Introduction
Ischemic brain injury is caused by insufficient
blood flow to the brain, is characterized by oxidative stress,
hypoxia, inflammation and glutamate excitotoxicity, which leads to
cell apoptosis and death (1).
Ischemic stroke has been indicated as a leading cause of death and
long-term disability worldwide (2).
Neonatal hypoxic-ischemic brain injury or neonatal stroke is a
major cause of hypoxic-ischemic encephalopathy and cerebral palsy
(3). The advances in therapy for the
purpose of decreasing ischemic brain injury have been limited
because the pathophysiological mechanisms remain unknown.
Therefore, it is of great importance to understand the pathological
mechanisms of ischemia-associated cell death in order to develop
effective therapies for ischemic brain injury.
microRNAs (miRNAs or miRs) are a family of small
RNAs (~22 nucleotides) that modify the expression of genes
implicated in biological processes, including tumor cell
differentiation, proliferation and apoptosis (4,5).
Previous studies have indicated that miRNAs are abundantly
expressed in the nervous system and have been initially identified
as crucial molecular mediators in the regulation of neuronal cell
survival (6–8). miR-497 was the first miRNA identified
to promote ischemic neuronal death by negatively regulating
anti-apoptotic proteins, B-cell lymphoma-2 (Bcl-2) and Bcl-2-like
protein 2 (9). miR-210, miR-29b and
miR-124 have also been demonstrated to participate in the
biological processes concerning the nervous system (10–12).
These findings suggest that several miRNAs may be
potential candidates for possible biomarkers or therapeutic targets
in stroke. In the present study, the expression of miR-196a was
explored during OGD in vitro and MCAO in vivo. The
role of miR-196a in ischemic brain injury was investigated.
Materials and methods
Primary culture of rat cortical
neurons
A total of 15 pregnant, female Sprague Dawley rats
(mean weight, 245±10 g) were obtain from the SLAC Laboratory Animal
Corporation (Shanghai, China). All rats housed at 20–25°C with 60%
humidity under 12 h light:dark cycles with free access to food and
water throughout the experiment. After 19–20 days, 115 neonatal
rats were born and cerebral cortexes were removed within 24 h by
aseptic decapitation. Then cortexes were placed in D-hanks solution
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and
sliced into 1-mm3 fragments. Subsequently, the cortical
pieces were dissociated using 0.25% trypsin (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) supplemented with 100 ng/ml DNase (Roche
Diagnostics, Basel, Switzerland) at 37°C for 35 min. The digestion
was terminated with Dulbecco's modified Eagle's medium (DMEM;
Hyclone; Logan, UT, USA) supplemented with 10% fetal bovine serum
(Gibco; Thermo Fisher Scientific, Inc.). Isolated cells were
suspended in neurobasal-A medium supplemented with 2% B27, 1%
glutamine (Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml
penicillin and 100 g/ml streptomycin, and seeded into 6-well plates
at 5×104 cell/well; the plates were precoated with 10
µg/ml poly-D-lysine (Sigma-Aldrich; Merck KGaA). Cells were
incubated in a humidified incubator containing 5% CO2
and 95% O2 at 37°C for 7 days. The present study and all
animal experiments were approved by the Ethics Committee at Beilun
People's Hospital of Ningbo (Ningbo, China).
OGD model
To construct an OGD model, glucose-free Earl's
balanced salt solution (EBSS) medium (consisting of 6,800 mg/l
NaCl, 400 mg/l KCl, 264 mg/l CaCl2, 200 mg/l
MgCl2, 2,200 mg/l NaHCO3 and 140 mg/l
NaH2PO4 mg/l; pH 7.2) was purged with
N2/CO2 (95/5%) at 37°C for 20 min, resulting
in an oxygen content of 1%. Cortical neuronal cell cultures were
washed three times with glucose-free EBSS medium and incubated at
37°C for 4 h in oxygen-free N2/CO2 (95/5%)
gas as previously described (13).
For reoxygenation, cells were incubated in fresh
neurobasal-A-medium supplemented with 2% B-27 in an atmosphere
containing 5% CO2 at 37°C for 12 and 24 h. The control
group was incubated in EBSS with 10 mM glucose at 37°C in 5%
CO2 for 12 and 24 h.
MCAO rat model
A total of 32 adult male Sprague Dawley rats (age,
10 weeks; mean weight, 289±10 g) were subjected to MCAO from the
SLAC Laboratory Animal Corporation. All rats housed at 20–25°C with
60% humidity under 12 h light:dark cycles with free access to food
and water throughout the experiment. Rats were anesthetized using
1% sodium pentobarbital (40 mg/kg; Shanghai Xitang Company,
Shanghai, China). Subsequently, the right common carotid artery,
external carotid artery and internal carotid artery were exposed
via a midline cervical incision. A piece of 4/0 monofilament nylon
suture with a heat-induced rounded tip was inserted through the
right internal carotid artery to the base of the middle cerebral
artery, which occluded the blood flow to the cortex and striatum.
Following 90 min of MCAO, the rats were allowed to recover for 24
h, as described previously (14).
Sham-operated mice underwent the internal carotid artery separation
without the suture insertion. Additionally, the rats were randomly
separated into four groups: Sham operation, MCAO model, MCAO model
treated with antagomiR control (5′-CAGUACUUUUGUGUAGUACAA-3′) and
antagomiR-196a (5′-CCCAACAACAUGAAACUACCUA-3′). The antagomiR
control and antagomiR-196a (both 80 mg/kg) were injected into the
caudal vein.
Cell transfection
A total of 2×105 cortical neurons were
plated in 6-well plates and cultured at 37°C for 24 h. miR-196
mimics, miR-196 inhibitor or high mobility group A1 (HMGA1) vectors
were transfected using Lipofectamine 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) at a final concentration of 30 mM. The
sequences for the miRNAs were as follows: NC control
(5′-CAGUACUUUUGUGUAGUACAA-3′), miR-155 mimic
(5′-UAGGUAGUUUCAUGUUGUUGGG-3′), miR-155 inhibitor
(5′-CCCAACAACAUGAAACUACCUA-3′). Neurons were harvested for terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labelling
(TUNEL) assay, western blot analysis or luciferase activity assay,
at the indicated time points.
Infarct volume analysis
A total of 24 h after reperfusion, the rats were
re-anesthetized and sacrificed. Then, the brain tissues of each
group were removed, frozen for 30 min at −20°C and sliced into four
coronal sections (1 mm thick). The sections were placed in a 1%
solution of 2,3,5-triphenyltetrazolium chloride (Sigma-Alrich;
Merck KGaA) for 10 min at 37°C and fixed in 4% formalin at 4°C for
24 h. The infarct region lacked staining and appeared white,
whereas the normal non-infarct tissue appeared red. Images were
captured of both sides of each stained coronal slice using a
digital camera and infarction was measured using ImageJ Pro 6.0
digital image analysis software (National Institutes of Health,
Bethesda, MD, USA). The infarct volume was expressed as a
percentage: Total infarct volume of ipsilateral structure/total
volume of contralateral structure ×100.
TUNEL assay
Coronal brain tissues were fixed in 10%
paraformaldehyde at 4°C for 24 h, dehydrated in ethanol (100, 95,
90, 80 and 70%; Beijing Solarbio Science & Technology Co.,
Ltd., Beijing, China) and embedded in paraffin. The specimens were
cut into 4-µm thick slices for TUNEL and immunocytochemistry
analysis. TUNEL assay was performed according to the manufacturer's
instructions of the TdT-mediated dUTP nick end labeling analysis
kit (Roche Diagnostics). All cell nuclei were indicated via blue
staining and the positive nuclei were indicated via green staining.
The number of positive nuclei was determined by manually counting
the positively labeled nuclei in five randomly selected fields
under a fluorescence microscope (magnification, ×400).
Immunocytochemistry
The 4-µm thick sections were deparaffinized and
rehydrated with ethanol (100, 95 and 75%), and exposed to the
antigen retrieval system (10 mM sodium citrate and 0.05% Tween 20;
pH 6.0) at room temperature for 15 min. Endogenous peroxidase was
blocked with 3% hydrogen peroxide for 15 min at room temperature.
Subsequently, the slides were rinsed in 0.01 mol/l PBS three times
and subsequently incubated with the primary mouse polyclonal HMGA1
antibody (1:200; cat. no. ab226112; Abcam, Cambridge, MA, USA) for
1 h at room temperature. Following washing with PBS twice for 5
min, the slides were incubated with horseradish peroxidase-labeled
goat anti-mouse antibody (1:100; cat. no. A0192; Beyotime Institute
of Biotechnology, Haimen, China) at room temperature for 1 h.
Samples were stained using a 3,3′-diaminobenzidine peroxidase
substrate kit (Beyotime Institute of Biotechnology) according to
the manufacturer's protocol. Positive nuclei were indicated as
brown in color. Slides were observed using a fluorescence
microscope (magnification, ×400) and the number of positive cells
was analyzed by Image J pro 6.0.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA obtained from rats and cells was extracted
using TRIzol Reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. A total of 1 µg total RNA
was reverse-transcribed into cDNA using a TaqMan microRNA Reverse
Transcription kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.) in a 15-µl total volume according to the manufacturer's
protocol. The reactions were incubated for 5 min at 95°C, followed
by 40 cycles consisting of a 15 sec interval at 95°C and 1 min
interval at 60°C. Expression levels of miR-196a were measured by
TaqMan MicroRNA Assay kits (Applied Biosystems; Thermo Fisher
Scientific, Inc.) in triplicate on an IQ5 real-time PCR system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) and normalized
using GAPDH as the internal control. The quantification was
performed by the comparative 2−ΔΔCq method (15). The primer sequences (5′→3′) were:
HMGA1, forward: TTACCGAGTACCCCACGCTA, reverse:
AGGCTGGGACAAATACTGGC; GAPDH, forward: GATGGTGAAGGTCGGTGTGA,
reverse: TGAACTTGCCGTGGGTAGAG; miR-196a RT-primer:
CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCCCAACAA, forward:
ACACTCCAGCTGGGTAGGTAGTTTCATGTT, reverse: TGGTGTCGTGGAGTCG.
Western blot analysis
Total proteins from cells were extracted with
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology) and protein concentration was tested using the BCA
assay (Beijing Solarbio Science & Technology Co., Ltd.). A
total of 30 µg protein were loaded and separated using 10%
Tris-glycine SDS-PAGE, and transferred to polyvinylidene fluoride
membranes (EMD Millipore, Billerica, MA, USA) using the
electrophoretic transfer system (Bio-Rad Laboratories, Inc.). The
membranes were blocked with 5% non-fat dry milk in Tris-HCl buffer
saline (pH 7.4) containing 0.1% Tween 20 for 1 h at room
temperature and incubated with primary antibodies against HMGA1 and
GAPDH (both 1:1,000; cat. no. ab181602; Abcam) overnight at 4°C.
Subsequently, the membranes were incubated with horseradish
peroxidase-conjugated secondary antibodies (1:2,500; cat. no.
A0192) at room temperature for 2 h followed by band detection with
a Super Signal Enhanced Chemiluminescence Kit (Pierce; Thermo
Fisher Scientific, Inc.). Quantification of the bands was performed
using densitometric analysis and the ImageJ Pro 6.0 analysis system
(National Institutes of Health).
Bioinformatics analysis
To predict the target genes of miR-196a, a series of
bioinformatics analyses were used. The authors predicted the target
genes for miR-196a with miRbase (http://www.mirbase.org/), TargetScan (http://www.targetscan.org/) and miRDB (mirdb.org/miRDB/). The predicted targets included
HMGA1.
Luciferase assay
Cortical neuronal cells were plated at a density of
4×103 cells/well in 96-well plates and cultured at 37°C
for 24 h. Cells were co-transfected with 40 ng recombinant
wild-type (WT) or mutated (MUT) luciferase vectors
[pGL3-HMGA1-3′-untranslated region (UTR)-WT or
pGL3-HMGA1-3′-UTR-Mut], pRL-TK vectors and miR-196a mimics or a
negative control (NC) mimics (Guangzhou RiboBio Co., Ltd.,
Guangzhou, China) at a final concentration of 45 nM using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. Luciferase assays
were performed with a dual luciferase reporter assay system
(Promega Corporation, Madison, WI, USA) 48 h following
transfection. Renilla luciferase activity was normalized to
that of Firefly luciferase.
Statistical analysis
All data were expressed as the mean ± standard
deviation of at least three separate experiments. Difference
between two groups was compared by Student's t-test. Difference
among multiple groups was analyzed by one-way analysis of variance
followed by a post-hoc Student-Neuman-Keuls test. P<0.05 was
considered to indicate a statistically significant difference.
Results
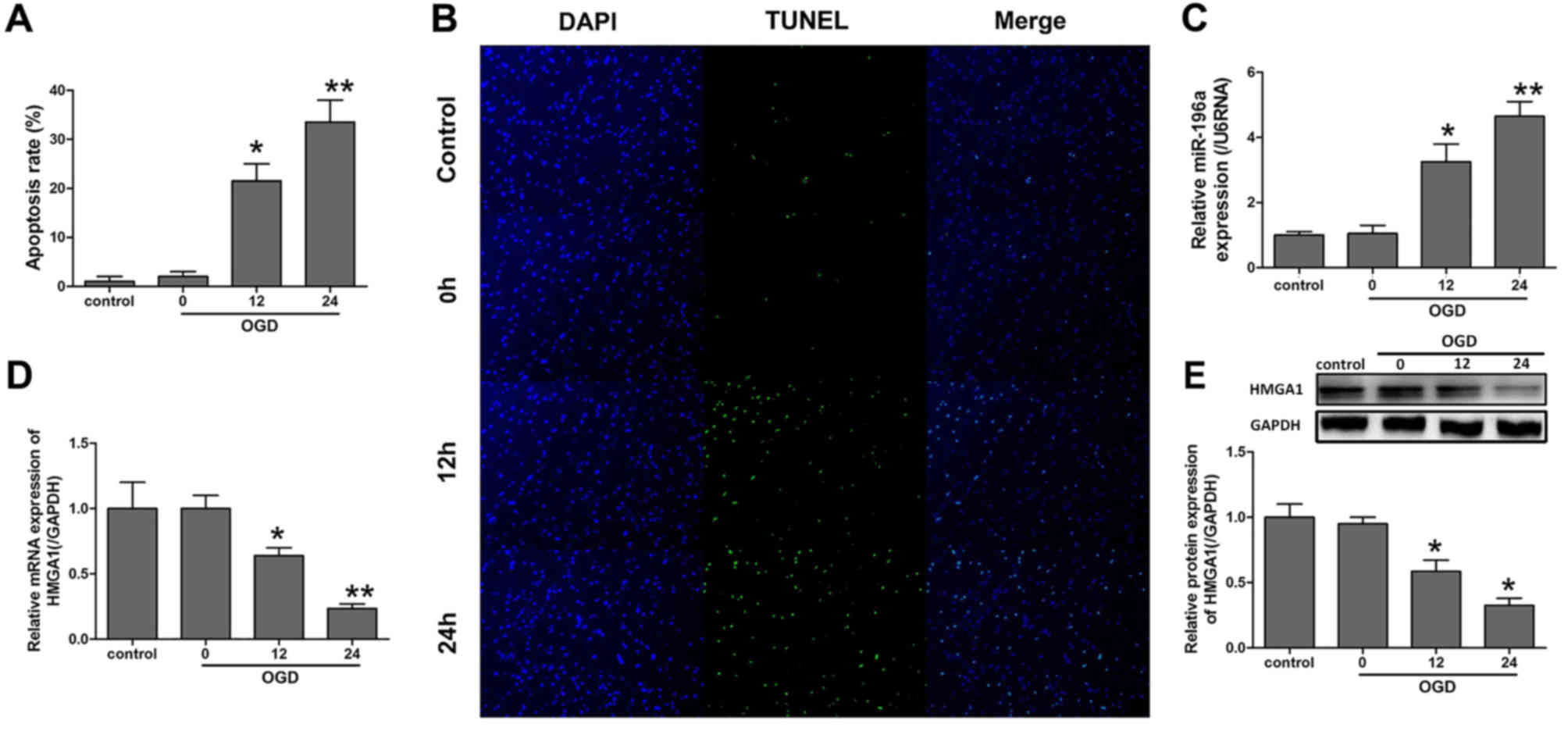
Expression of miR-196a and HMGA1 was
changed in cortical neurons subjected to OGD
To investigate the role of miR-196a and HMGA1 in
ischemic brain injury, an OGD model of cortical neurons was
established. RT-qPCR was performed to evaluate the gene expression
levels (Fig. 1). As Fig. 1B demonstrated, OGD induced apoptosis
and the difference between the control and OGD groups at 12 and 24
h was significant (P<0.05; Fig.
1A). Notably, the miR-196a expression was elevated
significantly following OGD at 12 and 24 h compared with the
control (P<0.05; Fig. 1C).
Conversely, the mRNA and protein expression levels of HMGA1 were
significantly downregulated after 12 and 24 h of OGD compared with
the control (P<0.05; Fig. 1D and
E).
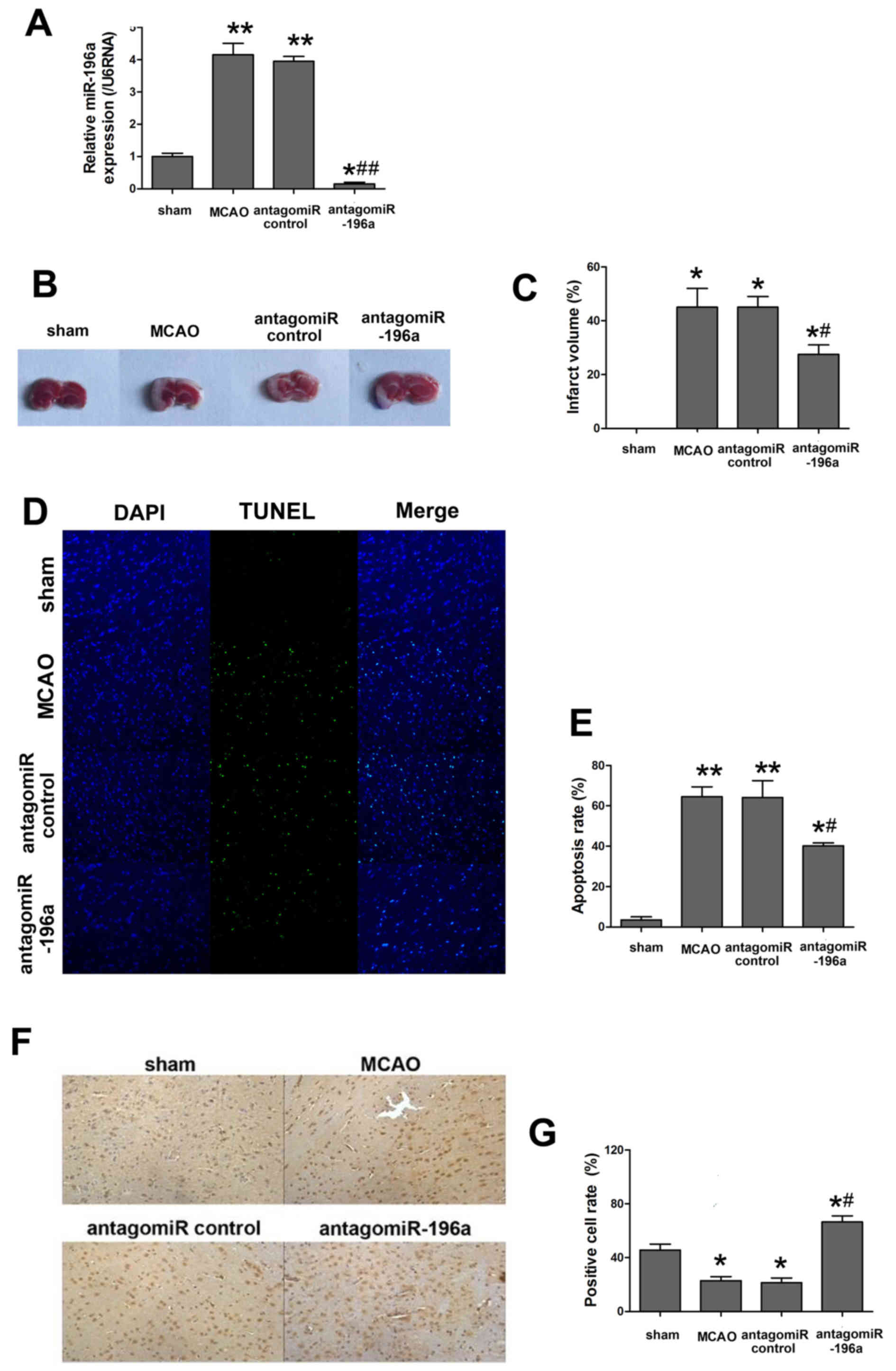
miR-196a reduces the neuronal rate of
apoptosis and infarction following ischemic brain injury in
vivo
A rat MCAO model was constructed to further explore
the effect of miR-196a on ischemic brain injury and its mechanism
(Fig. 2). The expression of miR-196a
was determined to assess the effects of antagomiR-196a. As
presented in Fig. 2A, miR-196a
expression in MCAO was significantly higher than the sham group
(P<0.01), whereas antagomiR-196a injection significantly
decreased the miR-196a expression compared with the antagomiR
control group (P<0.05). The infarct volume and cortical neuron
apoptosis rate was increased in the MCAO group compared with the
sham and this difference was indicated as statistically significant
(P<0.05; Fig. 2B-E), which
indicated the successful establishment of the in vivo model.
As expected, knockout of miR-196a by intracerebroventricular
injection of antagomiR-196a resulted in a significant decrease in
miR-196a expression compared with the MCAO group (P<0.05;
Fig. 2A) and significantly improved
the recovery of brain function as the infarct volume and the
apoptosis rate were significantly decreased compared with the MCAO
groups (P<0.05; Fig. 2B-E).
Furthermore, TUNEL staining indicated that the proportion of
apoptotic cells was decreased in miR-196a antagomiR-injected rat
brains compared with the antagomir control (Fig. 2D) and the difference was
statistically significant (Fig. 2E).
Considering the abnormal expression of HMGA1 in cells undergoing
OGD (Fig. 1E), the expression of
HMGA1 using immunocytochemistry in rat brains subjected to MCAO was
evaluated. Consistent with the previous study, the expression of
HMGA1 was markedly downregulated in the MCAO group compared with
that in the sham group and antagomir-196a injection significantly
enhanced the HMGA1 expression to sham group (P<0.05; Fig. 2F and G, respectively).
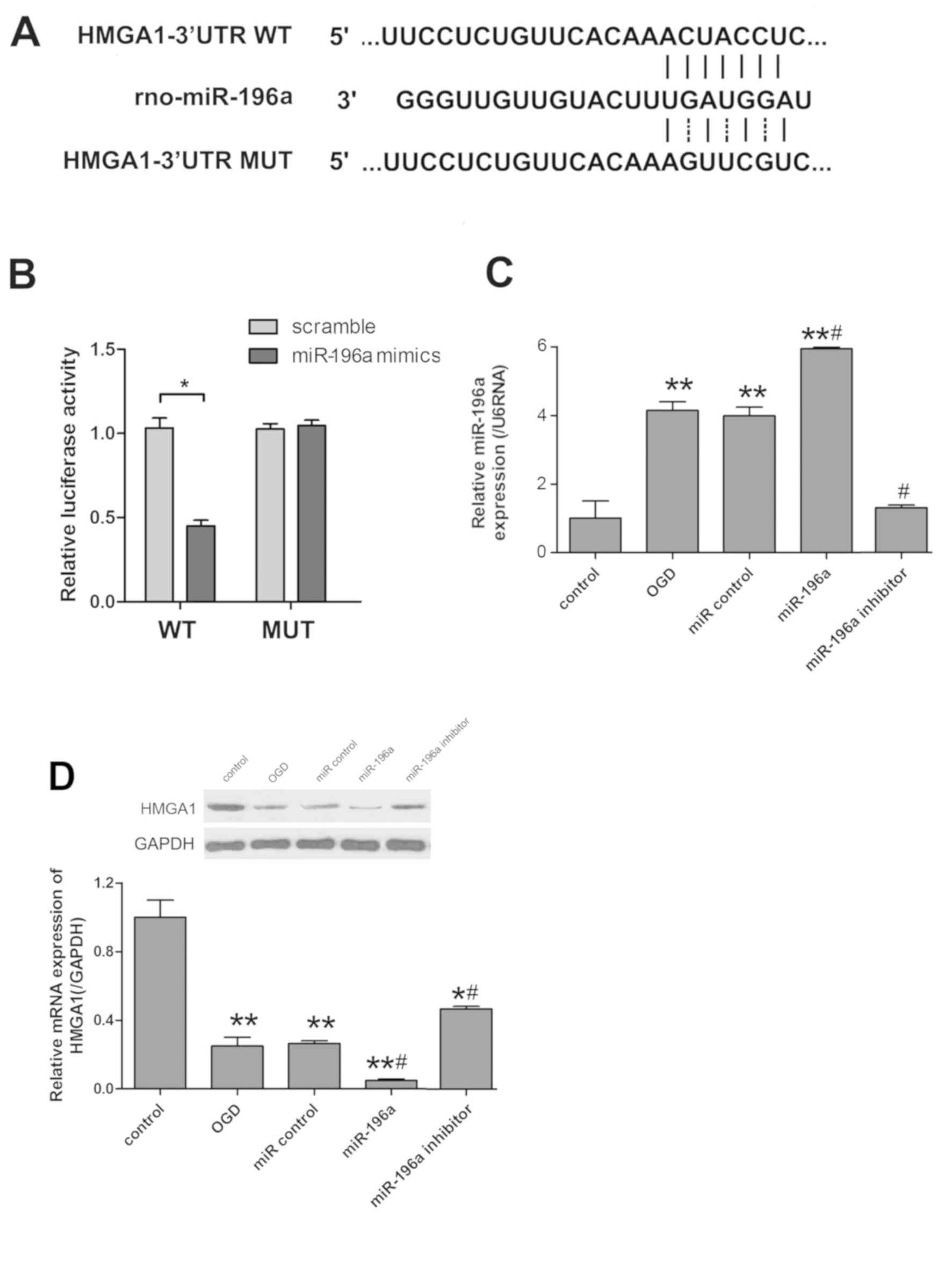
miR-196a directly targets HMGA1 and
inhibits its expression
To identify the target genes of miR-196a in
regulating ischemic brain injury, candidate genes were investigated
using miRBase, TargetScan 5.1 and micoRNA databases. HMGA1 was
selected as a potential candidate gene due to its abnormal
expression in cortical neuronal cells subjected to OGD and in rat
brains following MCAO. miR-196a mediated HMGA1 expression by
directly binding to the 3′UTR of HMGA1 in cortical neuronal cells
(Fig. 3A). Luciferase assay was
performed to indicate whether miR-196a targeted HMGA1.
Co-transfection of miR-196a mimics significantly inhibited the
luciferase activity produced by the reporter construct that
contained the wild-type 3′UTR segment of HMGA1 (P<0.05), whereas
no significant effect was observed with the construct containing
the mutated segment of HMGA1 3′UTR (Fig.
3B). RT-qPCR demonstrated that miR-196a expression was
significantly increased in the miR-196a mimics group, but was
reduced in miR-196a inhibitor group when compared with the OGD
group (P<0.05; Fig. 3C). In
addition, western blot analysis was applied to observe the protein
expression levels of HMGA1. The results revealed that transfection
of miR-196a mimics significantly decreased HMGA1 in OGD-induced
cells, whereas miR-196a inhibitor increased HMGA1 compared with the
OGD group (P<0.05; Fig. 3D).
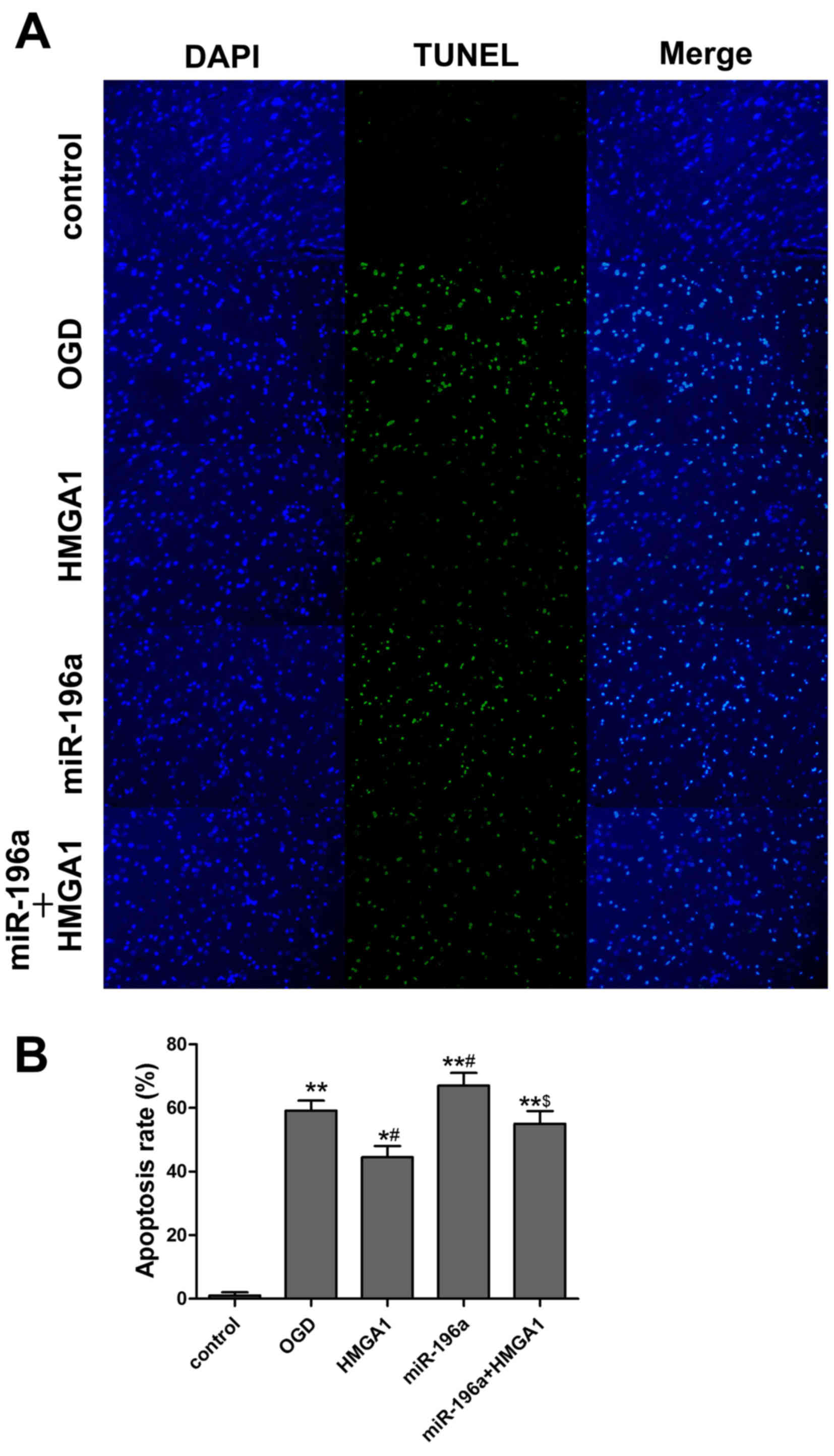
HMGA1 overexpression suppresses cell
apoptosis in cortical neurons subjected to OGD
To directly determine the effect of HMGA1 on
OGD-induced neuronal damage, neuronal cells were transfected with
pcDNA3.1/HMGA1 vector and the TUNEL assay was performed to evaluate
the apoptosis of cortical neurons under different treatments
(Fig. 4A). Compared with the
control, HMGA1 overexpression significantly suppressed the
apoptosis induced by OGD (P=0.043; Fig.
4B).
HMGA1 reverses the induction of
apoptosis by miR-196a
In accordance with the findings in the in
vivo study, miR-196a overexpression was indicated to promote
the apoptosis in OGD neurons. To explore whether miR-196a targets
HMGA1, neuronal cells were co-transfected with HMGA1 vector and
miR-196a mimics. As expected, HMGA1 overexpression significantly
reversed the induction of apoptosis by miR-196a (P<0.05;
Fig 4B).
Discussion
miRNA binding is mediated by argonaute proteins
within the RNA-induced silencing complex, which is dependent on the
degree of sequence complementarity and leads to either cleavage of
the target mRNA or a reduction in its translational efficiency
(16). A subset of miRNAs are
abundantly expressed in the human brain (17) and have critical roles in the
pathophysiology of brain seizures, ischemia and trauma (18,19).
miR-196a was initially identified as an oncogene
that was associated with apoptosis, invasion and proliferation
(20). Increasing evidence has
suggested the aberrant expression of miR-196a is a frequent event
in various cancers, including head and neck squamous cell
carcinomas, laryngeal cancer, pancreatic cancer and gastric cancer
(21–24). In addition, miR-196a is a putative
diagnostic biomarker or therapeutic target for various human
diseases, including adrenomyeloneuropathy, chronic hepatitis and
Huntington's Disease (25–27). To date, limited understanding exists
regarding the association between miR-196a and ischemic brain
injury. In the present study, miR-196a expression was significantly
increased during OGD in vitro and MCAO in vivo. To
determine whether miR-196a has a role in ischemic brain injury, its
expression was downregulated and the effects explored. The results
demonstrated that knockdown of endogenous miR-196a expression
significantly reduced brain infarct size and protected against
ischemic-induced brain cell apoptosis. To identify the target genes
of miR-196a in regulating cell apoptosis, miRBase and TargetScan
5.1 were used to search for candidate genes. Due to the low
expression of HMGA1 in the current study, HMGA1 was selected as a
potential target gene for miR-196a. However, whether miR-196a
directly targets HMGA1 required further elucidation. Luciferase
assay results indicated that miR-196a significantly repressed
luciferase activity in the cortical neuron cells transiently
transfected with wide-type HMGA1 3′UTR compared with cells
transfected with mutated HMGA1 3′UTR, which suggested that HMGA1
may be a direct target of miR-196a. In addition, western blot
analysis revealed that knockdown of miR-196a significantly
increased the protein expression of HMGA1. The present study
indicated that HMGA1 may be a physiological target of miR-196a and
regulate its expression at the transcriptional level.
HMGA1 proteins are non-histone proteins that
regulate chromatin structure and gene expression during
embryogenesis, tumourigenesis and immune responses (28,29).
HMGA1 is considered as a key hub for several oncogenic pathways,
including the Wnt/ß-catenin, Notch pathways and
Ras/extracellular-related kinase signaling (30,31).
Previous results have indicated that HMGA1 represses p53 apoptotic
activity by promoting the cytoplasmic relocalization of the p53
proapoptotic activator homeodomain interacting protein kinase 2
(32). Furthermore, the
anti-apoptotic effect of HMGA1 has also been associated with the
deregulation of Bcl-2 (33). In
addition, ischemic brain injury has been suggested to be associated
with widespread molecular and biochemical changes, inflammatory
responses, oxidative stress, free radical production and neuronal
apoptosis (34–36). Notably, neuronal apoptosis is the
prominent cause of cell death in secondary brain damage, which
greatly affects the functional outcome of ischemic injury (37). It was hypothesized that miR-196a
targeted HMGA1 and regulated neuronal apoptosis following OGD and
MCAO. However, the underlying mechanism requires further
exploration and further studies are required to explore whether
HMGA1 regulates p53 or Bcl-2.
In conclusion, the present study demonstrated that
miR-196a was induced by ischemia and may contribute to the
pathogenesis of ischemic brain injury by directly targeting HMGA1.
Accordingly, the inhibition of miR-196a may become a potential
therapeutic option for ischemia-associated brain damage. However,
further studies using neuron-specific miR-196a are warranted.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JH designed the study and collected the data. WS
analyzed the data and drafted the manuscript. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
The present study and all animal experiments were
approved by the Ethics Committee at Beilun People's Hospital of
Ningbo (Ningbo, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lo EH, Dalkara T and Moskowitz MA:
Mechanisms, challenges and opportunities in stroke. Nat Rev
Neurosci. 4:399–415. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Elkind MS: Outcomes after stroke: Risk of
recurrent ischemic stroke and other events. Am J Med. 122 (4 Suppl
2):S7–S13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nelson KB: Perinatal ischemic stroke.
Stroke. 38 (2 Suppl):S742–S745. 2007. View Article : Google Scholar
|
|
4
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kosik KS: The neuronal microRNA system.
Nat Rev Neurosci. 7:911–920. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kole AJ, Swahari V, Hammond SM and
Deshmukh M: miR-29b is activated during neuronal maturation and
targets BH3-only genes to restrict apoptosis. Genes Dev.
25:125–130. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yadav S, Pandey A, Shukla A, Talwelkar SS,
Kumar A, Pant AB and Parmar D: MiR-497 and miR-302b regulate
ethanol induced neuronal cell death through BCL2 and cyclin D2. J
Biol Chem. 286:37347–3757. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yin KJ, Deng Z, Huang H, Hamblin M, Xie C,
Zhang J and Chen YE: miR-497 regulates neuronal death in mouse
brain after transient focal cerebral ischemia. Neurobiol Dis.
38:17–26. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jiang Y, Li L, Tan X, Liu B, Zhang Y and
Li C: miR-210 mediates vagus nerve stimulation-induced antioxidant
stress and anti-apoptosis reactions following cerebral
ischemia/reperfusion injury in rats. J Neurochem. 134:173–181.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi G, Liu Y, Liu T, Yan W, Liu X, Wang Y,
Shi J and Jia L: Upregulated miR-29b promotes neuronal cell death
by inhibiting Bcl2L2 after ischemic brain injury. Exp Brain Res.
216:225–230. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Clark AM, Goldstein LD, Tevlin M, Tavaré
S, Shaham S and Miska EA: The microRNA miR-124 controls gene
expression in the sensory nervous system of Caenorhabditis elegans.
Nucleic Acids Res. 38:3780–3793. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Q, Gong Q, Wu Q and Shi J:
Neuroprotective effects of Dendrobium alkaloids on rat cortical
neurons injured by oxygen-glucose deprivation and reperfusion.
Phytomedicine. 17:108–115. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi GD, OuYang YP, Shi JG, Liu Y, Yuan W
and Jia LS: PTEN deletion prevents ischemic brain injury by
activating the mTOR signaling pathway. Biochem Biophys Res Commun.
404:941–945. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peng B, Chen Y and Leong KW: MicroRNA
delivery for regenerative medicine. Adv Drug Deliv Rev. 88:108–122.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bak M, Silahtaroglu A, Møller M,
Christensen M, Rath MF, Skryabin B, Tommerup N and Kauppinen S:
MicroRNA expression in the adult mouse central nervous system. RNA.
14:432–444. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu DZ, Tian Y, Ander BP, Xu H, Stamova
BS, Zhan X, Turner RJ, Jickling G and Sharp FR: Brain and blood
microRNA expression profiling of ischemic stroke, intracerebral
hemorrhage, and kainate seizures. J Cereb Blood Flow Metab.
30:92–101. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ziu M, Fletcher L, Rana S, Jimenez DF and
Digicaylioglu M: Temporal differences in microRNA expression
patterns in astrocytes and neurons after ischemic injury. PLoS One.
6:e147242011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schimanski CC, Frerichs K, Rahman F,
Berger M, Lang H, Galle PR, Moehler M and Gockel I: High miR-196a
levels promote the oncogenic phenotype of colorectal cancer cells.
World J Gastroenterol. 15:2089–2096. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tsai KW, Liao YL, Wu CW, Hu LY, Li SC,
Chan WC, Ho MR, Lai CH, Kao HW, Fang WL, et al: Aberrant expression
of miR-196a in gastric cancers and correlation with recurrence.
Genes Chromosomes Cancer. 51:394–401. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu M, Du Y, Gao J, Liu J, Kong X, Gong Y,
Li Z, Wu H and Chen H: Aberrant expression miR-196a is associated
with abnormal apoptosis, invasion, and proliferation of pancreatic
cancer cells. Pancreas. 42:1169–1181. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Saito K, Inagaki K, Kamimoto T, Ito Y,
Sugita T, Nakajo S, Hirasawa A, Iwamaru A, Ishikura T, Hanaoka H,
et al: MicroRNA-196a is a putative diagnostic biomarker and
therapeutic target for laryngeal cancer. PLoS One. 8:e714802013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Severino P, Brüggemann H, Andreghetto FM,
Camps C, Klingbeil Mde F, de Pereira WO, Soares RM, Moyses R,
Wünsch-Filho V, Mathor MB, et al: MicroRNA expression profile in
head and neck cancer: HOX-cluster embedded microRNA-196a and
microRNA-10b dysregulation implicated in cell proliferation. BMC
Cancer. 13:5332013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shah N and Singh I: MicroRNA profiling
identifies miR-196a as differentially expressed in childhood
adrenoleukodystrophy and adult adrenomyeloneuropathy. Mol
Neurobiol. 54:1392–1403. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hong XL, Cao H, Zhao F, Pan XF, Zhang K,
Xu QH, Zhao ZX and Li G: MiR-196a-2 gene polymorphism and the
antiviral therapy of chronic hepatitis C. Zhonghua Shi Yan He Lin
Chuang Bing Du Xue Za Zhi. 24:470–472. 2010.(In Chinese).
PubMed/NCBI
|
|
27
|
Fu MH, Li CL, Lin HL, Tsai SJ, Lai YY,
Chang YF, Cheng PH, Chen CM and Yang SH: The potential regulatory
mechanisms of miR-196a in huntington's disease through
bioinformatic analyses. PLoS One. 10:e01376372015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shah SN and Resar L: High mobility group
A1 and cancer: Potential biomarker and therapeutic target. Histol
Histopathol. 27:567–579. 2012.PubMed/NCBI
|
|
29
|
Moussavi Nik SH, Newman M and Lardelli M:
The response of HMGA1 to changes in oxygen availability is
evolutionarily conserved. Exp Cell Res. 317:1503–1512. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xing J, Cao G and Fu C: HMGA1 interacts
with β-catenin to positively regulate Wnt/β-catenin signaling in
colorectal cancer cells. Pathol Oncol Res. 20:847–851. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Treff NR, Pouchnik D, Dement GA, Britt RL
and Reeves R: High-mobility group A1a protein regulates Ras/ERK
signaling in MCF-7 human breast cancer cells. Oncogene. 23:777–785.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pierantoni GM, Rinaldo C, Mottolese M, Di
Benedetto A, Esposito F, Soddu S and Fusco A: High-mobility group
A1 inhibits p53 by cytoplasmic relocalization of its proapoptotic
activator HIPK2. J Clin Invest. 117:693–702. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tang D, Kang R, Cheh CW, Livesey KM, Liang
X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, et
al: HMGB1 release and redox regulates autophagy and apoptosis in
cancer cells. Oncogene. 29:5299–5310. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Keller JN, Kindy MS, Holtsberg FW, St
Clair DK, Yen HC, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins
JB and Mattson MP: Mitochondrial manganese superoxide dismutase
prevents neural apoptosis and reduces ischemic brain injury:
Suppression of peroxynitrite production, lipid peroxidation, and
mitochondrial dysfunction. J Neurosci. 18:687–697. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chan PH: Reactive oxygen radicals in
signaling and damage in the ischemic brain. J Cereb Blood Flow
Metab. 21:2–14. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kriz J: Inflammation in ischemic brain
injury: Timing is important. Crit Rev Neurobiol. 18:145–157. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fluiter K, Opperhuizen AL, Morgan BP, Baas
F and Ramaglia V: Inhibition of the membrane attack complex of the
complement system reduces secondary neuroaxonal loss and promotes
neurologic recovery after traumatic brain injury in mice. J
Immunol. 192:2339–2348. 2014. View Article : Google Scholar : PubMed/NCBI
|