Introduction
Traumatic brain injury (TBI) is a serious
neurological condition (1,2). Millions of people suffer from
TBI-related disabilities worldwide, costing billions of dollars
annually (3). TBI-related nerve
injury is attributed to both primary and secondary injury
mechanisms (4,5). Primary injury manifests as immediate
damage, whilst secondary injury is a long-term process, accompanied
by oxidative stress, neuronal apoptosis and an inflammatory
response (2,6–8),
ultimately leading to neuronal death. There is growing evidence
that inflammation and oxidative stress serve pivotal roles in the
course of secondary brain injury (10–12).
Thus, anti-inflammatory treatments that combat oxidative stress are
predicted to be effective therapeutic strategies for improving
outcomes following TBI.
The inflammatory response and oxidative stress are
important adverse pathological events that occur following TBI. It
is widely accepted that NF-κB performs an essential role in the
inflammatory response, triggering the expression of tumor necrosis
factor (TNF), pro-inflammatory interleukins (ILs) and interferons
in response to inflammation induction (13,14).
Nuclear factor erythroid 2-related factor 2 (Nrf2) is an inhibitor
of oxidative stress. In unstressed conditions, Nrf2 is retained in
the cytoplasm by binding to kelch-like ECH-associated protein 1
(15,16). However, following Nrf2 activation, an
increase in the expression of downstream cytoprotective proteins,
such as heme oxygenase 1 (HO-1) and quinine oxidoreductase 1
(NQO-1), occurs, enhancing the cell's resistance to oxidative
stress (17). In addition, genetic
knockout of Nrf2 exacerbates oxidative stress injury following TBI
in mice (18). Thus, targeting NF-κB
and Nrf2 is an attractive strategy for identifying novel treatments
for TBI.
Edaravone (EDA; 3-methyl-1-phenyl-2-pyrazolin-5-one)
is a free radical scavenger that has been demonstrated to improve
neurological function in mice following cerebral hemorrhage
(19). Multiple studies also
indicate that EDA exerts a neuroprotective effect mediated by
quenching oxidative stress in a model of ischemia/reperfusion
injury (20–22). Furthermore, EDA has a protective
effect in multiple in vitro models of brain function,
mediated by the inhibition of oxidative stress. However, few
studies have addressed the role of oxidative stress inhibition by
EDA in regards to TBI. One study demonstrated that EDA attenuates
the inflammatory response in a rat transient focal ischemia model
by regulating NF-κB (23). However,
the mechanism by which EDA attenuates the inflammatory response in
a TBI mouse model remains poorly understood. In addition, it is
also not known whether EDA has a role in protecting neurological
function following TBI by regulating Nrf2.
The objective of the present study was to
investigate the ability of EDA to attenuate a TBI-induced
inflammatory response and oxidative stress injury in mice. The
results indicated that EDA likely suppressed the inflammatory
response and oxidative stress following TBI by regulating NF-κB and
Nrf2. Collectively, these findings suggested that EDA may be an
effective novel treatment for TBI.
Materials and methods
Experimental animals
A total of 60 male C57BL/6 mice (20–25 g) were
obtained from the Experimental Animal Center of Nanjing Medical
University. The animal study protocols were approved by the Animal
Care and Use Committee of Wenzhou Medical University. Mice were
housed under standard conditions, including adequate temperature,
standard humidity and a 12-h light/dark cycle. All the animals were
allowed free access to food and water and acclimatized for at least
7 days before any experiment.
Reagents and chemicals
EDA was purchased from the Simcere Pharmaceutical
Group. Anti-β-actin (cat. no. 3700s) and anti-NF-κB (cat. no.
8242s) antibodies were purchased from Cell Signaling Technology,
Inc. Anti-Nrf2 (cat. no. ab31163) and anti-histone3 (cat. no.
ab1791), antibodies were purchased from Abcam. Anti-mouse secondary
antibodies and anti-rabbit secondary antibodies were purchased from
Multi Sciences Biotech Co.
TBI model
The TBI model was established as previously
described (24). In brief, the mice
were anesthetized by intraperitoneal injection of chloral hydrate
(400 mg/kg) and placed onto a stereotaxic frame (David Kopf
Instruments; Fig. 1C). A portable
drill was used to penetrate the right parieto-temporal cortex in
order to allow removal of the bone flap. A pneumatic cylinder
(velocity of 4 m/s; depth, 1 mm) was then used to apply a
controlled cortical impact. Following the blow, the scalp was
sutured closed, and the mice were returned to cages for recovery.
Mice subjected to the same procedures without impact were the sham
group. EDA (3 mg/kg) was administered by intraperitoneal injection
1 h post-TBI. This dose of EDA administration was based on previous
studies of neuroprotection by EDA in an intracerebral hemorrhage
mouse model (9,17). Furthermore, according to our previous
studies, inflammation and oxidative stress levels were
significantly upregulated following TBI and peaked 24 h
post-surgery. Therefore, the 24 h timepoint was selected to examine
the anti-inflammatory and anti-oxidative stress capacity of
EDA.
Neurological evaluation
The 10-point Neurological severity score (NSS)
(24) was used to assess mice 24 h
following TBI. NSS tests were as follows: i) Ability and initiative
to exit circle of 30 cm diameter with a time limit of 3 min; ii)
paresis of upper and/or lower limb of the contralateral side; iii)
alertness, initiative and motor ability to walk straight once
placed on the floor; iv) innate startle reflex where the mouse
bounces in response to a loud hand clap; v) seeking behavior
involving physiological behavior as a sign of ‘interest’ in the
environment; vi) ability to balance on a beam of 7 mm width for at
least 10 sec; vii) ability to grip on a beam of 5 mm width for at
least 10 sec; viii) ability to cross a 30 cm long beam of 3 cm
width; ix) ability to cross a 30 cm long beam of 2 cm width; and x)
ability to cross a 30 cm long beam of 1 cm width. One point is
given for failing to perform each of the tasks.
Hematoxylin and eosin (H&E) and
Nissl staining
At 24 h and 7 days following TBI, mice were
anesthetized and perfused with saline. Brain tissue was collected
and fixed in 4% (w/v) paraformaldehyde (PFA) for 24 h at 4°C, then
embedded in paraffin wax. Sections (5 µm) were mounted on
poly-L-lysine-coated slides for histopathological examination.
H&E and Nissl staining was then performed according to the
manufacturer's instructions. Images were captured by light
microscope using a Nikon Eclipse 80i (Nikon Corporation) with
Nissl-positive cells automatically counted in five randomly
selected fields per sample and quantified using Integrated
Performance Primitives v9.0 software (Intel Corporation).
Terminal deoxynucleotidyl transferase
dUTP nick end labelling (TUNEL) staining
An in situ cell death detection kit (cat. no.
1168481710; Roche Diagnostics) was used to determine the presence
of apoptotic cells in sections. In brief, the sections (5 µm) were
cut from the same blocks as the H&E staining, then were
deparaffinized, rehydrated then washed briefly with distilled water
before digestion for 15 min using proteinase K at room temperature.
The sections were then washed and incubated with a prepared TUNEL
reaction mixture in a dark humidifying box for 1 h at 25°C. The
sections were then washed and DAPI was added to stain the cell
nuclei, prior to being sealed with a coverslip. Brain slices
treated with 10 U/ml DNase I buffer for 10 min at room temperature
prior to incubation with the TUNEL reaction mixture provided a
positive control for the study. The negative control was brain
slices incubated with the TUNEL reagent without the addition of TdT
enzyme. Positive cells were identified and analyzed using
fluorescence microscopy by an investigator blinded to the
experimental groups. TUNEL-positive cells were counted in five
randomly selected fields in each sample and quantified using
Integrated Performance Primitives v9.0 software (Intel
Corporation).
Measurement of superoxide dismutase
(SOD), glutathione peroxidase (GPx), hydrogen peroxide
(H2O2), IL-6 and TNF-α concentration
Cortical tissue was homogenized and centrifuged at
12,000 × g for 15 min. The supernatant was collected for the
spectrophotometric measurement of SOD (cat. no. S0109), GPx (cat.
no. S0056), H2O2 (cat. no. S0038), IL-6 (cat.
no. F1083) and TNF-α (cat. no. F1163) using the respective kits
(Beyotime Institute of Biotechnology), according to the
manufacturer's instructions. In brief, supernatants were added to
96-well plates that had been coated with specific murine monoclonal
antibodies raised against SOD, GPx, H2O2,
IL-6 and TNF-α then incubated for 90 min at 37°C. Biotin-labeled
specific antibodies for SOD, GPx and H2O2
were added and incubated for a further 60 min at 37°C. Finally,
3,3′5,5-tetramethylybenzidine substrate was added and incubated for
25 min at 37°C. Stock concentrations of murine SOD, GPx and
H2O2 were serially diluted to create standard
titration curves. A microplate reader was used to measure optical
densities at 450 nm.
Western blot analysis
Protein extracts were obtained using the Nuclear and
Cytoplasmic Protein Extraction kit (Beyotime Institute of
Biotechnology), total protein concentration was quantified using
the bicinchoninic acid assay kit (cat. no. p0012s; Beyotime
Institute of Biotechnology) according to the manufacturer's
instructions. A total of 80 µg of protein was loaded per lane, then
separated via SDS-PAGE on a 10% of gel. The separated proteins were
transferred to polyvinylidene-difluoride membranes (EMD Millipore).
After blocking for 2 h at 25°C in 5% skimmed milk, the membranes
were incubated with primary antibodies anti-Nrf2 (1:1,000),
anti-histone 3 (1:1,000), anti-β-actin (1:3,000) and anti-NFκB
(1:1,000) overnight at 4°C. The membranes were then washed with
Tris-buffered saline and Polysorbate 20 and incubated with
secondary antibodies (1:10,000) for 1 h at room temperature. The
protein bands were visualized using enhanced chemiluminescence
(cat. no. P0018AS; Beyotime Institute of Biotechnology) and the
ChemiDicTM XRS+ Imaging System (Bio-Rad Laboratories, Inc.), and
the densities of the immunoreactive bands were analysed using Image
J software (National Institutes of Health). β-actin was used as the
loading control for quantification.
Reverse transcription quantitative PCR
(RT-qPCR)
Total RNA was extracted from cells and ipsilateral
cortex samples with TRIzol (Invitrogen; Thermo Fisher Scientific,
Inc.). The concentration of total RNA was detected using a nucleic
acid protein analyzer (Beckman Coulter, Inc.). To avoid RNA
degradation, reverse transcription was performed immediately with
the Prime Script RT-PCR kit (Takara Bio, Inc.) according to the
manufacturer's instructions. qPCR was performed on the Eppendorf
Real Plex 4 instrument (Eppendorf) using real-time SYBR Green (cat.
no. 1708882AP; Bio-Rad Laboratories, Inc.). The specific primers
designed by Invitrogen (Thermo Fisher Scientific, Inc.) with the
following sequences: NQO1 forward, 5′-CATTCTGAAAGGCTGGTTTGA-3′ and
reverse, 5′-CTAGCTTTGATCTGGTTGTCAG-3′; HO-1 forward,
5′-ATCGTGCTCGCATGAACACT-3′ and reverse,
5′-CCAACACTGCATTTACATGGC-3′; TNF-α forward, 5′-TGATCCGCGACGTGGAA-3′
and reverse, 5′-ACCGCCTGGAGTTCTGGAA-3′; IL-6 forward,
5′-CCAAGAGGTGAGTGCTTCCC-3′ and reverse,
5′-CTGTTGTTCAGACTCTCTCCCT-3′ and β-actin forward,
5′-CCGTGAAAAGATGACCCAGA-3′ and reverse, 5′-TACGACCAGAGGCATACAG-3′.
The thermocycling conditions were as follows: 95°C for 10 min
followed by 40 cycles of 95°C for 15 sec then 60°C for 1 min. mRNA
levels were quantified using the 2−ΔΔCq method (25).
Immunohistochemistry
Following deparaffinization and rehydration, brain
tissue sections and cells were mounted on coverslips and fixed with
4% PFA for 48 h at 4°C. These samples then were then blocked with
5% bovine serum albumin in PBS for 30 min at 37°C then incubated
overnight at 4°C with primary antibody anti-NF-κB (1:300).
Following washing with PBS (7 min washes for a total of three
times), the slides were incubated for 2 h at room temperature with
the secondary antibody (1:1,000), using the same antibodies as for
western blot analysis. Following washing in PBS, the slides were
stained with DAPI for 7 min. Fluorescence was detected using a
confocal laser microscope (Nikon Corporation). Analysis was
performed by blinded observers using Integrated Performance
Primitives v9.0 software (Intel Corporation).
Statistical analysis
Data are presented as the mean ± standard error of
the mean with experiments repeated at least three times. GraphPad
Pro 5.0 (GraphPad Software, Inc.) was used to process data.
Statistical significance was assessed between two groups using
student's t-test, and between multiple groups using one-way
analysis of variance followed by Dunnett's post hoc test. P<0.05
was considered to indicate statistical significance.
Results
EDA treatment decreases
neurofunctional deficits in mice following TBI
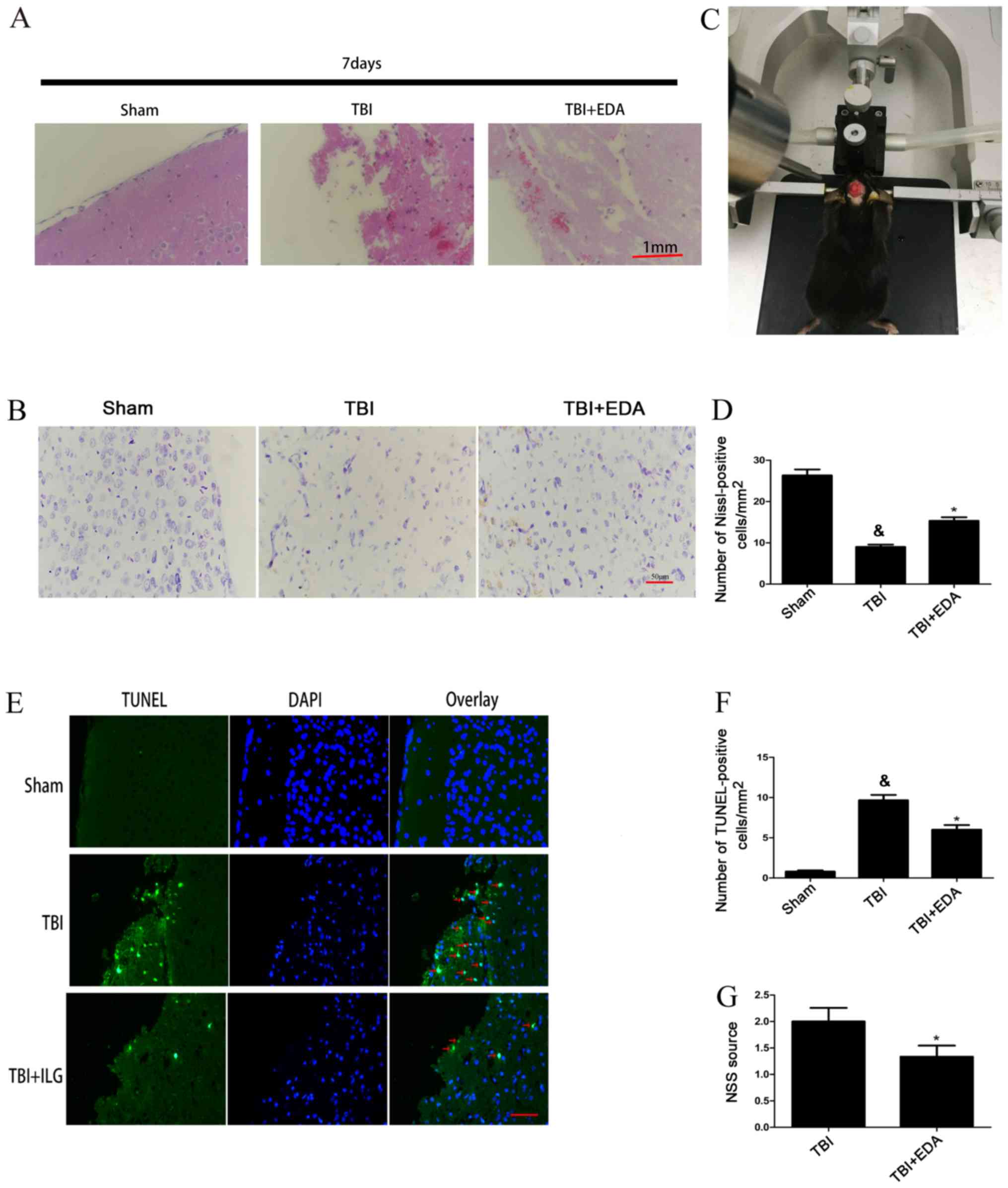
H&E staining was used to investigate neuronal
survival. Compared with the TBI group, smaller lesion cavity
volumes were observed following EDA treatment (Fig. 1A). Furthermore, treatment with EDA
resulted in marked preservation of the number of Nissl-positive
stained neurons in the treatment group compared with the
non-treated group (Fig. 1B and D).
TUNEL-positive apoptotic cells were present at a higher frequency
in the TBI group compared with the sham group, and the apoptotic
index of the EDA group was decreased compared with the TBI group
(Fig. 1E and F). The neuroprotective
role of EDA was investigated 24 h following TBI. The NSS score of
mice in the TBI group was significantly higher compared with the
EDA group (Fig. 1G).
EDA suppresses the TBI-induced
inflammatory response
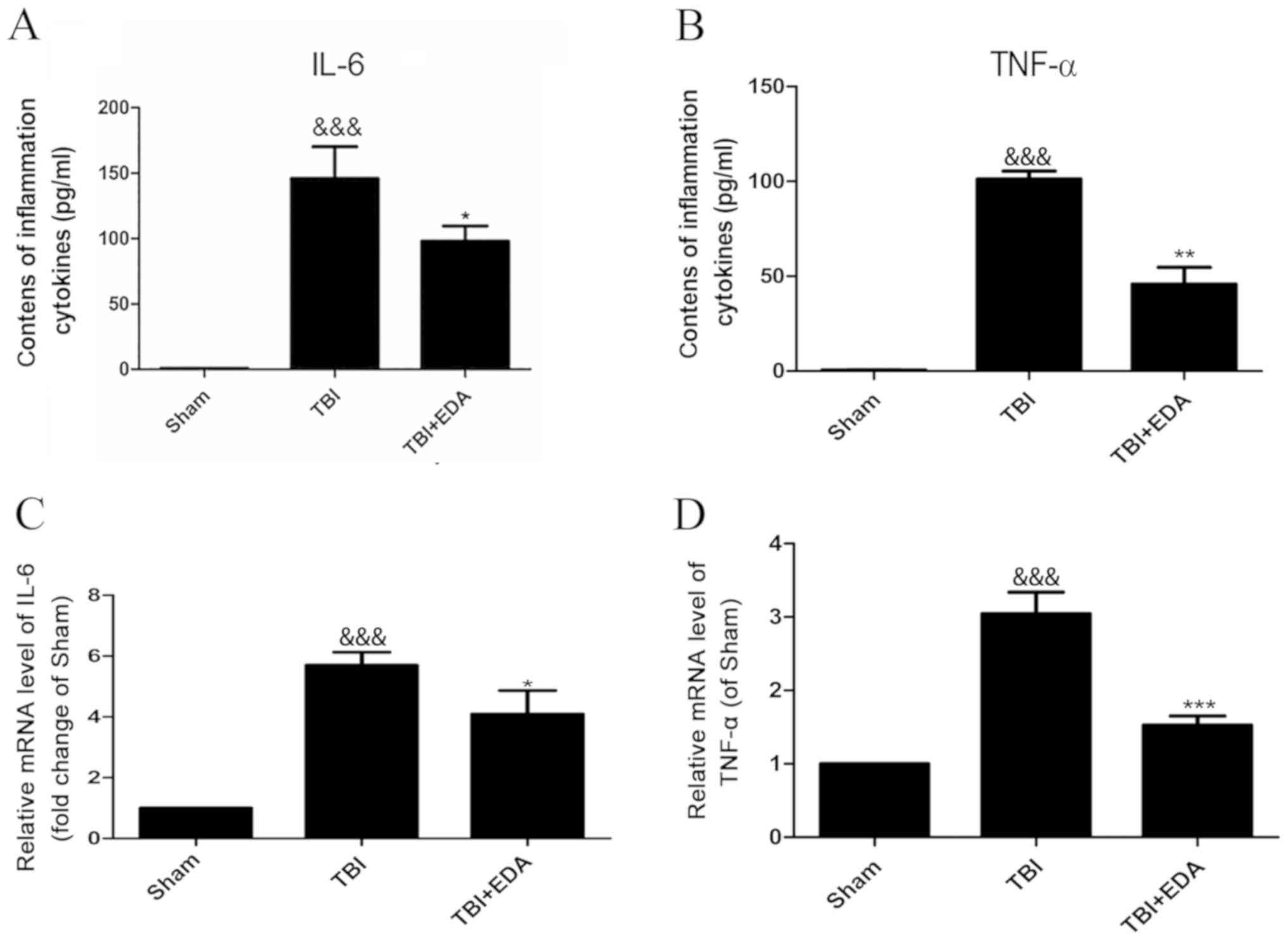
IL-6 and TNF-α levels in the cortex of brain tissue
samples were investigated to determine whether the EDA protective
effect was due to its ability to suppress inflammation induced by
TBI. IL-6 and TNF-α concentration in brain tissue (Fig. 2A and B) and mRNA expression levels
(Fig. 2C and D) were higher in the
TBI group compared with the sham group. However, both cytokine
concentration and mRNA expression significantly decreased following
EDA treatment (Fig. 2).
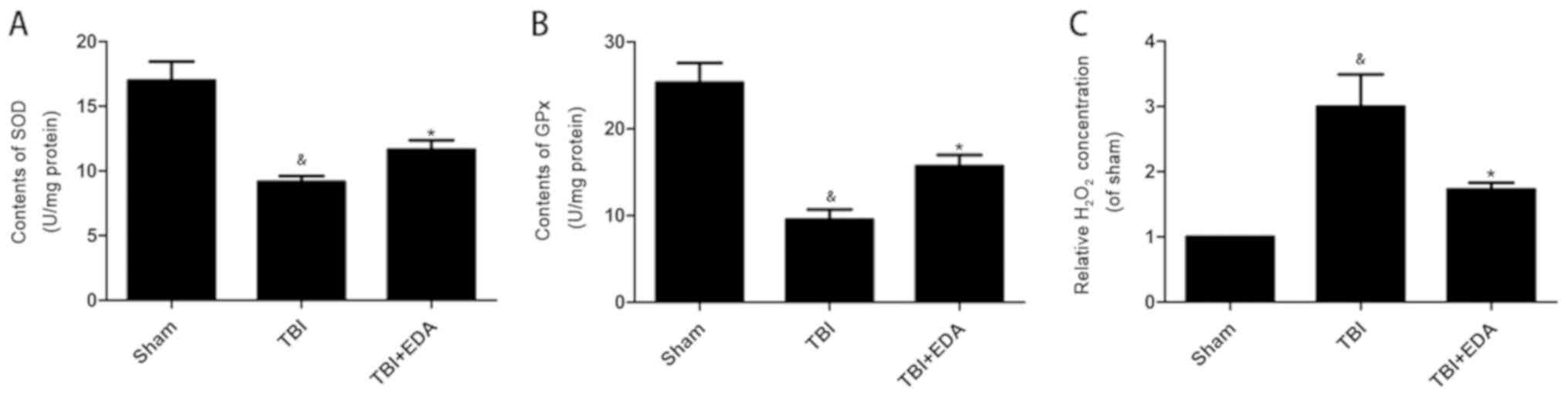
EDA suppresses oxidative stress levels
following TBI
SOD, GPx and H2O2 levels in
brain cortex samples were investigated to determine whether the EDA
protective effect was due to its ability to suppress oxidative
stress induced by TBI. H2O2 levels were
significantly higher in the TBI group compared with the sham group
(Fig. 3C), whilst EDA treatment
resulted in a significant decrease in levels in the brain (Fig. 3C). The levels of important
antioxidant enzymes SOD and GPx were significantly decreased in the
TBI group compared with the sham group in the brain. However, SOD
and GPx levels were higher in the EDA treated group compared with
the TBI group (Fig. 3A and B).
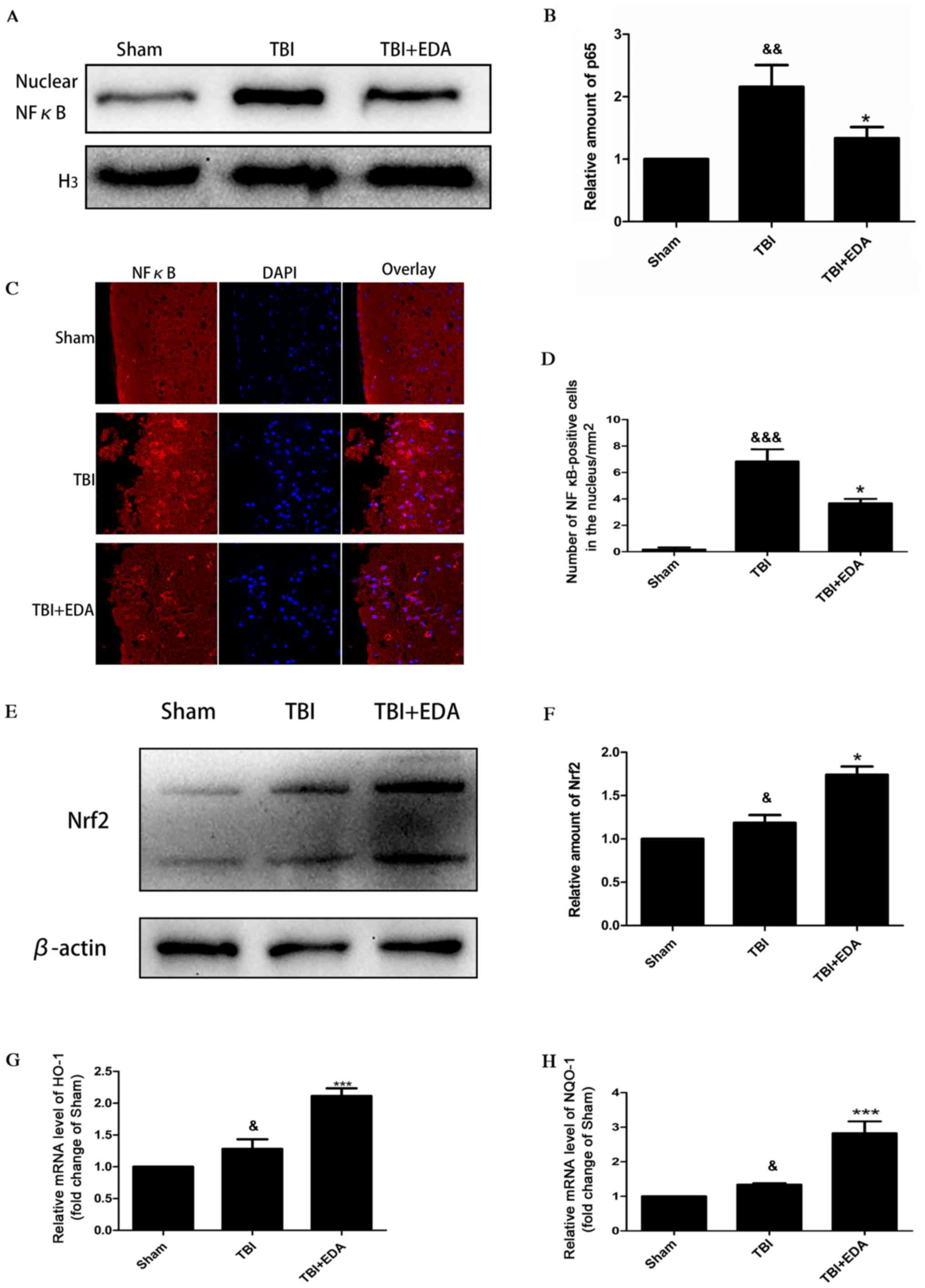
EDA inhibits NF-кB protein
translocation from the cytoplasm to the nucleus and upregulates
Nrf2 protein expression following TBI
NF-κB activity is essential to induce an
inflammatory response and it was demonstrated that EDA treatment
significantly decreased inflammatory cytokine expression (Fig. 2). Thus, the effects of EDA on NF-κB
activation following TBI were further examined. Following EDA
treatment, reduced expression of NF-κB was detected in the nucleus
compared with untreated controls (Fig.
4A and B). This was confirmed by immunofluorescence analysis of
NF-κB (Fig. 4C and D). To validate
that EDA significantly increased antioxidant enzyme activity
following TBI, Nrf2 expression was determined by western blot
analysis. Both TBI and EDA treatment induced higher Nrf2 expression
compared with the sham group (Fig.
4E). In addition, the EDA-treated group exhibited significantly
increased Nrf2 expression compared with the TBI group (Fig. 4F). Due to Nrf2 being activated by
EDA, the expression of genes downstream to Nrf2 were investigated.
RT-qPCR analysis demonstrated that HO-1 and NQO-1 mRNA expression
was significantly increased following TBI compared with the sham
group (Fig. 4G and H). EDA
administration further elevated HO-1 and NQO-1 mRNA expression
compared with the TBI group (Fig. 4G and
H).
Discussion
TBI is a serious public health problem that can lead
to permanent injury and neurological disorders (26). The associated secondary brain injury
involves a series of pathological processes, including the
inflammatory response (27) and
oxidative stress (28), and is
considered to be the leading cause of death and disability. Due to
the lack of effective TBI treatments, new and effective therapies
are urgently required. EDA is a novel free radical scavenger that
exerts a number of beneficial biological functions, involving
mechanisms that are anti-apoptotic (29), anti-inflammatory (23) and that alleviate stress on the
endoplasmic reticulum (17). A
previous study demonstrated that EDA can reduce oxidative stress,
has anti-inflammatory effects and improves neurological deficits
following concussion in vivo (30). The present results confirmed for the
first time, to the best of our knowledge, that EDA treatment exerts
a protective role in TBI by inhibiting the activation of
NF-κB-mediated inflammation and promoting the Nrf2 antioxidant
pathway.
NF-κB has a critical role in the inflammatory
response induced by TBI, and previous research has demonstrated
that EDA suppresses the systemic inflammatory response in a
transient focal ischemia rat model (23). A study determined that EDA inhibits
the activity of NF-κB induced by ischemia/reperfusion injury
(19). In addition, EDA treatment
confers neuroprotective effects following TBI in rats (31). Based on the literature, the present
study hypothesized that EDA may alleviate a TBI-induced
inflammatory response by regulating the NF-κB protein. The present
findings demonstrated that EDA treatment inhibited the
translocation of NF-кB from the cytoplasm to the nucleus following
TBI.
Oxidative stress occurs immediately following TBI.
Recent studies have identified that EDA treatment regulates
oxidative stress-related indicators in vivo and in
vitro (9,20). It was also demonstrated that EDA
attenuates hippocampal damage in an infant mouse model of
pneumococcal meningitis via the Nrf2/HO-1 pathway (9). Therefore, the present study
investigated the effect of EDA on these oxidative stress-related
indicators. It was established that treatment with EDA reduced
H2O2 level and elevated SOD and GPx activity
in mice following TBI. Since EDA can activate the Nrf2/ARE pathway
in the brains of mice, it was hypothesized that the protective
effects of EDA following TBI may be mediated via the Nrf2/ARE
signaling pathway. As predicted, the present results demonstrated
that EDA exerts neuroprotective effects following TBI via the
Nrf2/ARE signaling pathway.
The present study has limitations, for example, a
key control group, such as TBI+vehicle, was missing in several
experiments, which will be included in future work. However,
numerous studies have demonstrated that vehicle administration
alone does not affect inflammation and oxidative stress indicators,
and has no protective effect on mice following TBI (32,33).
In conclusion, the present study demonstrated, for
the first time, that EDA exerted neuroprotective effects following
TBI, resulting in improved neurological function, attenuated
neuronal apoptosis and inhibition of inflammation and oxidative
stress. The beneficial effects of EDA treatment were achieved, at
least partially, by promotion of Nrf2 expression and inhibition of
NF-κB translocation into the nucleus. These findings provide
evidence for the potential clinical application of EDA in TBI
treatment.
Acknowledgements
Not applicable.
Funding
Funding was provided by the Zhejiang Provincial
Natural Science Funding (grant no. LY19H150001) and Project of
Wenzhou Science and Technology Bureau (grant nos. Y20160155 and
Y20170234).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
MZ performed the experiments and drafted the
manuscript. CHT performed analysis and interpretation of data. FFW
acquired data and supervised the research. LYG revised the
manuscript. JX and HYZ were responsible for acquisition of funding
and supervision of the research group. DQC was responsible for the
conception and design of the present study and financial support.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The animal study protocols were approved by the
Animal Care and Use Committee of Wenzhou Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Delbary-Gossart S, Lee S, Baroni M,
Lamarche I, Arnone M, Canolle B, Lin A, Sacramento J, Salegio EA,
Castel MN, et al: A novel inhibitor of p75-neurotrophin receptor
improves functional outcomes in two models of traumatic brain
injury. Brain. 139:1762–1782. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Scott G, Zetterberg H, Jolly A, Cole JH,
De Simoni S, Jenkins PO, Feeney C, Owen DR, Lingford-Hughes A,
Howes O, et al: Minocycline reduces chronic microglial activation
after brain trauma but increases neurodegeneration. Brain.
141:459–471. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang ZG, Cheng Y, Yu XC, Ye LB, Xia QH,
Johnson NR, Wei X, Chen DQ, Cao G, Fu XB, et al: bFGF protects
against blood-brain barrier damage through junction protein
regulation via PI3K-Akt-rac1 pathway following traumatic brain
injury. Mol Neurobiol. 53:7298–7311. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lu J, Frerich JM, Turtzo LC, Li S, Chiang
J, Yang C, Wang X, Zhang C, Wu C, Sun Z, et al: Histone deacetylase
inhibitors are neuroprotective and preserve NGF-mediated cell
survival following traumatic brain injury. Proc Natl Acad Sci USA.
110:10747–10752. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu Z, Wang H, Shi X, Li L, Zhou M, Ding
H, Yang Y, Li X and Ding K: DL-3-n-Butylphthalide (NBP) Provides
Neuroprotection in the mice models after traumatic brain injury via
Nrf2-ARE signaling pathway. Neurochem Res. 42:1375–1386. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bramlett HM and Dietrich WD: Long-term
consequences of traumatic brain injury: Current status of potential
mechanisms of injury and neurological outcomes. J Neurotrauma.
32:1834–1848. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gyoneva S and Ransohoff RM: Inflammatory
reaction after traumatic brain injury: Therapeutic potential of
targeting cell-cell communication by chemokines. Trends Pharmacol
Sci. 36:471–480. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang L, Liu Z, Ren H, Zhang L, Gao S, Ren
L, Chai Z, Meza-Romero R, Benedek G, Vandenbark AA, et al:
DRα1-MOG-35-55 treatment reduces lesion volumes and improves
neurological deficits after traumatic brain injury. Metab Brain
Dis. 32:1395–1402. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li Z, Ma QQ, Yan Y, Xu FD, Zhang XY, Zhou
WQ and Feng ZC: Edaravone attenuates hippocampal damage in an
infant mouse model of pneumococcal meningitis by reducing HMGB1 and
iNOS expression via the Nrf2/HO-1 pathway. Acta Pharmacol Sin.
37:1298–1306. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kumar A, Stoica BA, Loane DJ, Yang M,
Abulwerdi G, Khan N, Kumar A, Thom SR and Faden AI:
Microglial-derived microparticles mediate neuroinflammation after
traumatic brain injury. J Neuroinflammation. 14:472017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen X, Wang H, Zhou M, Li X, Fang Z, Gao
H, Li Y and Hu W: Valproic acid attenuates traumatic brain
injury-induced inflammation in vivo: Involvement of autophagy and
the Nrf2/ARE signaling pathway. Front Mol Neurosci. 11:1172018.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mayeux JP, Teng SX, Katz PS, Gilpin NW and
Molina PE: Traumatic brain injury induces neuroinflammation and
neuronal degeneration that is associated with escalated alcohol
self- administration in rats. Behav Brain Res. 279:22–30. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ishii T, Kwon H, Hiscott J, Mosialos G and
Koromilas AE: Activation of the I kappa B alpha kinase (IKK)
complex by double-stranded RNA-binding defective and catalytic
inactive mutants of the interferon-inducible protein kinase PKR.
Oncogene. 20:1900–1912. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gupta SC, Sundaram C, Reuter S and
Aggarwal BB: Inhibiting NF-κB activation by small molecules as a
therapeutic strategy. Biochim Biophys Acta. 1799:775–787. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vomund S, Schäfer A, Parnham MJ, Brüne B
and von Knethen A: Nrf2, the master regulator of anti-oxidative
responses. Int J Mol Sci. 18:E27722017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fabrizio FP, Sparaneo A, Trombetta D and
Muscarella LA: Epigenetic versus genetic deregulation of the
KEAP1/NRF2 axis in solid tumors: Focus on methylation and noncoding
RNAs. Oxid Med Cell Longev. 2018:24920632018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tian A, Ma H, Zhang R, Cui Y and Wan C:
Edaravone improves spatial memory and modulates endoplasmic
reticulum stress- mediated apoptosis after abdominal surgery in
mice. Exp Ther Med. 14:355–360. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pan H, Wang H, Zhu L, Mao L, Qiao L and Su
X: Depletion of Nrf2 enhances inflammation induced by oxyhemoglobin
in cultured mice astrocytes. Neurochem Res. 36:2434–2441. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chiazza F, Chegaev K, Rogazzo M, Cutrin
JC, Benetti E, Lazzarato L, Fruttero R and Collino M: A nitric
oxide-donor furoxan moiety improves the efficacy of edaravone
against early renal dysfunction and injury evoked by
ischemia/reperfusion. Oxid Med Cell Longev. 2015:8046592015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jami MS, Salehi-Najafabadi Z, Ahmadinejad
F, Hoedt E, Chaleshtori MH, Ghatrehsamani M, Neubert TA, Larsen JP
and Møller SG: Edaravone leads to proteome changes indicative of
neuronal cell protection in response to oxidative stress. Neurochem
Int. 90:134–141. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lei S, Zhang P, Li W, Gao M, He X, Zheng
J, Li X, Wang X, Wang N, Zhang J, et al: Pre- and posttreatment
with edaravone protects CA1 hippocampus and enhances neurogenesis
in the subgranular zone of dentate gyrus after transient global
cerebral ischemia in rats. ASN Neuro. 6:17590914145584172014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou S, Yu G, Chi L, Zhu J, Zhang W, Zhang
Y and Zhang L: Neuroprotective effects of edaravone on cognitive
deficit, oxidative stress and tau hyperphosphorylation induced by
intracerebroventricular streptozotocin in rats. Neurotoxicology.
38:136–145. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fujiwara N, Som AT, Pham LD, Lee BJ,
Mandeville ET, Lo EH and Arai K: A free radical scavenger edaravone
suppresses systemic inflammatory responses in a rat transient focal
ischemia model. Neurosci Lett. 633:7–13. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Choi YK, Maki T, Mandeville ET, Koh SH,
Hayakawa K, Arai K, Kim YM, Whalen MJ, Xing C, Wang X, et al: Dual
effects of carbon monoxide on pericytes and neurogenesis in
traumatic brain injury. Nat Med. 22:1335–1341. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bedi SS, Aertker BM, Liao GP, Caplan HW,
Bhattarai D, Mandy F, Mandy F, Fernandez LG, Zelnick P, Mitchell
MB, et al: Therapeutic time window of multipotent adult progenitor
therapy after traumatic brain injury. J Neuroinflammation.
15:842018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lozano D, Gonzales-Portillo GS, Acosta S,
de la Pena I, Tajiri N, Kaneko Y and Borlongan CV:
Neuroinflammatory responses to traumatic brain injury: Etiology,
clinical consequences, and therapeutic opportunities.
Neuropsychiatr Dis Treat. 11:97–106. 2015.PubMed/NCBI
|
|
28
|
Liu Y, Bao Z, Xu X, Chao H, Lin C, Li Z,
Liu Y, Wang X, You Y, Liu N and Ji J: Extracellular
Signal-Regulated Kinase/Nuclear Factor-Erythroid2-like2/Heme
Oxygenase-1 Pathway-Mediated Mitophagy Alleviates Traumatic Brain
Injury-Induced Intestinal Mucosa Damage and Epithelial Barrier
Dysfunction. J Neurotrauma. 34:2119–2131. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang WW, Bai F, Wang J, Zheng RH, Yang
LW, James EA and Zhao ZQ: Edaravone inhibits pressure
overload-induced cardiac fibrosis and dysfunction by reducing
expression of angiotensin II AT1 receptor. Drug Des Devel Ther.
11:3019–3033. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Higashi Y, Hoshijima M, Yawata T, Nobumoto
A, Tsuda M, Shimizu T, Saito M and Ueba T: Suppression of oxidative
stress and 5-lipoxygenase activation by edaravone improves
depressive-like behavior after concussion. J Neurotrauma.
31:1689–1699. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang GH, Jiang ZL, Li YC, Li X, Shi H, Gao
YQ, Vosler PS and Chen J: Free-radical scavenger edaravone
treatment confers neuroprotection against traumatic brain injury in
rats. J Neurotrauma. 28:2123–2134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rowe RK, Harrison JL, Zhang H, Bachstetter
AD, Hesson DP, O'Hara BF, Greene MI and Lifshitz J: Novel TNF
receptor-1 inhibitors identified as potential therapeutic
candidates for traumatic brain injury. J Neuroinflammation.
15:1542018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gao YY, Zhang ZH, Zhuang Z, Lu Y, Wu LY,
Ye ZN, Zhang XS, Chen CL, Li W and Hang CH: Recombinant milk fat
globule-EGF factor-8 reduces apoptosis via integrin β3/FAK/PI3K/AKT
signaling pathway in rats after traumatic brain injury. Cell Death
Dis. 9:8452018. View Article : Google Scholar : PubMed/NCBI
|