Introduction
Aging is a natural as well as undeniable
developmental process in human life, and ‘biological age’ is
usually affected by many factors including environment, heredity,
lifestyle, as well as any kind of disease (1). Developing reliable age-related measures
is the main goal of gerontology (2).
Biomarkers related to biological age have been identified in the
research to detect the age of a person, and these signatures are
significant for geriatric evaluation, which might be in favor of
the adaptation of habits to assist healthy aging (3,4). Many
aging biomarkers have been investigated, for example, telomere
length, and mitochondrial DNA (5,6).
Nevertheless, all of these signatures have a relatively low
precision (7).
Methylated DNA is biologically and chemically more
stable in relevance to mRNA. DNA methylation (DNAm) has been
demonstrated to be an especially promising signature of aging
(8–10). Bekaert et al (11) have predicted the person's age based
on four age-associated DNAm biomarkers. Previous studies have
demonstrated that the age-related epigenetic drift may be closely
related to disease progression (12)
and human evolution (13).
Therefore, it is of great interest and importance to uncover the
specific dynamics of DNAm landscape in aging.
As known, complicated diseases are believed to be
induced by the perturbations of biological networks, other than
single genes. Nevertheless, in a previous research, only two
conditions were considered (that is to say, there is only one
biological network) (14).
Therefore, simultaneously measuring network dynamics in the
progression of a disease is very important to understand the
molecular mechanisms underlying the given disease. Of note, with
the development of high throughput techniques, a great deal of
protein interactions are collected, however, a number of
interactions are yet not measured (15). This problem might be solved, to some
extent, by utilizing modules or sub-networks within the complicated
network (16). Hence, it is
crucially important to detect significant modules in order to
better understand the biological events related to aging.
In the present study, with the goal of detecting
dynamically controlled modules related to aging, inference of
multiple differential modules (iMDM) was utilized to analyze the
DNA methylation data of aging at three age groups, in order to
obtain the connectivity alterations of modules in the aging
process. By utilizing iMDM to multiple sub-networks, candidate
methylated genes were detected. We believe that our results can
provide foundation for experimental verification in a future study,
and expound the molecular mechanisms of aging.
Materials and methods
This module-based approach takes as input
methylation data collected from control and disease cases, and is
implemented based on the following steps: establishment of multiple
differential expression networks (DENs) for each case; extraction
of statistically significant multiple differential modules (DMs) in
multiple DENs; quantification of the connectivity changes of the
common multiple DMs; pathway and GO analyses for the genes in the
common modules.
Microarray data
The microarray data of E-GEOD-64490 on aging were
downloaded from the EMBL-EBI database (https://www.ebi.ac.uk/arrayexpress/experiments/E-GEOD-64490/),
relying on the A-GEOD-13534 - Illumina Human-Methylation450
BeadChip (HumanMethylation450_15017482_v.1.1). In this study, gene
microarray data of 48 samples were included. In the DNAm age, there
were 14 samples with age <70 years, 29 samples of 70–80 years of
age, and 5 samples of >80 years of age. After the probes were
retrieved, we mapped the probes to the human gene symbols, and
finally obtained 20,417 genes.
Protein-protein interaction network
(PPIN) downloaded from STRING database
The human background PPIN covering 787,896
interactions and 16,730 genes was retrieved from STRING database
(http://string-db.org/), and then only the common
part of the microarray data and genes in the background PPIN were
taken to construct the informative PPIN. Finally, 698,580
interactions were obtained.
Construction of DENs
For each age group, DEN was established based on the
differential expression in the aging conditions. Firstly, on the
basis of the absolute value of the Pearson's correlation
coefficient (PCC) of any two genes, significant edges were selected
to establish a binary co-expression network. Herein, we only
selected the edges having PCC higher than the predefined value of
0.8 in order to construct the binary co-expression network.
Secondly, we utilized one-side t-test to calculate the gene
expressions in each age group. Using the P-value of differential
gene expression in each age condition, we assigned the weight value
to the interaction of the binary co-expression network. In view of
multiple DENs, the same nodes were included but there were
different edges; this was determined as Hk = (V,
Ek) (1≤k≤M), where V is the node set of the
co-expression network, Ek is represented by a
3-dimensional matrix A = (aijk)n × n × M, where
aijk stands for the weight on the edge e(i,j), in
network Hk, and M denotes the number of DEN.
Identifying candidate modules across
multiple DENs
The unique and common modules across multiple DENs
(that is to say, multiple candidate modules) were identified.
Module algorithm (17) was developed
to select gene modules with the same gene content, but different
connectivity among multiple interaction networks. This module
algorithm was applied to extract the candidate modules. The
specific steps are described below.
Prioritization of informative
genes
In order to obtain the informative genes, the genes
in the multiple DENs were ranked using degree characteristics.
Then, we obtained the sort order for each gene among all DENs based
on the mean value of z-scores across all DENs. We selected and
determined the top 10% genes as informative genes.
Module identification
Starting with each informative gene, module
identification iteratively included genes whose addition led to the
maximum reduction in the graph entropy-based function until no
reduction was found.
Refining candidate modules
We removed the multiple candidate modules having
node sizes <5. Jaccard index was used to merge the overlapping
multiple candidate modules. In the present analysis, we set the
threshold of the Jaccard index ≥0.5.
Statistical significance for multiple
candidate modules
After obtaining the candidate modules, we
implemented statistical significance for multiple candidate modules
according to the null score distribution of candidate modules
produced based on randomized networks. After that,
Benjamini-Hochberg was utilized to adjust the P-values for multiple
testing relying on false discovery rate (FDR) (18). FDR<0.05 was selected to be the
significance threshold.
Connectivity dynamics of common
multiple DMs and MCDS were used to further quantify the
connectivity change of component modules
Analogously, we calculated the statistical
significance of Module Connectivity Dynamic Score (MCDS) for a DM
as that for multiple DMs. Concretely, we firstly computed the null
distribution for MCDS scores according to the random multiple DMs.
Then, we calculated the P-value for an MCDS on the basis of the
null distribution. Benjamini-Hochberg was then used for correction.
The DMs with greater connectivity were identified based on
FDR<0.05.
Enrichment analysis for the genes in
dynamic modules
To investigate the significant biological processes,
DAVID was employed for pathway and GO annotation of genes in the
dynamic modules. Expression Analysis Systematic Explorer (EASE)
test was used to evaluate the significant pathways. Significant GO
and pathway categories were determined according to
FDR<0.05.
Results
Construction of multiple DENs
In addition to the age group <70 years, two
conditions were also included, age group 70–80 years, and age group
>80 years, multiple=2. Thus, a total of two DENs were
constructed, and 2-DMs were identified. Significant genes and
biological processes were then extracted after the significance of
DMs and MCDS was evaluated. These significant genes and biological
processes across the two conditions might provide novel insights on
the molecular mechanism of aging progression.
After we mapped the microarray data to the
background PPIN, an informative PPIN was extracted, which included
15,248 genes. To increase the network confidence, only interactions
with scores ≥0.8 of the informative PPIN were selected in order to
build DENs. Finally, we constructed two DENs, and each of them
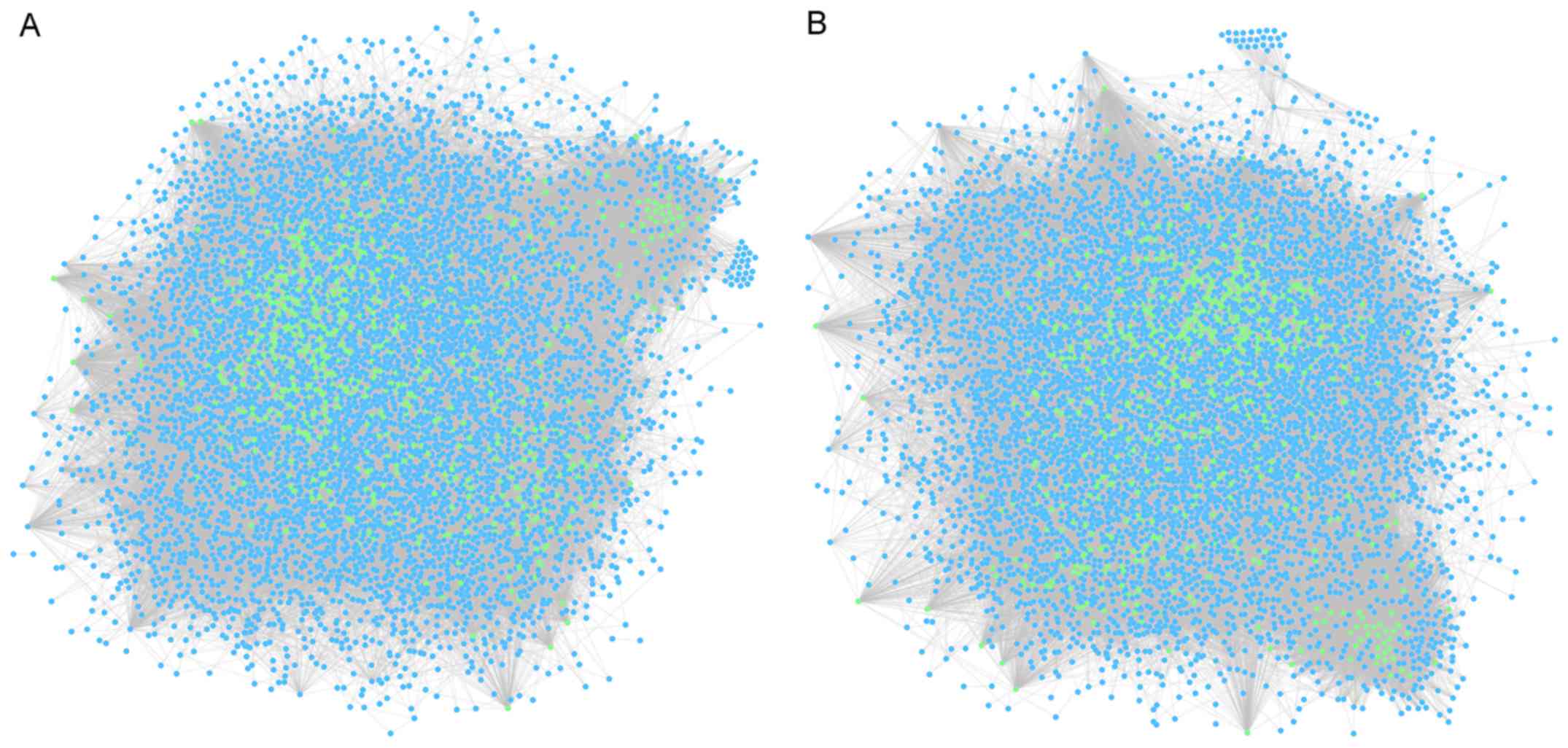
covered 87,702 edges, as well as 6,576 nodes. The DEN compositions
are shown in Fig. 1.
Identification of multiple candidate
modules in two DENs and statistical significance of candidate
modules
Using the z-score distribution of 6,576 nodes in the
DENs, 657 informative genes were screened out. Afterwards, module
search and refinement were conducted. Subsequently, a total of 162
candidate modules in the DENs of two aging conditions were
identified. When FDR was set as 0.05, we found that there were 148
significant modules.
Common 2-DMs used to uncover dynamics
during the development of aging
When FDR was set as 0.05, we found only module 493
observed in the DENs of aging group 70–80 years, and aging group
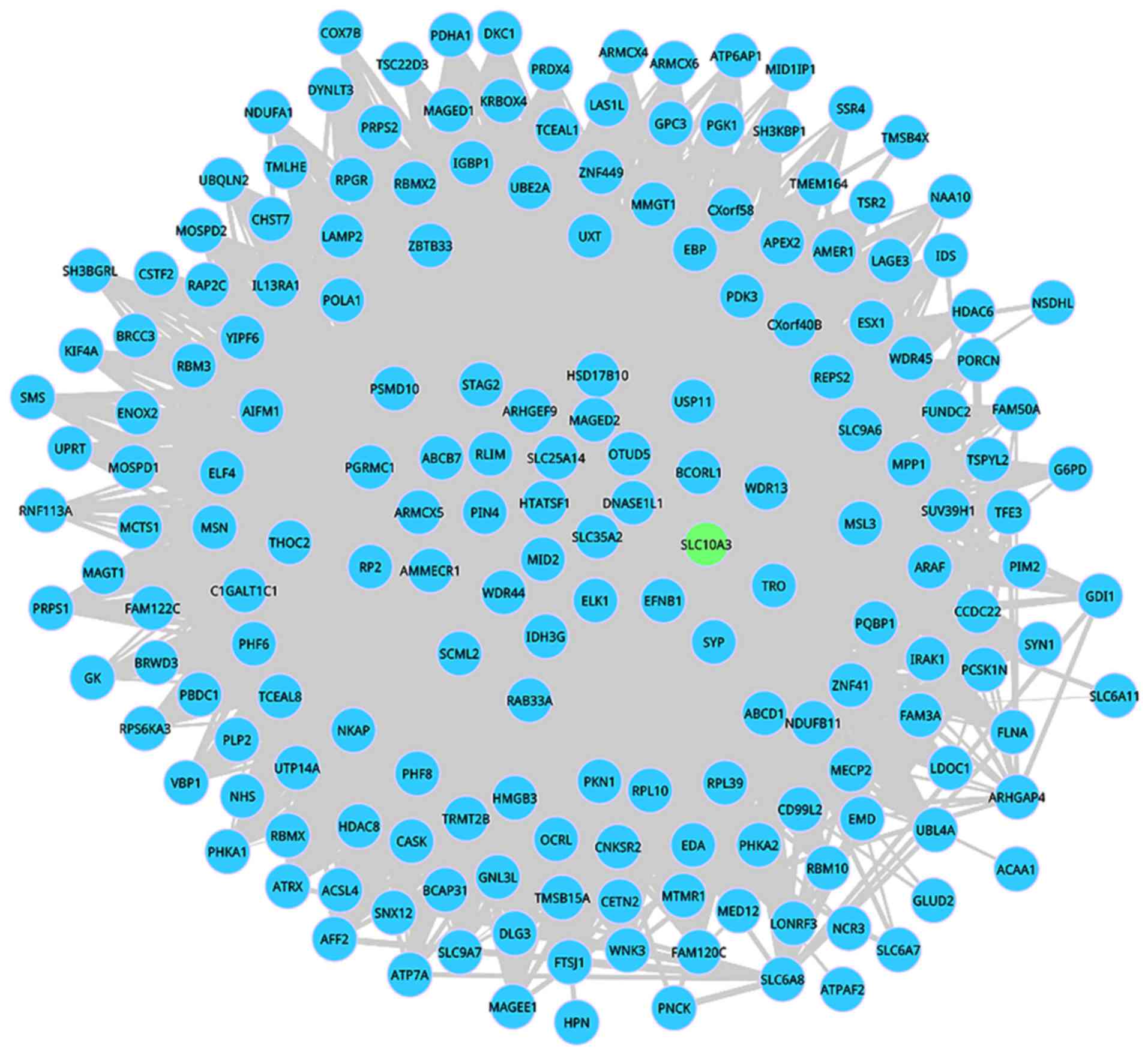
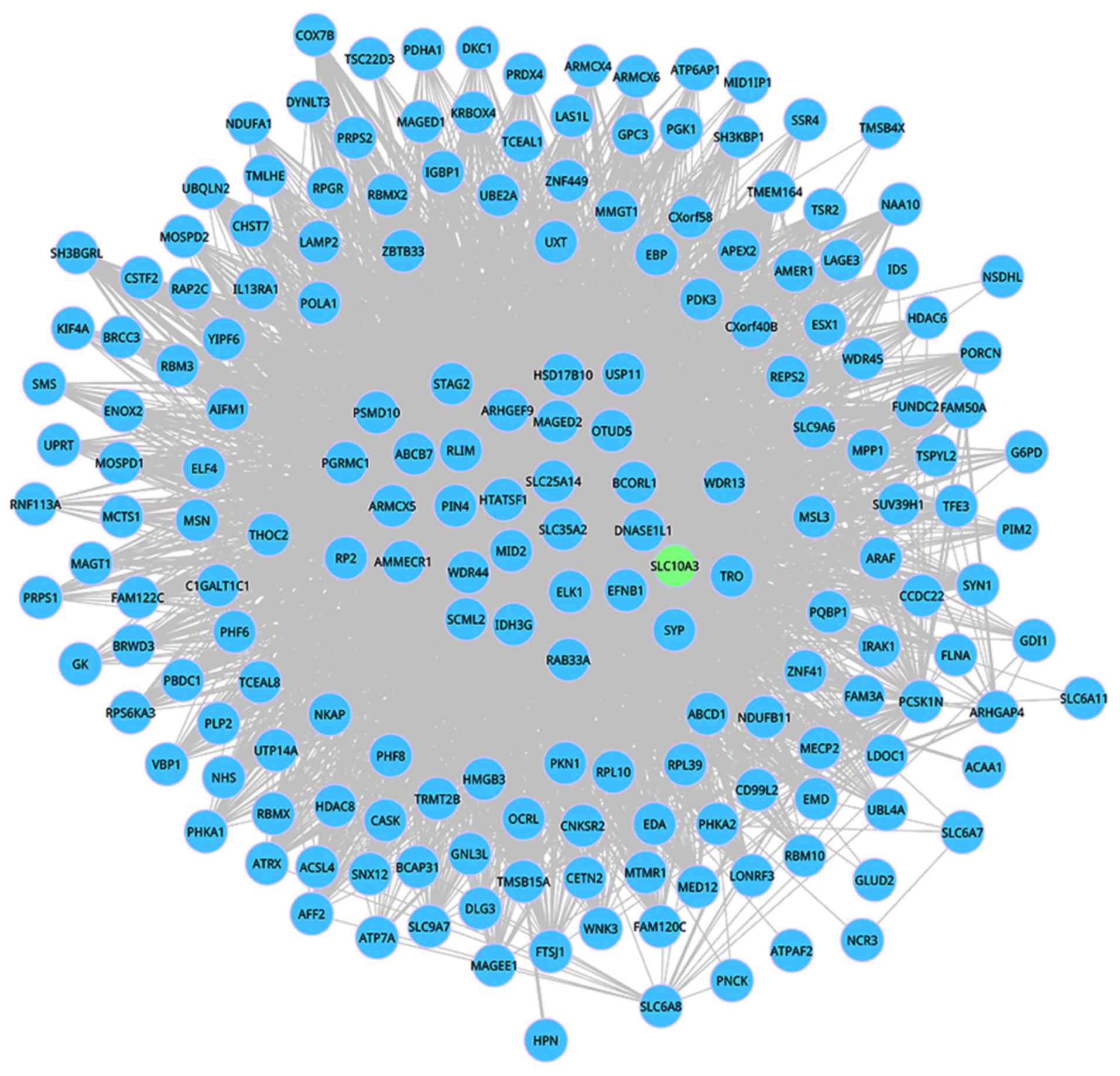
>80 years was dynamic. This dynamic DM 493 including 187 nodes
and 4,006 interactions is shown in Figs.
2 and 3. Among these 187 nodes,
only one informative gene SLC10A3 appeared in this network. In the
aging development, the connectivity of the interactions in this
dynamic module was significantly changed, which further suggested
that network rewired exerts key functions during the aging
progression.
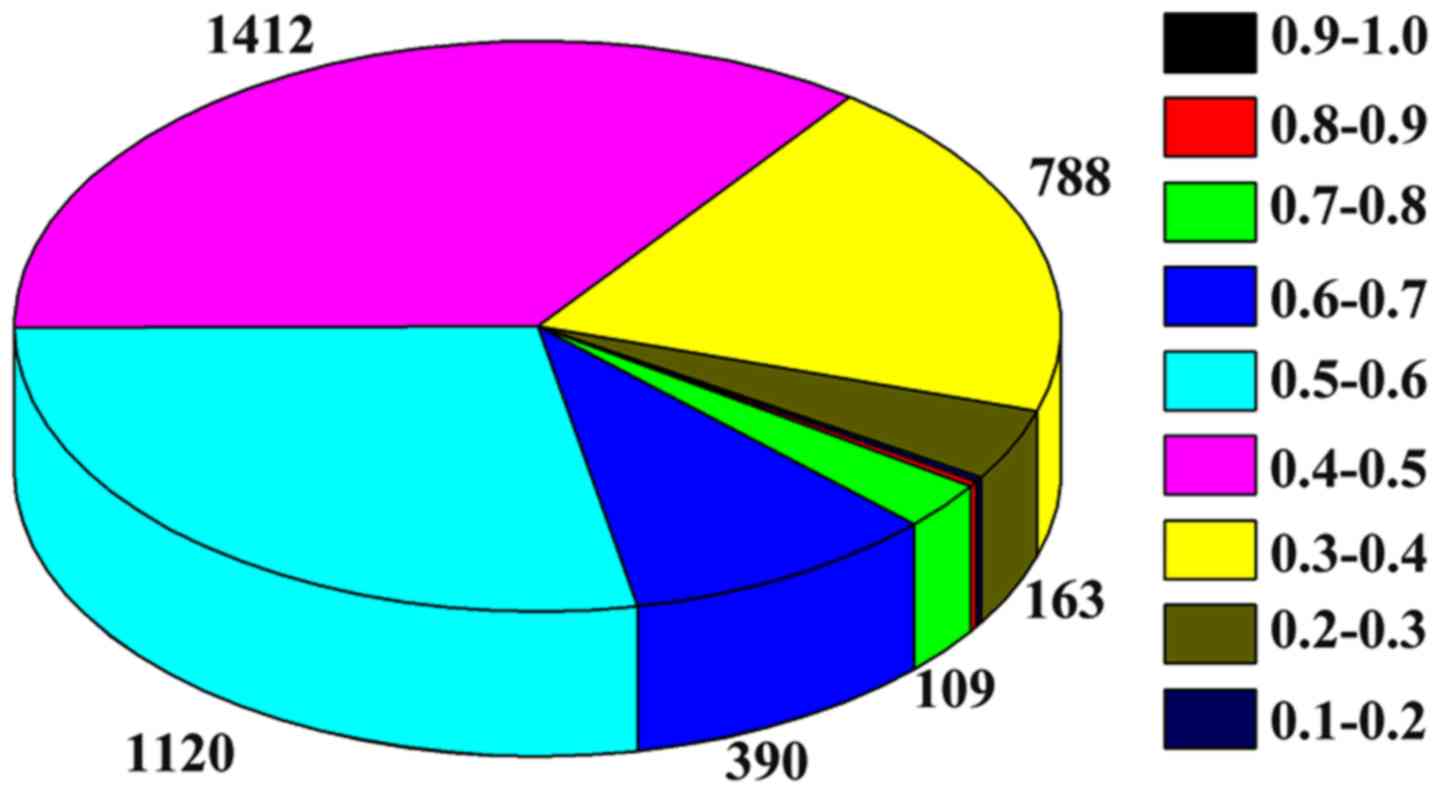
Because the weight of edges in the DENs is a
measurement of evaluating the differential expression between
control and disease, the average edge weight acts as a way to
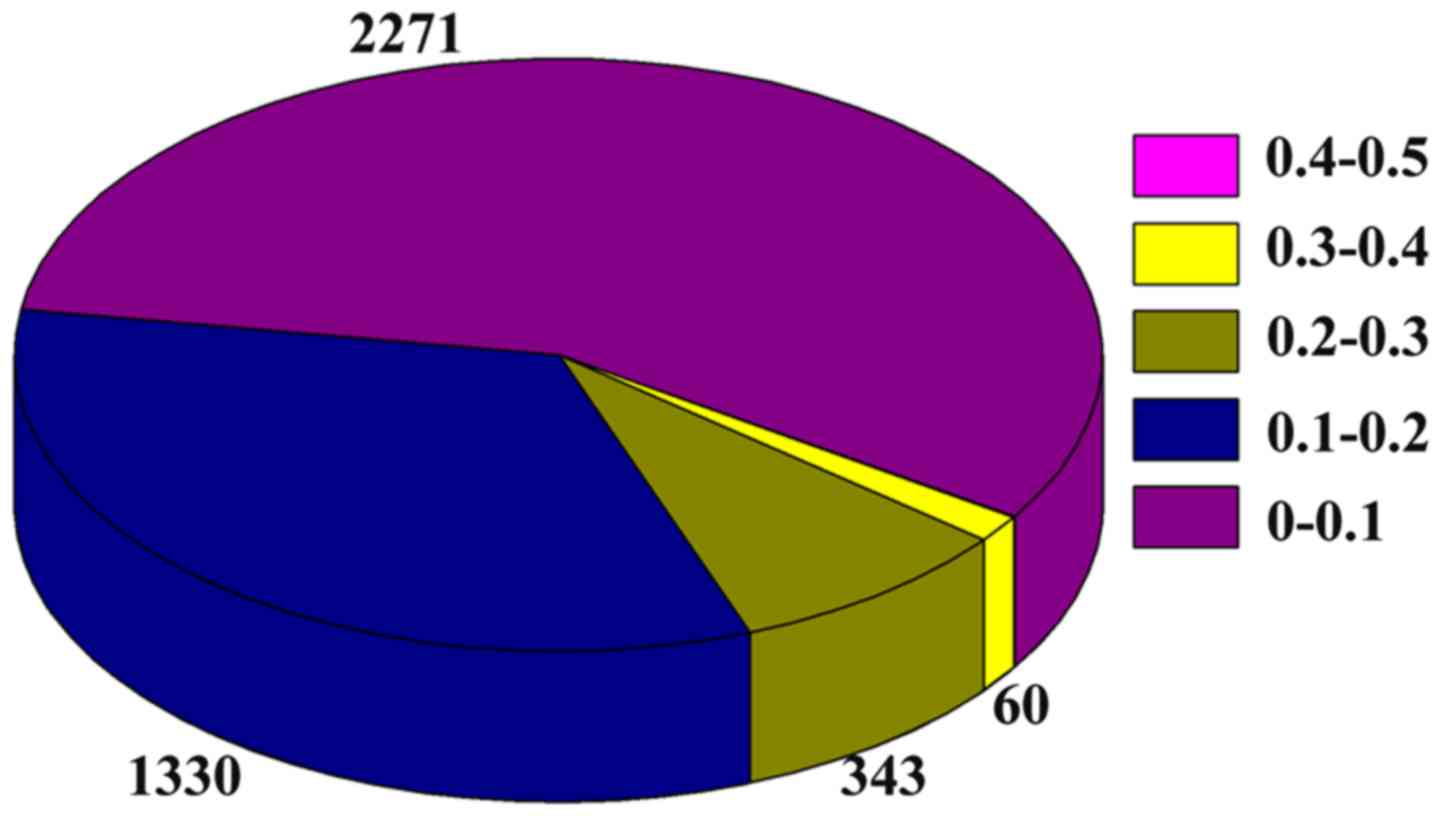
assess the differential activity of the module. Figs. 4 and 5
show the weight distribution of the interactions in the dynamic
2-DMs for the aging groups 70–80 years and >80 years,
respectively. As shown in Fig. 4,
the majority of interactions in the network of the aging group
70–80 years are distributed in the weight distribution of 0.3–0.4,
0.4–0.5, and 0.5–0.6. However, the majority of interactions in the
network of the aging group >80 years are distributed in the
weight distribution of 0–0.1, and 0.1–0.2, as shown in Fig. 5.
Enrichment analysis
To investigate the biological processes, we
implemented GO and pathway enrichment analysis for the genes in the
dynamic module 493. Based on FDR<0.05, no GO terms were
identified. However, overall 7 significant pathway terms were
enriched in this dynamic module, including pentose phosphate
pathway, carbon metabolism, citrate cycle (TCA cycle), chromosomal
instability, ateroid biosynthesis, PPAR signaling pathway, and
immune response.
Discussion
Aging is a major risk factor for chronic, metabolic
and neurodegenerative disorders. Developing reliable age-related
modules and functional categories is the main goal of gerontology,
helpful to understand the potential mechanisms and provide
signatures for assisting healthy aging. In our study, in order to
explore the molecular events indicative of aging, the available DNA
methylation data and DEN were integrated to investigate the altered
modules and biological functions related to aging. Significantly, 1
dynamic dysregulated module (module 493) was extracted. GO
enrichment results demonstrated that genes in module 493 were not
involved in any category. Significantly, overall 7 significant
pathway terms were enriched in the dynamic dysregulated module,
including pentose phosphate pathway, carbon metabolism, citrate
cycle (TCA cycle), chromosomal instability, ateroid biosynthesis,
PPAR signaling pathway, and immune response.
Carbon metabolism that transforms carbon through
pentose phosphate pathway, as well as TCA cycle to energy is
essential for the physiology of brain, heart, liver, and kidneys
(19). Significantly, mitochondria
are responsible for ATP production, and progressive mitochondrial
dysfunction, occurring with aging, leads to growing generation of
reactive oxygen species (ROS), which in turn lead to further
mitochondrial deterioration, as well as cellular damage (20). Furthermore, deletions and mutations
of aged mitochondrial DNA have been reported to be related to aging
(21). Of note, endurance training,
as well as alternate-day-fasting has been suggested to enhance
healthspan via avoiding mitochondrial degeneration (22). Thus, carbon metabolism, pentose
phosphate pathway, and citrate cycle (TCA cycle) might exert
significant functions in the progression of aging.
Chromosomal instability is one form of genomic
instability, resulting from defects in chromosomal segregation, DNA
damage, as well as telomere stability. Genetic evidence has
demonstrated that the enhancement of the faithful chromosomal
segregation can extend longevity in mammals (23). Telomere shortening is found in normal
aging in mice and human (24). The
premature aging of telomerase deficient mice can be reverted if
telomerase is genetically reactivated in the aged mice (25). Accordingly, chromosomal instability
is highly associated with aging progression.
Aging is implicated to be related to oxidative
stress, which is regarded as the effector of the cascade of
degenerative events (26). Oxidative
stress is connected to the enhanced intracellular levels of ROS,
and excessive production of ROS induces neuroinflammation, and
neuronal death (27). Moderate
increase of ROS has been demonstrated to serve as a molecular
signal downstream effect which lure endogenous defense mechanisms,
ending up with the increased stress resistance, as well as extended
life span (28). Significantly,
specific targets for a balanced ROS regulation are PPARs (29). In our study, the genes in the dynamic
module participated in PPAR signaling pathway. Accordingly, PPAR
signaling pathway might be highly related to the aging
progression.
Immune system is an important component of the
inflammatory process and is the natural response to infection. The
recognition and elimination of senescent and hyperploid cells is an
important role of the immune system (30). Inflammation plays important roles in
the progression of obesity and diabetes, which are correlated with
human aging (31). Overexpression of
NF-κB pathway is observed in aging (32). Moreover, inhibiting NF-κB signaling
pathway prevents age-related characteristics in mouse models of
accelerated aging (33). Thus, we
infer that aging might be modulated by NF-κB-driven inflammatory
responses.
Taken together, this study has established
interesting management options (dynamic DMs and the corresponding
pathways) for the assessing the quality of life in the aging
population. However, this study had several limitations. The number
of samples was small which might cause a high false-positive rate
of the findings. Moreover, no experimental confirmation was done
in vivo or in vitro based on patient tissue or an
animal model. Thus, suture studies with larger number of samples,
using an animal model or patient tissues, are needed to confirm our
preliminary findings obtained from bioinformatics analysis.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
GL was involved in the conception of the study,
acquired the data and drafted the manuscript; KYL analyzed the
data; ZPQ was involved in the conception of the study and revised
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Xu C, Qu H, Wang G, Xie B, Shi Y, Yang Y,
Zhao Z, Hu L, Fang X, Yan J, et al: A novel strategy for forensic
age prediction by DNA methylation and support vector regression
model. Sci Rep. 5:177882015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sanders JL, Boudreau RM, Penninx BW,
Simonsick EM, Kritchevsky SB, Satterfield S, Harris TB, Bauer DC
and Newman AB; Health ABC Study, : Association of a Modified
Physiologic Index with mortality and incident disability: The
Health, Aging, and Body Composition Study. J Gerontol A Biol Sci
Med Sci. 67:1439–1446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Song Z, von Figura G, Liu Y, Kraus JM,
Torrice C, Dillon P, Rudolph-Watabe M, Ju Z, Kestler HA, Sanoff H,
et al: Lifestyle impacts on the aging-associated expression of
biomarkers of DNA damage and telomere dysfunction in human blood.
Aging Cell. 9:607–615. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zbieć-Piekarska R, Spólnicka M, Kupiec T,
Parys-Proszek A, Makowska Ż, Pałeczka A, Kucharczyk K, Płoski R and
Branicki W: Development of a forensically useful age prediction
method based on DNA methylation analysis. Forensic Sci Int Genet.
17:173–179. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin J, Epel E, Cheon J, Kroenke C,
Sinclair E, Bigos M, Wolkowitz O, Mellon S and Blackburn E:
Analyses and comparisons of telomerase activity and telomere length
in human T and B cells: Insights for epidemiology of telomere
maintenance. J Immunol Methods. 352:71–80. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cortopassi GA, Shibata D, Soong NW and
Arnheim N: A pattern of accumulation of a somatic deletion of
mitochondrial DNA in aging human tissues. Proc Natl Acad Sci USA.
89:7370–7374. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meissner C and Ritz-Timme S: Molecular
pathology and age estimation. Forensic Sci Int. 203:34–43. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Horvath S, Zhang Y, Langfelder P, Kahn RS,
Boks MP, van Eijk K, van den Berg LH and Ophoff RA: Aging effects
on DNA methylation modules in human brain and blood tissue. Genome
Biol. 13:R972012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Day K, Waite LL, Thalacker-Mercer A, West
A, Bamman MM, Brooks JD, Myers RM and Absher D: Differential DNA
methylation with age displays both common and dynamic features
across human tissues that are influenced by CpG landscape. Genome
Biol. 14:R1022013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Numata S, Ye T, Hyde TM, Guitart-Navarro
X, Tao R, Wininger M, Colantuoni C, Weinberger DR, Kleinman JE and
Lipska BK: DNA methylation signatures in development and aging of
the human prefrontal cortex. Am J Hum Genet. 90:260–272. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bekaert B, Kamalandua A, Zapico SC, Van de
Voorde W and Decorte R: Improved age determination of blood and
teeth samples using a selected set of DNA methylation markers.
Epigenetics. 10:922–930. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Teschendorff AE, Jones A, Fiegl H, Sargent
A, Zhuang JJ, Kitchener HC and Widschwendter M: Epigenetic
variability in cells of normal cytology is associated with the risk
of future morphological transformation. Genome Med. 4:242012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
West J, Widschwendter M and Teschendorff
AE: Distinctive topology of age-associated epigenetic drift in the
human interactome. Proc Natl Acad Sci USA. 110:14138–14143. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma X, Gao L, Karamanlidis G, Gao P, Lee
CF, Garcia-Menendez L, Tian R and Tan K: Revealing pathway dynamics
in heart diseases by analyzing multiple differential networks. PLoS
Comput Biol. 11:e10043322015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nibbe RK, Chowdhury SA, Koyutürk M, Ewing
R and Chance MR: Protein-protein interaction networks and
subnetworks in the biology of disease. Wiley Interdiscip Rev Syst
Biol Med. 3:357–367. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu Y, Jing R, Jiang L, Jiang Y, Kuang Q,
Ye L, Yang L, Li Y and Li M: Combination use of protein-protein
interaction network topological features improves the predictive
scores of deleterious non-synonymous single-nucleotide
polymorphisms. Amino Acids. 46:2025–2035. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ma X, Gao L and Tan K: Modeling disease
progression using dynamics of pathway connectivity. Bioinformatics.
30:2343–2350. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: A practical and powerful approach to
multiple testing. J R Stat Soc B. 57:289–300. 1995.
|
|
19
|
Sauer U, Lasko DR, Fiaux J, Hochuli M,
Glaser R, Szyperski T, Wüthrich K and Bailey JE: Metabolic flux
ratio analysis of genetic and environmental modulations of
Escherichia coli central carbon metabolism. J Bacteriol.
181:6679–6688. 1999.PubMed/NCBI
|
|
20
|
Harman D: The free radical theory of
aging: Effect of age on serum copper levels. J Gerontol.
20:151–153. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Park CB and Larsson NG: Mitochondrial DNA
mutations in disease and aging. J Cell Biol. 193:809–818. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Safdar A, Bourgeois JM, Ogborn DI, Little
JP, Hettinga BP, Akhtar M, Thompson JE, Melov S, Mocellin NJ,
Kujoth GC, et al: Endurance exercise rescues progeroid aging and
induces systemic mitochondrial rejuvenation in mtDNA mutator mice.
Proc Natl Acad Sci USA. 108:4135–4140. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Baker DJ, Dawlaty MM, Wijshake T,
Jeganathan KB, Malureanu L, van Ree JH, Crespo-Diaz R, Reyes S,
Seaburg L, Shapiro V, et al: Increased expression of BubR1 protects
against aneuploidy and cancer and extends healthy lifespan. Nat
Cell Biol. 15:96–102. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Blasco MA: Telomere length, stem cells and
aging. Nat Chem Biol. 3:640–649. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jaskelioff M, Muller FL, Paik JH, Thomas
E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A,
Cadiñanos J, et al: Telomerase reactivation reverses tissue
degeneration in aged telomerase-deficient mice. Nature.
469:102–106. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Finkel T and Holbrook NJ: Oxidants,
oxidative stress and the biology of ageing. Nature. 408:239–247.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mattson MP: Calcium and neurodegeneration.
Aging Cell. 6:337–350. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ristow M and Schmeisser S: Extending life
span by increasing oxidative stress. Free Radic Biol Med.
51:327–336. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zolezzi JM, Silva-Alvarez C, Ordenes D,
Godoy JA, Carvajal FJ, Santos MJ and Inestrosa NC: Peroxisome
proliferator-activated receptor (PPAR) γ and PPARα agonists
modulate mitochondrial fusion-fission dynamics: Relevance to
reactive oxygen species (ROS)-related neurodegenerative disorders?
PLoS One. 8:e640192013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Senovilla L, Vitale I, Martins I, Tailler
M, Pailleret C, Michaud M, Galluzzi L, Adjemian S, Kepp O,
Niso-Santano M, et al: An immunosurveillance mechanism controls
cancer cell ploidy. Science. 337:1678–1684. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Barzilai N, Huffman DM, Muzumdar RH and
Bartke A: The critical role of metabolic pathways in aging.
Diabetes. 61:1315–1322. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Adler AS, Sinha S, Kawahara TLA, Zhang JY,
Segal E and Chang HY: Motif module map reveals enforcement of aging
by continual NF-kappaB activity. Genes Dev. 21:3244–3257. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tilstra JS, Robinson AR, Wang J, Gregg SQ,
Clauson CL, Reay DP, Nasto LA, St Croix CM, Usas A, Vo N, et al:
NF-κB inhibition delays DNA damage-induced senescence and aging in
mice. J Clin Invest. 122:2601–2612. 2012. View Article : Google Scholar : PubMed/NCBI
|