaemia. J Infect.
27:301–304. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Speert DP, Bond M, Woodman RC and Curnutte
JT: Infection with Pseudomonas cepacia in chronic
granulomatous disease: Role of nonoxidative killing by neutrophils
in host defense. J Infect Dis. 170:1524–1531. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Holmes A, Govan J and Goldstein R:
Agricultural use of burkholderia (Pseudomonas) cepacia: A
threat to human health? Emerg Infect Dis. 4:221–227. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Whitby PW, Pope LC, Carter KB, LiPuma JJ
and Stull TL: Species-specific PCR as a tool for the identification
of burkholderia gladioli. J Clin Microbiol. 38:282–285.
2000.PubMed/NCBI
|
|
18
|
Bauernfeind A, Schneider I, Jungwirth R
and Roller C: Discrimination of burkholderia gladioli from
other burkholderia species detectable in cystic fibrosis
patients by PCR. J Clin Microbiol. 36:2748–2751. 1998.PubMed/NCBI
|
|
19
|
Coenye T, Schouls LM, Govan JR, Kersters K
and Vandamme P: Identification of Burkholderia species and
genomovars from cystic fibrosis patients by AFLP fingerprinting.
Int J Syst Bacteriol. 4:1657–1666. 1999. View Article : Google Scholar
|
|
20
|
Vermis K, Coenye T, Mahenthiralingam E,
Nelis HJ and Vandamme P: Evaluation of species-specific recA-based
PCR tests for genomovar level identification within the
Burkholderia cepacia complex. J Med Microbiol. 51:937–940.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Baldwin A, Mahenthiralingam E, Thickett
KM, Honeybourne D, Maiden MC, Govan JR, Speert DP, Lipuma JJ,
Vandamme P and Dowson CG: Multilocus sequence typing scheme that
provides both species and strain differentiation for the
Burkholderia cepacia complex. J Clin Microbiol.
43:4665–4673. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Payne GW, Vandamme P, Morgan SH, Lipuma
JJ, Coenye T, Weightman AJ, Jones TH and Mahenthiralingam E:
Development of a recA gene-based identification approach for the
entire burkholderia genus. Appl Environ Microbiol.
71:3917–3927. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maeda Y, Shinohara H, Kiba A, Ohnishi K,
Furuya N, Kawamura Y, Ezaki T, Vandamme P, Tsushima S and Hikichi
Y: Phylogenetic study and multiplex PCR-based detection of
burkholderiaplantarii, burkholderiaglumae and burkholderia
gladioli using gyrB and rpoD sequences. Int J Syst Evol
Microbiol. 56:1031–1038. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Slinger R, Yan L, Myers R, Ramotar K, St
Denis M and Aaron SD: Pyrosequencing of a recA gene variable region
for burkholderia cepacia complex genomovar identification.
Diagn Microbiol Infect Dis. 58:379–384. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tabacchioni S, Ferri L, Manno G, Mentasti
M, Cocchi P, Campana S, Ravenni N, Taccetti G, Dalmastri C,
Chiarini L, et al: Use of the gyrB gene to discriminate among
species of the Burkholderia cepacia complex. FEMS Microbiol
Lett. 281:175–182. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rose TM, Henikoff JG and Henikoff S:
CODEHOP (COnsensus-DEgenerate Hybrid Oligonucleotide Primer) PCR
primer design. Nucleic Acids Res. 31:3763–3766. 2003. View Article : Google Scholar : PubMed/NCBI
|