Introduction
Myocardial ischemia-reperfusion (MI/R) injury is one
of the leading causes of morbidity and mortality worldwide,
particularly in developed countries (1). Although a number of pharmacological
agents have been developed for the treatment of myocardial disease,
few effective strategies for MI/R injury prevention are available
(2). It is therefore necessary to
elucidate the underlying molecular mechanism associated with the
development of MI/R injury in order to develop an effective
treatment. Oxidative stress serves a critical role in the
pathophysiology of MI/R injury (3)
and reactive oxygen species (ROS) overproduction caused by I/R
injury induces oxidative stress, leading to DNA injury and changes
in protein kinase activation, which in turn trigger apoptosis,
cardiac dysfunction and even heart failure (4–6).
Mitochondrial dysfunction occurs in the early stage of ischemia and
may result in mitochondrial calcium overload, mitochondrial
membrane depolarization, mitochondrial permeability transition pore
(mPTP) opening, the release of pro-apoptotic proteins and
cytochrome c and even cardiomyocyte death (7,8). As
such, molecules and compounds that are able to attenuate oxidative
stress and improve mitochondrial dysfunction may be potential
candidates for cardioprotective treatment following MI/R
injury.
Apurinic/apyrimidinic endonuclease 1 (APE1) is a
multifunctional protein that serves a vital role in the cellular
response to DNA injury and oxidative stress (9). APE1 also serves a role in controlling
cellular processes, including apoptosis, proliferation,
inflammation and angiogenesis, and is present in survival pathways
(10,11). A number of studies have reported that
APE1 expression is associated with a number of pathological
conditions, including cancer, neurodegenerative diseases and
cardiovascular disease (9,11,12).
Leak et al (13) revealed
that decreased APE1 expression and endonuclease activity cause
oxidative base lesions and apurinic/apyrimidinic sites, triggering
ischemic cell death. The role of APE1 in the cardiovascular system
(14,15), as well as cytoplasmic and
mitochondrial localizations of this protein have also been reported
(16,17). APE1 may be associated with decreased
mitochondrial fragmentation and may improve mitochondrial function
(18). Mitochondrial 8-oxoguanine
glycosylase, which is able to remove 8-hydroxy-2′-deoxyguanosine to
prevent further DNA damage, improves mitochondrial function and
decreases apoptotic events in H9c2 cells under oxidative stress
conditions (19). Based on the
results of the aforementioned studies, it was hypothesized that
APE1 may serve a therapeutic role in MI/R injury, potentially via
regulating oxidative stress and mitochondrial function.
The aim of the present study was to demonstrate the
effects of APE1 on H9c2 cells with hypoxia/reoxygenation
(H/R)-induced injury and to explore the underlying mechanisms. The
results of the present study demonstrate that APE1 is able to
reduce oxidative stress and maintain mitochondrial function in H/R
conditions in vitro, effectively protecting cells from MI/R
injury.
Materials and methods
Materials and regents
Dulbecco's Modified Eagle's Medium (DMEM) and fetal
bovine serum (FBS) was purchased from Gibco (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). An Annexin V-fluorescein
isothiocyanate (FITC) apoptosis detection kit was purchased from BD
Biosciences (San Jose, CA, USA). Rabbit polyclonal antibodies
against APE1 (ALX-210-723-R100) and rabbit monoclonal antibodies
against NAPDH oxide 2 (NOX2; ALX-804-196-T050) were purchased from
Enzo Life Sciences, Inc. (Farmingdale, NY, USA). Rabbit polyclonal
antibodies against B-cell lymphoma 2 (Bcl-2; ab59348) and rabbit
monoclonal antibodies against Bcl-2-associated X protein (Bax;
ab32503) were purchased from Abcam (Cambridge, UK). Rabbit
monoclonal antibody against tubulin (2146) was obtained from Cell
Signaling Technology, Inc. (Danvers, MA, USA). A JC-1 Mitochondrial
Membrane Potential Detection kit was supplied by Beyotime Institute
of Biotechnology (Haimen, China). 2′,7′-dichlorofluorescein acetyl
acetate (DCFH-DA) was purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). The detection kits for superoxide dismutase
(SOD; cat. no. A001-1-1) and glutathione (GSH; cat. no. A005) were
purchased from Nanjing Jiancheng Institute of Biotechnology
(Nanjing, China).
Cell culture and H/R injury model
The H9c2 cardiomyocyte line (rat embryonic
cardiomyoblasts) was obtained from the Cell Bank of Type Culture
Collection of the Chinese Academy of Sciences (Shanghai Institute
of Biotechnology, Shanghai, China). Cells were maintained in DMEM
supplemented with 10% (v/v) FBS and 1% (v/v)
penicillin/streptomycin in a humidified atmosphere containing 5%
CO2 at 37°C. In order to induce H/R injury, the cells
were placed in an anaerobic chamber containing 2.5% O2,
5% CO2 and 92.5% N2 at 37°C for 3, 6, 12 or
24 h, followed by reoxygenation under normoxic conditions (95% air
and 5% CO2) at 37°C for 6 h as previously described
(20). Control cells were maintained
under normal culture conditions as above.
Generation and transfection of
lentiviruses expressing APE1
APE1 cDNA was first synthesized and constructed into
a lentivirus expressing vector using a lentivirus expressing system
(cat. no. K532000; Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocols. Human wild type APE1
cDNA was cloned into pcDNA3.1-HA vectors (Invitrogen; Thermo Fisher
Scientific, Inc.). cDNAs were then cloned into the third generation
lentivirus (LV) vector pBOB with XbaI and BamHI sites. LVs
expressing APE1 (LV-APE1) or empty vector (LV-Scramble, as
controls) amplification were prepared by transient transfection in
HEK 293 T cells (cat. no. K1538 Thermo Fisher Scientific, Inc.).
Following growth to 80–90% confluence, HEK 293T cells were
transfected with recombinant LV-APE1 [virus preservation solution
(0.5 µl)/DMEM (1 ml); MOI=5] or LV-Scramble adenovirus at 37°C for
90 min. The virus solution was then removed and 10% DMEM was added
to culture the HEK 293T cells at 37°C. When cell morphology became
rounded, with cells still demonstrating adherence to the wall, the
supernatant and cells were collected. Following repeated freezing
and thawing three times (37–70°C) to lyse cells, the supernatant
was centrifuged (1,006 × g) at 4°C, for 5 min and the virus
supernatant was collected and stored at −80°C. Subsequently, the
amplified virus (MOI=20) was transfected to H9c2 cells. The cells
were treated with a reagent as indicated for further experiments
after 24 h. The infection efficiency (95–100%) after 24 h was
assessed using western blotting as described later.
MTT assessment of H9c2 cell
viability
Cell viability was assessed using an MTT kit.
Briefly, H9c2 cells were seeded into 96-well-plates at
2×105 cells/well. Following H/R and LV-Scramble or
LV-APE1 transfection, 100 µl of MTT solution (0.5 mg/ml) was added
to each well incubated for an additional 4 h at 37°C. Dimethyl
sulfoxide (DMSO, 100 ml/well) was added to dissolve the formazan
crystals. Absorbance at 570 nm was measured using an epoch
microplate spectrophotometer (BioTek Instruments, Inc., Winooski,
VT, USA). The control group was considered to be 100% viable and
results were expressed as a percentage of the control group.
Detection of lactate dehydrogenase
(LDH) release
LDH levels in the cell supernatant were assessed
using a commercially available kit (cat. no. A020-2) according to
the manufacturer's protocol (Nanjing Jiancheng Bioengineering
Institute). Briefly, cell supernatants were collected, mixed with
reagents including matrix buffer, coenzyme
1,2,4-dinitrophenylhydrazine and NaOH solution from the LDH assay
kit and incubated for 5 min at room temperature. Absorbance was
detected at 450 nm using a microplate reader. Results were
expressed as a percentage of the LDH level in the control
group.
Measurement of intracellular ROS
generation
Intracellular ROS generation was detected by flow
cytometry following staining with cell-permeable fluorogenic probe
DCFH-DA as previously described (21). Briefly, H9c2 cells (1×105
cells/well) were seeded into 6-well plates overnight and exposed to
H/R for 12 h or transfected with LV-Scramble or LV-APE1 followed by
H/R treatment. Cells were stained with the non-fluorescent DCFH-DA
probe (10 µM) for 20 min at 37°C and washed in PBS once. Cells were
re-suspended in PBS (500 µl) and the intracellular accumulation of
DCF was measured using an Olympus X51 fluorescence microscope
(Olympus Corp., Tokyo, Japan, magnification 200×). The fluorescence
intensity was analyzed using a flow cytometer. The fluorescence
intensity in control group was arbitrarily assigned a value of 100%
and the results were calculated as a percentage of the control
group.
Measurement of mitochondrial membrane
potential (MMP)
Decrease in MMP, a marker for mitochondrial
dysfunction, is one of the earliest events that result in apoptosis
(22). In the present study, MMP was
measured using a JC-1 assay kit as previously described (23). Briefly, H9c2 cells were seeded into
6-well plates at a density of 1×105 cells/well and
treated as above. Cells were washed twice with PBS and incubated
with JC-1 for 20 min at 37°C. Carbonyl cyanide
m-chlorophenylhydrazone, included in the JC-1 assay kit, was used
as a positive control. The fluorescent signals were detected using
confocal laser scanning microscopy (LSM 700; Carl Zeiss AG,
Oberkochen, Germany) with 530 and 630 nm as red excitation and
emission wavelengths, respectively, and 488 and 530 nm as the green
excitation and emission wavelengths, respectively. Red fluorescence
was attributable to a potential-dependent aggregation in
mitochondria. Green fluorescence, reflecting the monomeric form of
JC-1, entered into the cytosol after mitochondrial membrane
depolarization.
Electron transport chain (ETC)
activity chain complex activity and ATP production capacity
ETC activity, including the activity of
NADH-ubiquinone oxidoreductase (complex I; cat. no. ab109721),
succinate dehydrogenase (complex II; cat. no. ab109908), coenzyme
Q-cytochrome c oxidoreductase (complex III; cat. no. ab109905) and
cytochrome c oxidase (complex IV; cat. no. ab109911), were analyzed
using commercial kits (Abcam) according to the manufacturer's
protocol. To measure the complex I, II, III or IV activity, the
detergent was added to cells to extract transmembrane proteins.
Absorbance was measured on a Spetramax M5 microplate reader
(Molecular Devices, LLC, Sunnyvale, CA, USA). ATP generation was
detected using a CellTiter-Glo luminescent ATP assay kit (Promega
Corp., Madison, WI, USA) according to the manufacturer's protocol.
The supernatant was collected and added to the substrate solution.
Luminescence was measured using a luminometer (Thermo Fisher
Scientific Inc.).
Flow cytometric analysis of
apoptosis
Apoptosis was analyzed using an Annexin V-FITC
apoptosis detection kit followed by flow cytometry according to the
manufacturer's protocol. In brief, H9c2 cells were transfected with
LV-Scramble or LV-APE1 followed by H/R treatment. At the last stage
of the treatment, floating and attached cells were harvested and
washed twice with cold PBS. The pellets were resuspended in 500 µl
1X binding buffer (provided in the kit) and incubated with Annexin
V-FITC (5 µl) and propidium iodide (PI; 10 µl) for 5 min in the
dark at room temperature. Apoptosis was then analyzed using a flow
cytometer. The number of cells in early (Annexin
V+/PI−) and late (Annexin
V+/PI+) apoptosis was expressed as a
percentage.
Measurement of SOD activity and GSH
level
H9c2 cells were grown on 6-well plates and treated
as above. Cells were harvested and cytosolic protein was extracted
in RIPA lysis buffer (Beyotime Institute of Biotechnology; cat. no.
P0013B). Protein concentration was quantified using a Bradford
Protein Assay (cat. no. 5000001; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). SOD activity was measured using a total SOD
assay kit with WST-8 according to the manufacturer's protocol. The
GSH expression was detected using a GSH assay kit according to the
manufacturer's protocol. The absorbance of each final solution was
measured at 450 (SOD) and 340 nm (GSH). SOD activity was calculated
according to the SOD standard control and expressed as U/mg
protein. GSH expression was expressed as nmol/mg protein.
Western blot analysis
H9c2 cells were treated as described, collected and
lysed in RIPA lysis buffer to measure the expressions of APE1,
NOX2, Bax and Bcl-2 proteins. The protein concentration was
determined using a protein assay kit according to the
manufacturer's protocol (Bio-Rad Laboratories, Inc.). Equal amounts
of protein (30 µg) were separated by 12% SDS-PAGE and transferred
to polyvinylidene fluoride membranes. Following blocking with 5%
nonfat milk for at room temperature 2 h, membranes were incubated
with the primary antibodies against APE1 (1:1,000), NOX2 (1:1,000),
Bax (1:2,000), Bcl-2 (1:2,000) or tubulin (1:2,000) overnight at
4°C, followed by incubation with horseradish peroxidase-conjugated
second antibodies (cat. no. 7076S; 1:5,000; Cell Signaling
Technology, Inc.) at 37°C for 1 h. Bands were visualized using an
enhanced chemiluminescence system (Beyotime Institute of
Biotechnology), scanned using a Bio-Rad gel imaging system (Bio-Rad
Laboratories, Inc.) and analyzed using Quantity One software v4.62
(Bio-Rad laboratories, Inc.).
Statistical analysis
All experiments were repeated independently at least
for three times. Data are expressed as the mean ± standard division
and analyses were performed using one-way analysis of variance
followed by the least significant difference (LSD) test using
GraphPad Prism v.6 (GraphPad Software, Inc., La Jolla, CA, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
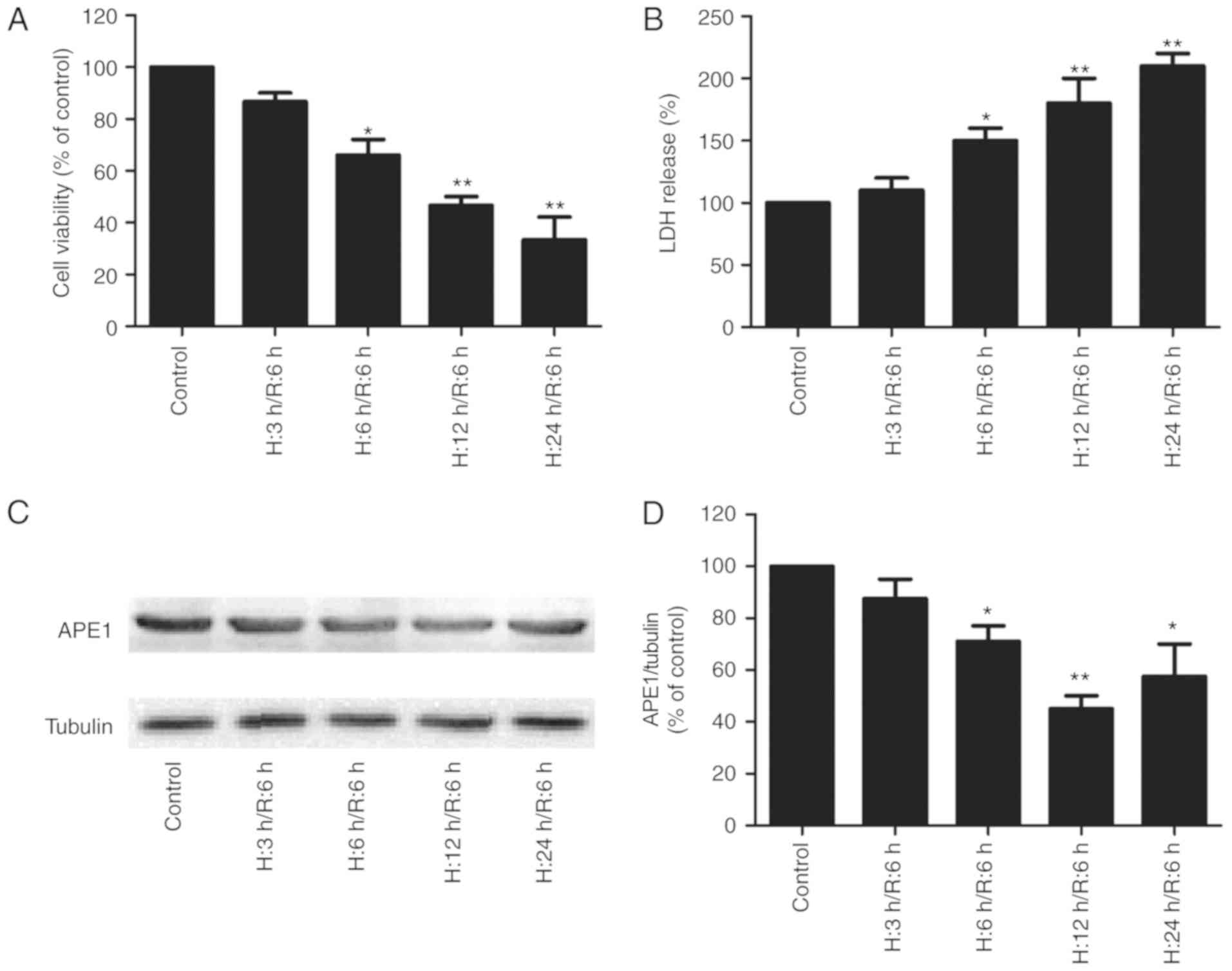
H/R treatment induces cytotoxicity and
decreases APE1 expression in H9c2 cardiomyocytes
To assess the potential effect of APE1 on MI/R
injury, APE1 expression in H9c2 cells treated with H/R was
measured. MTT results revealed that viability was significantly
reduced in cells exposed to 6, 12 or 14 h hypoxia followed by 6 h
reoxygenation compared with the control (Fig. 1A). LDH release is a biomarker of cell
injury (24) and it was observed
that H/R treatment significantly increased LDH release in a
time-dependent manner (Fig. 1B). The
results of western blotting revealed that APE1 expression decreased
with increased hypoxia duration, reaching the lowest level when
cells were exposed to 12 h of hypoxia followed by 6 h of
reoxygenation (Fig. 1C and D). Based
on these results, 1 h hypoxia followed by 6 h reoxygenation was
selected for use in subsequent experiments. These results suggest a
potential role of APE1 in MI/R injury.
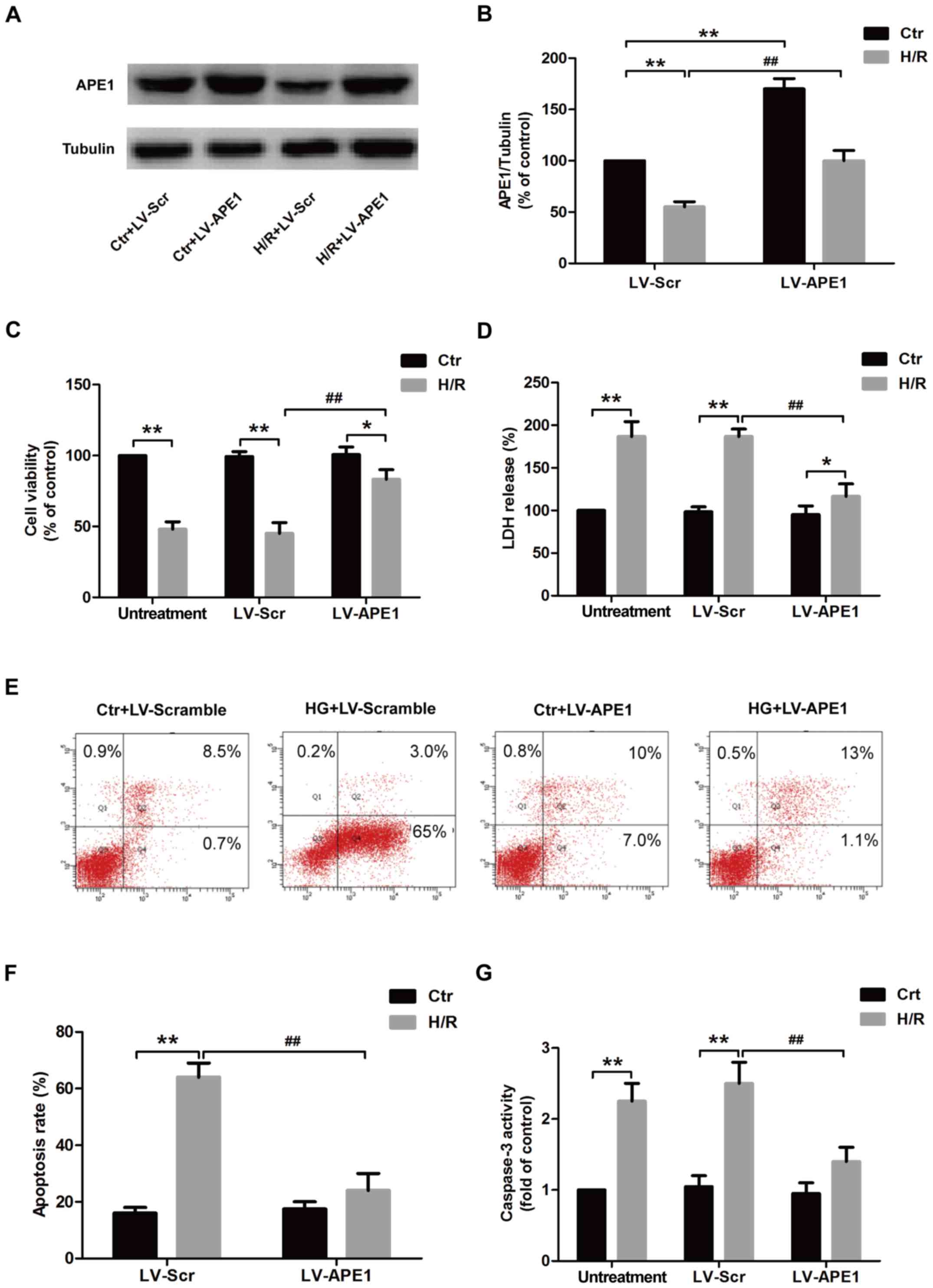
APE1 overexpression alleviates
H/R-induced cytotoxicity and apoptosis in H9c2 cardiomyocytes
To further investigate the role of APE1 in
H/R-induced injury in H9c2 cardiomyocytes, cells were transfected
with LV-APE1 or LV-Scramble. The results of western blotting
revealed that LV-APE1 transfection significantly increased the
expression of APE1 protein compared with LV-Scramble transfection
in the control and H/R treatment groups (Fig. 2A and B). Cell viability was also
significantly increased in LV-APRE1 transfected H9c2 cells compared
with LV-Scramble transfected H9c2 cells under H/R treatment, while
LV-APE1 or LV-Scramble had no effect on cell viability in control
group (Fig. 2C). In the untreatment
group, transfection with LV-APE1 or LV-Scramble had no effect on
LDH release, whereas LV-APE1 transfection in the H/R treatment
group, significantly decreased LDH release compared with
LV-Scramble-transfected cells, indicating that APE1 overexpression
reverses H/R-induced H9c2 cell damage (Fig. 2D). The effect of APE1 on apoptosis in
H/R-treated H9c2 cells was further investigated using flow
cytometry (Fig. 2E). Annexin
V-FITC/PI apoptosis detection results revealed that H/R treatment
caused a significant increase in apoptosis compared with control
cells in LV-Scramble transfection groups (Fig. 2F). However, transfection with LV-APE1
significantly reduced H/R-induced apoptosis compared with the
LV-Scramble transfection group in H/R groups (Fig. 2F). No significant differences in cell
viability were observed in control cells transfected with LV-APE1
or LV-Scramble. Furthermore, H/R treatment significantly increased
the activity of caspase-3 compared with the control in the
untreatment and LV-Scramble transfection groups, whereas LV-APE1
transfection significantly decreased the activity of caspase-3
compared with LV-Scramble transfection in the H/R treatment group
(Fig. 2G). These results indicate
that APE1 overexpression promotes proliferation, attenuates
cytotoxicity and inhibits apoptosis in H9c2 cells, alleviating H/R
injury.
APE1 overexpression alleviates
H/R-induced oxidative stress in H9c2 cardiomyocytes
Oxidative stress serves an important role in the
pathogenesis of MI/R injury (25).
The effect of APE1 on oxidative stress in H/R conditions was
investigated in the present study. In cells transfected with
LV-Scramble, H/R treatment significantly increased intracellular
ROS production compared with the control group (Fig. 3A and B), while LV-APE1 transfection
reduced intracellular ROS levels compared with the LV-Scramble
group. NOX2 enzymes serve a critical role in ROS production
(26). The results of western
blotting revealed that H/R treatment increased the expression of
NOX2 protein in the LV-Scramble transfection group, whereas LV-APE1
transfection significantly decreased this upregulation (Fig. 3C and D). The effect of APE1 on SOD
activity and GSH expression was also assessed. The results from SOD
and GSH detection kits indicated that, under H/R conditions,
LV-APE1 transfection significantly increased SOD activity (Fig. 3E) and GSH levels (Fig. 3F). In the control group, no
significant differences in SOD activity and GSH expression were
observed following transfection with LV-Scramble or LV-APE1. These
results suggest that APE1 reverses H/R-induced oxidative stress by
attenuating oxidative stress and enhancing antioxidant defense.
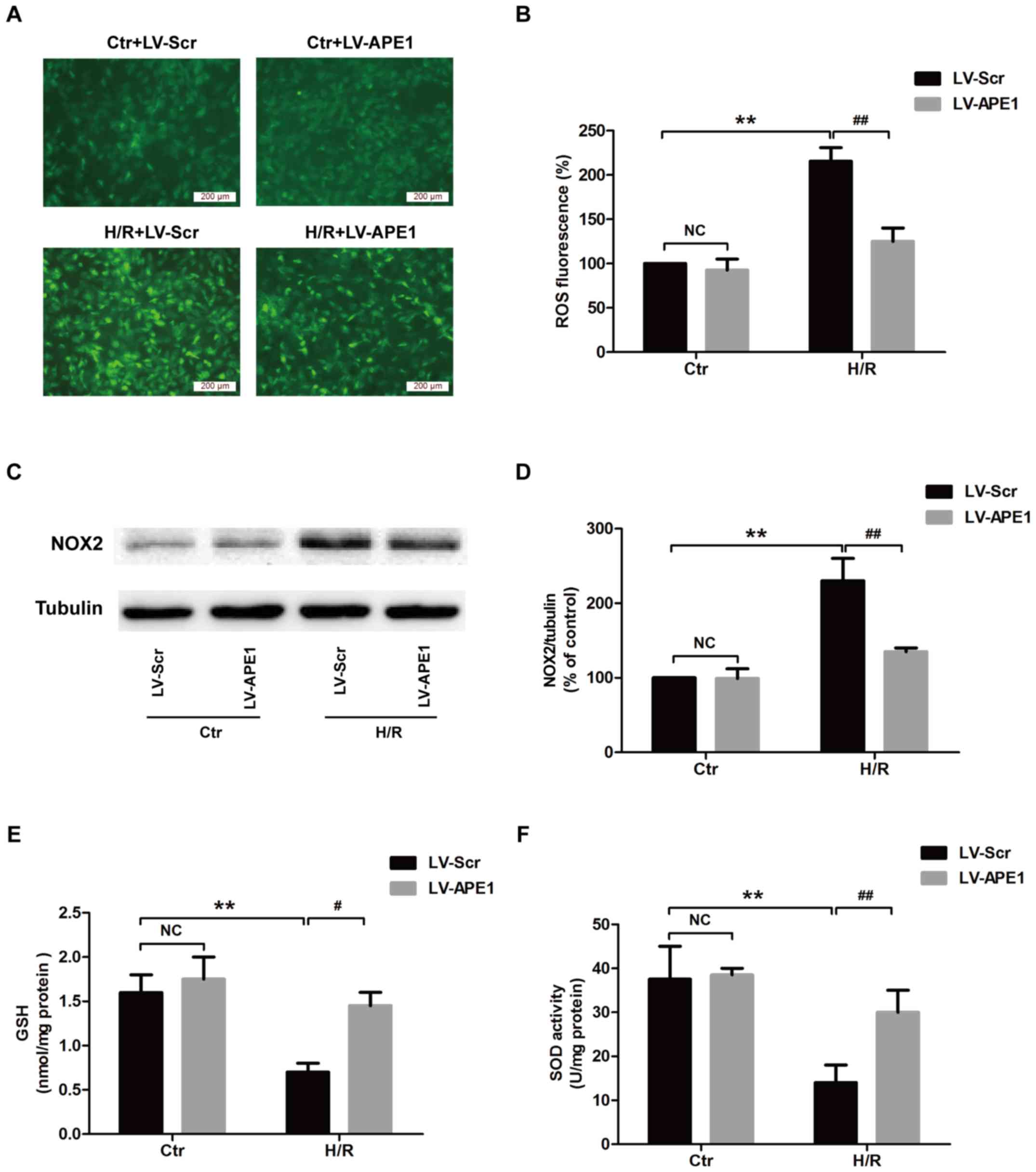 | Figure 3.Effects of APE1 overexpression on
oxidative stress in H/R-treated H9c2 cells. H9c2 cells were
transfected with LV-APE1 or LV-Scr and subjected to H/R. (A) ROS
production was measured using DCFH-DA staining (magnification,
×200) and (B) quantified. (C) The expression of NOX2 protein was
detected using western blotting and (D) quantified. (E) GSH
expression and (F) SOD activity were measured using kits.
#P<0.05, **P<0.01 and ##P<0.01, as
indicated. APE1, apurinic/apyrimidinic endonuclease/redox factor 1;
H/R, 12 h hypoxia/6 h reoxygenation; LV, lentivirus; Scr, scramble;
ROS, reactive oxygen species; DCFH-DA, 2′,7′-dichlorofluorescein
acetyl acetate; NOX2, NAPDH oxide 2; GSH, glutathione; SOD,
superoxide dismutase; Ctr, control. |
APE1 overexpression ameliorates
H/R-induced mitochondrial dysfunction in H9c2 cardiomyocytes
Mitochondrial dysfunction is reflected in the
structure, function and mitochondria number of cardiomyocytes
(27). Mitochondrial dysfunction
leads to diminished energy generation, loss of myocyte
contractility, altered electrical properties and eventual
cardiomyocyte death (27). A
reduction in MMP is an early indicator of apoptosis induction. In
the present study, JC-1 staining result reveled that green
fluorescence was significantly increased in the LV-Scramble group
and red fluorescence was significantly decreased in the H/R group
compared with the control group (Fig.
4A). However, under H/R conditions, green fluorescence was
significantly decreased and red fluorescence was significantly
increased in the LV-APE1 group compared with the LV-Scramble group.
These results indicate that APE1 reversed the H/R-induced
downregulation of MMP in H9c2 cells. ATP generation is associated
with the ETC including complex I–IV activities (22). The effect of APE1 on the activity of
these enzymes was assessed and it was demonstrated that H/R
treatment reduces the activities of complex I (Fig. 4B), complex II (Fig. 4C), complex III (Fig. 4D) and complex IV (Fig. 4E) in LV-Scramble-transfected H9c2
cells, while these changes were blocked by LV-APE1 transfection
under H/R condition. Furthermore, in the LV-Scramble-transfected
H9c2 cells, ATP generation was reduced in the H/R treatment groups
compared with control group, while in H/R-treated H9c2 cells
(Fig. 4F). ATP generation was
increased in the LV-APE1 transfection group compared with the
LV-Scramble transfection group (Fig.
4G), suggesting that APE1 reduces the H/R-induced
downregulation of mitochondrial ATP production. Finally, it was
demonstrated that LV-APE1 transfection significantly ameliorated
H/R-induced increase in the Bax/Bcl-2 ratio in H9c2 cells (Fig. 4H). These results suggest that APE1
improves mitochondrial dysfunction in H/R-treated H9c2 cells.
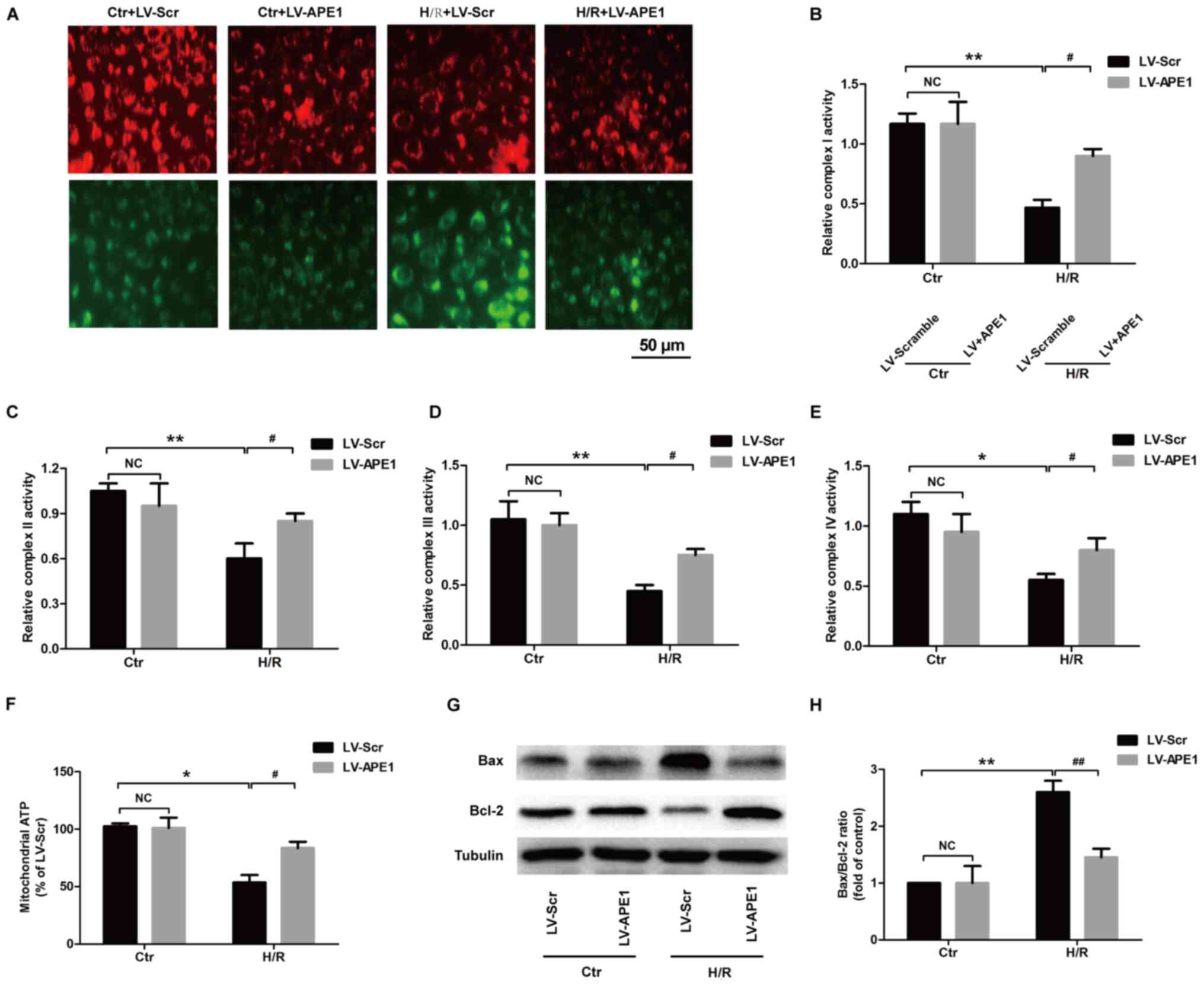 | Figure 4.Effects of APE1 overexpression on
mitochondrial function in H/R-treated H9c2 cells. H9c2 cells were
transfected with LV-APE1 or LV-Scr and subjected to H/R. (A) The
mitochondrial membrane potential was detected using JC-1 staining.
(B) Complex I, (C) complex II, (D) complex III and (E) complex IV
activities were detected using microplate assay kits. (F) ATP
production was assessed using the CellTiter-Glo luminescent ATP
assay kit. (G) Bax/Bcl-2 ratio was measured using western blotting
and (H) quantified. *P<0.05, #P<0.05, **P<0.01,
and ##P<0.01, as indicated. APE1,
apurinic/apyrimidinic endonuclease/redox factor 1; H/R, 12 h
hypoxia/6 h reoxygenation; LV, lentivirus; Scr, scramble; Bcl-2,
B-cell lymphoma 2; Bax, Bcl-2-associated X protein; Ctr,
control. |
Discussion
I/R injury within the myocardium is widely accepted
as a primary contributor for the development of ischemic
cardiovascular diseases (28).
During this process, H/R-induced injury to myocardial cells
triggers a number of mechanisms to alleviate cellular damage caused
by oxidative stress (29) and
mitochondrial dysfunction (30). In
the present study, it was demonstrated that H/R decreases cell
viability, increases LDH release, apoptosis and oxidative stress
and causes mitochondrial dysfunction in H9c2 cardiomyocytes.
APE1 is a multifunctional protein that serves
important roles in DNA repair and redox regulation, as well as
anti-oxidant and anti-apoptosis activities (9). While APE1 downregulation is consistent
with the DNA repair failure coinciding with apoptotic cell death,
APE1 upregulation may repair the cells after an injury (31). Previous studies have reported that
the multifunctional enzyme APE1 is associated with the progression
of a number of human diseases, including cancer, cardiovascular
disease and neurodegenerative diseases, making it a promising focus
of research into the treatment and management of human diseases
(10,11). Jin et al (15) reported that serum APE1 levels were
higher in s patients with coronary artery disease compared with
control patient, which may be a result of protective endogenous
APE1 release. It has also been reported that APE1 activation is
required to protect cells from oxidative injuries (32,33). The
results of the present study indicate that H/R treatment
significantly decreases the expression of APE1 protein, suggesting
that APE1 downregulation may serve a role in the development of
MI/R injury. Considering that APE1 is associated with
cardiovascular disease, apoptosis, oxidative stress and I/R injury,
the lack of APE1 in H/R-induced myocardial injury may be expected.
Further investigation revealed that APE1 overexpression
significantly increased cell viability, reduced LDH release,
decreased apoptosis and reduced caspase-3 activity, protecting
cells against H/R treatment-induced injury and apoptosis. These
results are consistent with previous reports that APE1 exhibits a
cytoprotective activity in normal endothelial cells (34) and that APE1 overexpression inhibits
hypoxia-induced endothelial cell apoptosis (35). These results indicate that APE1
overexpression attenuates H/R-induced injury to H9c2
cardiomyoblasts, providing a potential novel strategy for the
treatment of MI/R injury.
MI/R causes an increase in oxidative stress during
reperfusion, resulting in further cardiomyocyte apoptosis and
mitochondrial dysfunction (2,22). A
number of reports have demonstrated that APE1 reduces intracellular
ROS production (36,37). This is consistent with the results of
the present study, in which H/R treatment significantly increased
ROS generation. However, APE1 overexpression remarkably decreased
the production of ROS in H/R-treated H9c2 cells. NOX is a family of
proteins that produces ROS when activated; NOX2 is the main source
of cytoplasmic ROS generation and serves a vital role in the
pathogenesis of MI/R injury (38).
In the present study, it was demonstrated that H/R treatment
obviously increased the expression of NOX2 in H9c2 cells, while
this effect was reversed by APE1 overexpression. In addition, the
role of SOD and GSH in the maintenance of cellular redox
homeostasis has been reported (39).
GSH depletion activates apoptotic signaling, resulting in eventual
cell death (40). The results of the
present study demonstrated that H/R treatment reduces the activity
of SOD and GSH expression in H9c2 cells. However, these effects
were blocked by APE1 overexpression. Taken together, these results
suggest that APE1 attenuates oxidative stress and enhances the
antioxidant defense, thereby reducing apoptosis and providing a
cardioprotective effect in MI/R injury.
MI/R causes excessive oxidative stress during
reperfusion (41), resulting in
cardiomyocyte apoptosis and mitochondrial dysfunction (2). These events trigger MMP dissipation, as
well as consequent mPTP opening and mitochondrial ETC complex
activity deletion, ultimately resulting in myocardial injury
(22). In addition, it has been
reported that APE1 localizes to the mitochondria in response to
oxidative stress (42,43). Notably, mitochondrial targeting of a
truncated APE1 form leads to increased survival in human umbilical
vein endothelial cells treated under H2O2
conditions (44). Siddiqui et
al (45) demonstrated that APE1
silencing in Huntington's disease cells resulted in exacerbated
mitochondrial dysfunction, suggesting that APE1 is critical for
mitochondrial function. Similarly, APE1 overexpression in the
present study increased MMP and ETC complex activities (complex
I–IV) as well as ATP production, alleviating H/R-induced
mitochondrial dysfunction. It has previously been reported that the
main regulators of mitochondrial apoptosis pathway are the Bcl-2
family proteins (46). Bax
translocation causes a loss of ATP content, mitochondrial swelling
and rupture of the outer mitochondrial membrane (47,48),
further increasing the release of ROS and cytochrome c, leading to
apoptosis. The anti-apoptotic members of the Bcl-2 protein family,
including Bcl-2, are able to promote cell survival (49). In the present study, APE1
overexpression decreased the expression of Bax and increased the
expression of Bcl-2 compared with the H/R alone group. These
results suggest that modulation of the Bcl-2 family may serve a
role in the mechanism by which APE1 improves mitochondrial
dysfunction in H/R injury. The scope of the present study did not
include investigating the effect of H/R injury on APE1 activity or
APE1's downstream target for regulating apoptosis. Future studies
should be expanded to include these experiments.
In conclusion, the results of the present study
demonstrate that APE1 overexpression alleviates H/R-induced damage
in H9c2 cardiomyocytes. The protective role of APE1 may be achieved
via attenuating oxidative stress and suppressing mitochondrial
damage, which provides an insight into the biological functions of
APE1 in MI/R injury.
Acknowledgements
Not applicable.
Funding
Funding information is not applicable.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JH analyzed and wrote the manuscript. HD and FL
participated in experiments and data analysis. J-CL and X-CY
conducted the statistical analysis to the data. WC made substantial
contributions to the conception and design of the study and given
final approval of the version to be published. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
Not applicable.
References
|
1
|
Ambrosio G and Tritto I: Reperfusion
injury: Experimental evidence and clinical implications. Am Heart
J. 138:S69–S75. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xia Z, Chen Y, Fan Q and Xue M: Oxidative
stress-mediated reperfusion injury: Mechanism and therapies. Oxid
Med Cell Longev. 2014:3730812014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun L, Fan H, Yang L, Shi L and Liu Y:
Tyrosol prevents ischemia/reperfusion-induced cardiac injury in
H9c2 cells: Involvement of ROS, Hsp70, JNK and ERK, and apoptosis.
Molecules. 20:3758–3775. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Selvaraju V, Joshi M, Suresh S, Sanchez
JA, Maulik N and Maulik G: Diabetes, oxidative stress, molecular
mechanism, and cardiovascular disease-an overview. Toxicol Mech
Methods. 22:330–335. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Armstrong SC: Protein kinase activation
and myocardial ischemia/reperfusion injury. Cardiovasc Res.
61:427–436. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tao L, Bei Y, Lin S, Zhang H, Zhou Y,
Jiang J, Chen P, Shen S, Xiao J and Li X: Exercise training
protects against acute myocardial infarction via improving
myocardial energy metabolism and mitochondrial biogenesis. Cell
Physiol Biochem. 37:162–175. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ertracht O, Malka A, Atar S and Binah O:
The mitochondria as a target for cardioprotection in acute
myocardial ischemia. Pharmacol Ther. 142:33–40. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Choi S, Joo HK and Jeon BH: Dynamic
regulation of APE1/Ref-1 as a therapeutic target protein. Chonnam
Med J. 52:75–80. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thakur S, Sarkar B, Cholia RP, Gautam N,
Dhiman M and Mantha AK: APE1/Ref-1 as an emerging therapeutic
target for various human diseases: Phytochemical modulation of its
functions. Exp Mol Med. 46:e1062014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tell G, Quadrifoglio F, Tiribelli C and
Kelley MR: The many functions of APE1/Ref-1: Not only a DNA repair
enzyme. Antioxid Redox Signal. 11:601–620. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thakur S, Dhiman M, Tell G and Mantha AK:
A review on protein-protein interaction network of APE1/Ref-1 and
its associated biological functions. Cell Biochem Funct.
33:101–112. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Leak RK, Li P, Zhang F, Sulaiman HH, Weng
Z, Wang G, Stetler RA, Shi Y, Cao G, Gao Y and Chen J:
Apurinic/apyrimidinic endonuclease 1 upregulation reduces oxidative
DNA damage and protects hippocampal neurons from ischemic injury.
Antioxid Redox Signal. 22:135–148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Aonuma T, Takehara N, Maruyama K, Kabara
M, Matsuki M, Yamauchi A, Kawabe J and Hasebe N:
Apoptosis-resistant cardiac progenitor cells modified with
apurinic/apyrimidinic endonuclease/redox factor 1 gene
overexpression regulate cardiac repair after myocardial infarction.
Stem Cells Transl Med. 5:1067–1078. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jin SA, Seo HJ, Kim SK, Lee YR, Choi S,
Ahn KT, Kim JH, Park JH, Lee JH, Choi SW, et al: Elevation of the
serum apurinic/apyrimidinic endonuclease 1/redox factor-1 in
coronary artery disease. Korean Circ J. 45:364–371. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tell G, Damante G, Caldwell D and Kelley
MR: The intracellular localization of APE1/Ref-1: More than a
passive phenomenon? Antioxid Redox Signal. 7:367–384. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tell G, Crivellato E, Pines A, Paron I,
Pucillo C, Manzini G, Bandiera A, Kelley MR, Di Loreto C and
Damante G: Mitochondrial localization of APE/Ref-1 in thyroid
cells. Mutat Res. 485:143–152. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Barchiesi A, Wasilewski M, Chacinska A,
Tell G and Vascotto C: Mitochondrial translocation of APE1 relies
on the MIA pathway. Nucleic Acids Res. 43:5451–5464. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Torres-Gonzalez M, Gawlowski T, Kocalis H,
Scott BT and Dillmann WH: Mitochondrial 8-oxoguanine glycosylase
decreases mitochondrial fragmentation and improves mitochondrial
function in H9C2 cells under oxidative stress conditions. Am J
Physiol Cell Physiol. 306:C221–C229. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li Y, Qiu L, Liu X, Hou Z and Yu B: PINK1
alleviates myocardial hypoxia-reoxygenation injury by ameliorating
mitochondrial dysfunction. Biochem Biophys Res Commun. 484:118–124.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen D, Jin Z, Zhang J, Jiang L, Chen K,
He X, Song Y, Ke J and Wang Y: HO-1 protects against
hypoxia/reoxygenation-induced mitochondrial dysfunction in H9c2
cardiomyocytes. PLoS One. 11:e01535872016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Halestrap AP and Richardson AP: The
mitochondrial permeability transition: A current perspective on its
identity and role in ischaemia/reperfusion injury. J Mol Cell
Cardiol. 78:129–141. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu Z, Lin S, Wu W, Tan H, Wang Z, Cheng C,
Lu L and Zhang X: Ghrelin prevents doxorubicin-induced
cardiotoxicity through TNF-alpha/NF-kappaB pathways and
mitochondrial protective mechanisms. Toxicology. 247:133–138. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Legrand C, Bour JM, Jacob C, Capiaumont J,
Martial A, Marc A, Wudtke M, Kretzmer G, Demangel C, Duval D, et
al: Lactate dehydrogenase (LDH) activity of the cultured eukaryotic
cells as marker of the number of dead cells in the medium
[corrected]. J Biotechnol. 25:231–243. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li M and Wilson DM III: Human
apurinic/apyrimidinic endonuclease 1. Antioxid Redox Signal.
20:678–707. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ushio-Fukai M: Localizing NADPH
oxidase-derived ROS. Sci STKE. 2006:re82006.PubMed/NCBI
|
|
27
|
Capetanaki Y: Desmin cytoskeleton: A
potential regulator of muscle mitochondrial behavior and function.
Trends Cardiovasc Med. 12:339–348. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ibanez B, Heusch G, Ovize M and Van de
Werf F: Evolving therapies for myocardial ischemia/reperfusion
injury. J Am Coll Cardiol. 65:1454–1471. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang Q, Shang M, Zhang M, Wang Y, Chen Y,
Wu Y, Liu M, Song J and Liu Y: Microvesicles derived from
hypoxia/reoxygenation-treated human umbilical vein endothelial
cells promote apoptosis and oxidative stress in H9c2
cardiomyocytes. BMC Cell Biol. 17:252016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kuznetsov AV, Javadov S, Sickinger S,
Frotschnig S and Grimm M: H9c2 and HL-1 cells demonstrate distinct
features of energy metabolism, mitochondrial function and
sensitivity to hypoxia-reoxygenation. Biochim Biophys Acta.
1853:276–284. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li P, Stetler RA, Leak RK, Shi Y, Li Y, Yu
W, Bennett MVL and Chen J: Oxidative stress and DNA damage after
cerebral ischemia: Potential therapeutic targets to repair the
genome and improve stroke recovery. Neuropharmacology. 134:208–217.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pines A, Perrone L, Bivi N, Romanello M,
Damante G, Gulisano M, Kelley MR, Quadrifoglio F and Tell G:
Activation of APE1/Ref-1 is dependent on reactive oxygen species
generated after purinergic receptor stimulation by ATP. Nucleic
Acids Res. 33:4379–4394. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shin JH, Choi S, Lee YR, Park MS, Na YG,
Irani K, Lee SD, Park JB, Kim JM, Lim JS and Jeon BH: APE1/Ref-1 as
a serological biomarker for the detection of bladder cancer. Cancer
Res Treat. 47:823–833. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jeon BH, Gupta G, Park YC, Qi B, Haile A,
Khanday FA, Liu YX, Kim JM, Ozaki M, White AR, et al:
Apurinic/apyrimidinic endonuclease 1 regulates endothelial NO
production and vascular tone. Circ Res. 95:902–910. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hall JL, Wang X, Van Adamson, Zhao Y and
Gibbons GH: Overexpression of Ref-1 inhibits hypoxia and tumor
necrosis factor-induced endothelial cell apoptosis through nuclear
factor-kappab-independent and -dependent pathways. Circ Res.
88:1247–1253. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guo Y, Chen J, Zhao T and Fan Z: Granzyme
K degrades the redox/DNA repair enzyme Ape1 to trigger oxidative
stress of target cells leading to cytotoxicity. Mol Immunol.
45:2225–2235. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Angkeow P, Deshpande SS, Qi B, Liu YX,
Park YC, Jeon BH, Ozaki M and Irani K: Redox factor-1: An
extra-nuclear role in the regulation of endothelial oxidative
stress and apoptosis. Cell Death Differ. 9:717–725. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Maslov LN, Naryzhnaia NV, Podoksenov IuK,
Prokudina ES, Gorbunov AS, Zhang I and Peĭ ZhM: Reactive oxygen
species are triggers and mediators of an increase in cardiac
tolerance to impact of ischemia-reperfusion. Ross Fiziol Zh Im I M
Sechenova. 101:3–24. 2015.(In Russian). PubMed/NCBI
|
|
39
|
Circu ML and Aw TY: Glutathione and
modulation of cell apoptosis. Biochim Biophys Acta. 1823:1767–1777.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Circu ML and Aw TY: Reactive oxygen
species, cellular redox systems, and apoptosis. Free Radic Biol
Med. 48:749–762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zweier JL and Talukder MA: The role of
oxidants and free radicals in reperfusion injury. Cardiovasc Res.
70:181–190. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Frossi B, Tell G, Spessotto P, Colombatti
A, Vitale G and Pucillo C: H(2)O(2) induces translocation of
APE/Ref-1 to mitochondria in the Raji B-cell line. J Cell Physiol.
193:180–186. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tomkinson AE, Bonk RT and Linn S:
Mitochondrial endonuclease activities specific for
apurinic/apyrimidinic sites in DNA from mouse cells. J Biol Chem.
263:12532–12537. 1988.PubMed/NCBI
|
|
44
|
Li MX, Wang D, Zhong ZY, Xiang DB, Li ZP,
Xie JY, Yang ZZ, Jin F and Qing Y: Targeting truncated APE1 in
mitochondria enhances cell survival after oxidative stress. Free
Radic Biol Med. 45:592–601. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Siddiqui A, Rivera-Sánchez S, Castro Mdel
R, Acevedo-Torres K, Rane A, Torres-Ramos CA, Nicholls DG, Andersen
JK and Ayala-Torres S: Mitochondrial DNA damage is associated with
reduced mitochondrial bioenergetics in Huntington's disease. Free
Radic Biol Med. 53:1478–1488. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chipuk JE, Moldoveanu T, Llambi F, Parsons
MJ and Green DR: The BCL-2 family reunion. Mol Cell. 37:299–310.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Saleh AM, Aljada A, El-Abadelah MM, Sabri
SS, Zahra JA, Nasr A and Aziz MA: The pyridone-annelated isoindigo
(5′-Cl) induces apoptosis, dysregulation of mitochondria and
formation of ROS in leukemic HL-60 cells. Cell Physiol Biochem.
35:1958–1974. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Abarikwu SO and Farombi EO: Atrazine
induces apoptosis of SH-SY5Y human neuroblastoma cells via the
regulation of Bax/Bcl-2 ratio and caspase-3-dependent pathway.
Pestic Biochem Physiol. 118:90–98. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Youle RJ and Strasser A: The BCL-2 protein
family: Opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View Article : Google Scholar : PubMed/NCBI
|