Introduction
A member of the caveolin family, caveolin-3 (CAV3),
is specifically expressed in muscle cells, including
cardiomyocytes, skeletal muscle cells and smooth muscle cells
(1,2). CAV3 interacts with and regulates
various signaling molecules in the caveolae of the cell membrane,
which include the insulin receptor (IR), IR substrate-1 (IRS-1),
G-protein-coupled receptors and protein kinase C (3–8). CAV3
gene mutations lead to a variety of clinical phenotypes including
limb-girdle muscular dystrophy, rippling muscle disease, distal
myopathy, hyperCKemia and cardiomyopathy (9,10).
CAV3-null mice have been demonstrated to exhibit insulin
resistance, characterized by reduced glucose uptake in skeletal
muscles, impaired glucose tolerance and elevated serum lipids
(4). Furthermore, the injection of
CAV3 proteins restored insulin signaling in skeletal muscles
(4). Studies have demonstrated that
CAV3-knockout mice have increased adiposity, postprandial
hyperinsulinemia, whole-body insulin resistance and whole-body
glucose intolerance (4,11,12).
More importantly, insulin-stimulated whole-body glucose uptake and
whole-body glycogen synthesis decreased in CAV3-knockout mice
compared with wild-type mice (12).
These results indicate that CAV3 serves an important role in
maintaining blood glucose balance.
Type 2 diabetes mellitus (T2DM) is the most common
type of diabetes. Insulin is produced in T2DM, but production is
either insufficient or the body does not respond with the
appropriate sensitivity, which leads to a high blood glucose
concentration. High blood glucose concentration is an important
factor in the development of T2DM (13). A previous study demonstrated that
among ~1,000 patients with T2DM, a variety of mutations were
present in the CAV3 gene of 50 patients that exhibited blood
glucose levels >20 mmol/l and no obvious genetic disease
(14). The results of full-gene
scans indicated that the total number of gene variations in the
CAV3 gene in patients with T2DM was 48%, compared with 7% in
healthy patients (14). A previous
study also assessed the transfection of wild-type CAV3 (WT) in
muscle cells and found that the PI3K/AKT signaling pathway were
activated, increasing the plasma membrane localization of glucose
transporter type 4 (GLUT-4) and increasing glucose uptake, cell
growth and proliferation (15). One
mutation of the CAV3 gene (P104L) in patients with myasthenia has
been revealed to be associated with the inhibition of
insulin-stimulated glucose uptake and glycogen synthesis in
myocytes (16). Muscles constitute
~40% of human body weight and are critical tissues for glucose
metabolism and important sites for insulin resistance in diabetic
patients (17,18). The aim of the current study was to
assess the extent to which the CAV3 gene mutations in patients with
T2DM impaired glycometabolism in muscle cells and subsequently
contributed to T2DM development. The CAV3 K15N (G/C) mutation was
assessed in the present study. The mutant was transfected into
C2C12 muscle cells and the effects were compared with cells
transfected with WT.
Materials and methods
Predicted secondary structure of the
mutant CAV3 protein
The CAV3 gene sequence was retrieved from the
National Center for Biotechnology Information (www.ncbi.nlm.nih.gov; Human CAC3: Chromosome 3,
location NC_000003.12; Mouse CAV3: Chromosome 6, location
NC_000072.6; Rat CAV3: Chromosome 4, location NC_005103.4), and
then the open reading frame sequence was searched by DNAstar 7.1
software (https://www.dnastar.com/), and
finally translated into amino acid sequence. Sequence alignment was
conducted using CLUSTAL 2.1 (www.clustal.org) and T-COFFEE 12.00 software
(http://tcoffee.crg.cat/). The secondary structure
of the CAV3 protein was predicted using PSIPRED 4.0 software
(bioinf.cs.ucl.ac.uk/psipred).
Cell culture
The mouse skeletal muscle cell line C2C12 (Type
Culture Collection of the Chinese Academy of Sciences) was cultured
in an incubator at 37°C, in DMEM (cat. no. 12800-017; Gibco; Thermo
Fisher Scientific, Inc.) supplemented with high glucose (25 mM
D-Glucose), 10% FBS (Invitrogen; Thermo Fisher Scientific, Inc.),
penicillin (100 U/ml) and streptomycin (100 µg/ml) at 5%
CO2. Cells in the logarithmic phase (the 4 and 5th
generations) were collected for transfection.
Stable transfection
A total of 0.5×105 cells/well were seeded
into 24-well plates in antibiotic-free DMEM containing 10% FBS.
When cells reached 80% confluence, 5,368 ng/µl of an expression
plasmid (EX-T1783-M98; GeneCopoeia, Inc.) containing wild-type CAV3
+ enhanced green fluorescent protein (eGFP; WT) or 1,289 ng/µl CAV3
K15N + eGFP (K15N) were transfected into C2C12 cells with
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) following manufacturer's protocol. The
transfection was as follows: 50 µl DMEM + 1.5 µl Lipofectamine 3000
+ 0.5 µg plasmid DNA + 1 µl P3000TM. The negative control (NC)
group was transfected with a vector carrying eGFP alone. The CAV3
genes in the expression vectors were homozygotes. At 24 h
post-transfection, cells were cultured in high-glucose DMEM
containing 400 µg/ml G418 for 7 days at 37°C prior to selection of
positive clones. To obtain a stable transfection cell line, cells
were subsequently transferred to high-glucose DMEM containing 200
µg/ml G418 at 37°C in a 5% CO2 incubator for 45 days.
After the cell line was constructed, the stability of the cell line
was assessed using western blot analysis 55 days after transfection
by analyzing whether the eGFP-CAV3 fluorescent protein was
successfully integrated into the cell genome.
Western blot analysis
Cells were stimulated with 100 nmol/l insulin (cat.
no. 16634; Sigma-Aldrich; Merck KGaA) for 30 min at 37°C. Cells
were subsequently rinsed with cold PBS and total protein was
extracted using 1X RIPA lysis buffer (cat. no. P0013; Beyotime
Institute of Biotechnology) and protease inhibitors (Beijing
Leagene Biotech Co., Ltd.). After 30 min of incubation on ice,
cells were centrifuged at 10,142 × g for 10 min at 4°C and
supernatant was collected for subsequent quantification. The
protein concentration was obtained by the BCA kit (cat. no. P0010S;
Beyotime Institute of Biotechnology). A total of 25 or 50 µg of
protein was separated by SDS-PAGE on a 12% gel and transferred to
PVDF membranes. Membranes were blocked with 5% dried skim milk and
TBS plus 0.01% Tween 20 for 1 h at room temperature and
subsequently incubated overnight at 4°C with mouse anti-GFP
monoclonal antibody (1:500; cat. no. J20625; Beijing Transgen
Biotech Co., Ltd.), mouse anti-CAV3 monoclonal antibody (1:100;
cat. no. sc-5310; Santa Cruz Biotechnology, Inc.), rabbit
anti-caveolin-1 (CAV1) polyclonal antibody (1:1,000; cat. no.
sc-894; Santa Cruz Biotechnology, Inc.), mouse anti-phosphorylated
(p)-AKT ser473 monoclonal antibody (1:1,000; cat. no. 12694; Cell
Signaling Technology, Inc.), mouse anti-AKT monoclonal antibody
(1:1,000; cat. no. 2920; Cell Signaling Technology, Inc.), goat
anti-AKT1 polyclonal antibody (1:1,000; cat. no. sc-1618; Santa
Cruz Biotechnology, Inc.), mouse anti-AKT2 monoclonal antibody
(1:1,000; cat. no. 5239; Cell Signaling Technology, Inc.), goat
anti-GLUT-4 polyclonal antibody (1:100; cat. no. sc-01608; Santa
Cruz Biotechnology, Inc.), rabbit anti-p glycogen synthase kinase 3
beta (GSK3β) monoclonal antibody (1:1,000; cat. no. 9323; Cell
Signaling Technology, Inc.) or rabbit anti-GSK3β monoclonal
antibody (1:1,000; cat. no. 9315; Cell Signaling Technology, Inc.).
After washing with TBST (50 mg Tris-HCl, pH 7.6, 150 mM NaCl, 0.2%
Tween 20), the membranes were incubated with Dylight™800-conjugated
secondary antibodies: Anti-mouse (1:1,000; cat. no. 5257P; Cell
Signaling Technology, Inc.), anti-rabbit (1:1,000; cat. no. 5151P;
Cell Signaling Technology, Inc.) and anti-goat (1:500; cat. no.
E032830-01; EarthOx, Inc.) at room temperature for 2 h. Protein
bands were subsequently visualized and quantified using a Li-Cor
Odyssey infrared imager (LI-COR Biosciences). Integrated
intensities of the 800 nm infrared signal for each band were
calculated using the Odyssey system.
Immunofluorescence
Cells were induced with 100 nmol/l insulin for 30
min at 37°C, washed with PBS three times, fixed with 4%
paraformaldehyde for 30 min at room temperature and blocked in 10%
donkey serum (cat. no. CJ-326; Beijing Biotopped Biotech Co., Ltd.)
in PBS at room temperature for 30 min. Mouse monoclonal anti-CAV3
(1:500; cat. no. sc-5310; Santa Cruz Biotechnology, Inc.) and goat
polyclonal anti-GLUT4 (1:500; cat. no. sc-01608; Santa Cruz
Biotechnology, Inc.) were used as primary antibodies. Alexa Fluor
594-conjugated donkey anti-goat (cat. no. A11058) and Alexa Fluor
647-conjugated donkey anti-mouse (cat. no. A31571; both 1:1,000;
Invitrogen; Thermo Fisher Scientific, Inc.) were used as secondary
antibodies. The cells were then washed with PBS three times and
incubated with 0.5 µg/ml DAPI (cat. no. c0065-10; Solarbio Inc.) at
37°C for 5 min in the dark. Cells were subsequently washed three
times in PBS and visualized under confocal laser scanning
microscopy at a magnification of ×40.
Glucose uptake and glycogen synthesis
assays
Stably transfected cells were seeded at a density of
1.25×105 in 60-mm culture dishes with high-glucose DMEM
containing 10% FBS under conventional culture conditions. After 2
days, cells were moved to low-glucose DMEM (5.55 mM D-Glucose; cat.
no. 31600-034; Gibco; Thermo Fisher Scientific, Inc.) for 12 h at
37°C and subsequently cultured in high-glucose DMEM containing 100
nmol/l insulin at 37°C. A total of 10 µl supernatant was removed
from the medium to measure glucose uptake at 12 and 24 h using the
glucose oxidase/hydrogen peroxide method (19) and a glucose assay kit (Nanjing
Jiancheng Bioengineering Institute) according to manufacturer's
protocol. Glycogen synthesis was detected at 24 h after the insulin
incubation with the glycogen assay kit (Nanjing Jiancheng
Bioengineering Institute) following manufacturer's protocol
(20,21).
Statistical analysis
All data are presented as the mean ± standard
deviation. An unpaired two-tailed Student's t-test was performed
using SPSS 20.0 software (IBM Corp.) for comparisons between two
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
K15N mutation alters the secondary
structure of the CAV3 protein
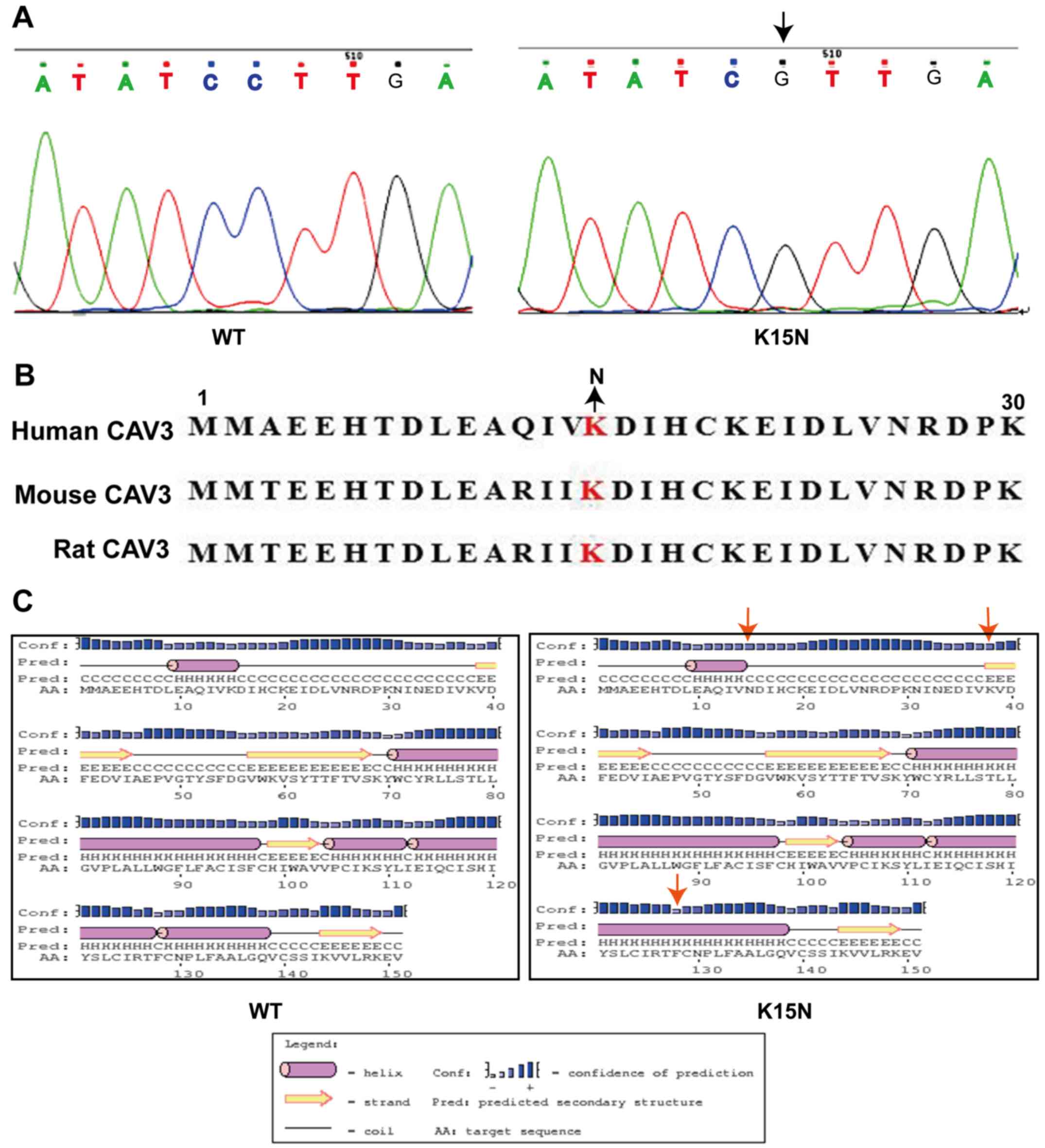
Two exons exist within the CAV3 gene. The K15N
mutation is located in the first exon and changes the 15th
nucleotide from G to C, replacing the protein's amino acid lysine
(AAG) with asparagine (AAC) (Fig.
1A). The 15th lysine is highly conserved in this protein region
among humans and other species (Fig.
1B). PSIPRED software predicted that the following three
changes would occur in the proteins' secondary structure caused by
the K15N mutation: One helix would change into one coil, a coil
would change into a strand at the N-terminus and a further coil
would change into a helix at the C-terminus (Fig. 1C).
Identification of stably transfected
cells
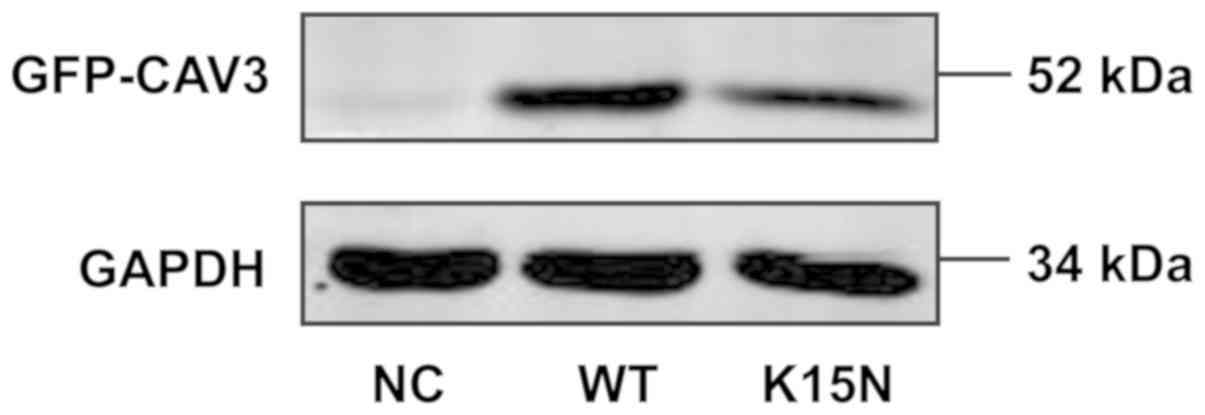
The molecular weight of the human CAV3 protein is 25
kDa and the molecular weight of the eGFP protein is 27 kDa. The
molecular weight of the recombinant eGFP-CAV3 protein was therefore
~52 kDa. As presented in Fig. 2,
recombinant proteins with a molecular weight of 52 kDa were
detected in the WT and K15N groups, respectively. However, no
recombinant protein was detected in the empty vector that contained
only eGFP. The results indicate that stable transfection was
successful and the cells could therefore be confidently used in the
subsequent experiments.
K15N mutation reduced recombinant CAV3
expression but did not affect the CAV1 protein levels
The recombinant protein consists of eGFP and CAV3,
therefore it can be detected by a eGFP antibody and a CAV3
antibody. After treatment with the CAV3 antibody, recombinant
CAV3-eGFP protein expression in the K15N group decreased and was
~41.54% lower compared with the expression level in the
WT-transfected group (P=0.037; Fig.
3A). However, no significant difference in CAV1 expression was
determined between the two groups (Fig.
3B).
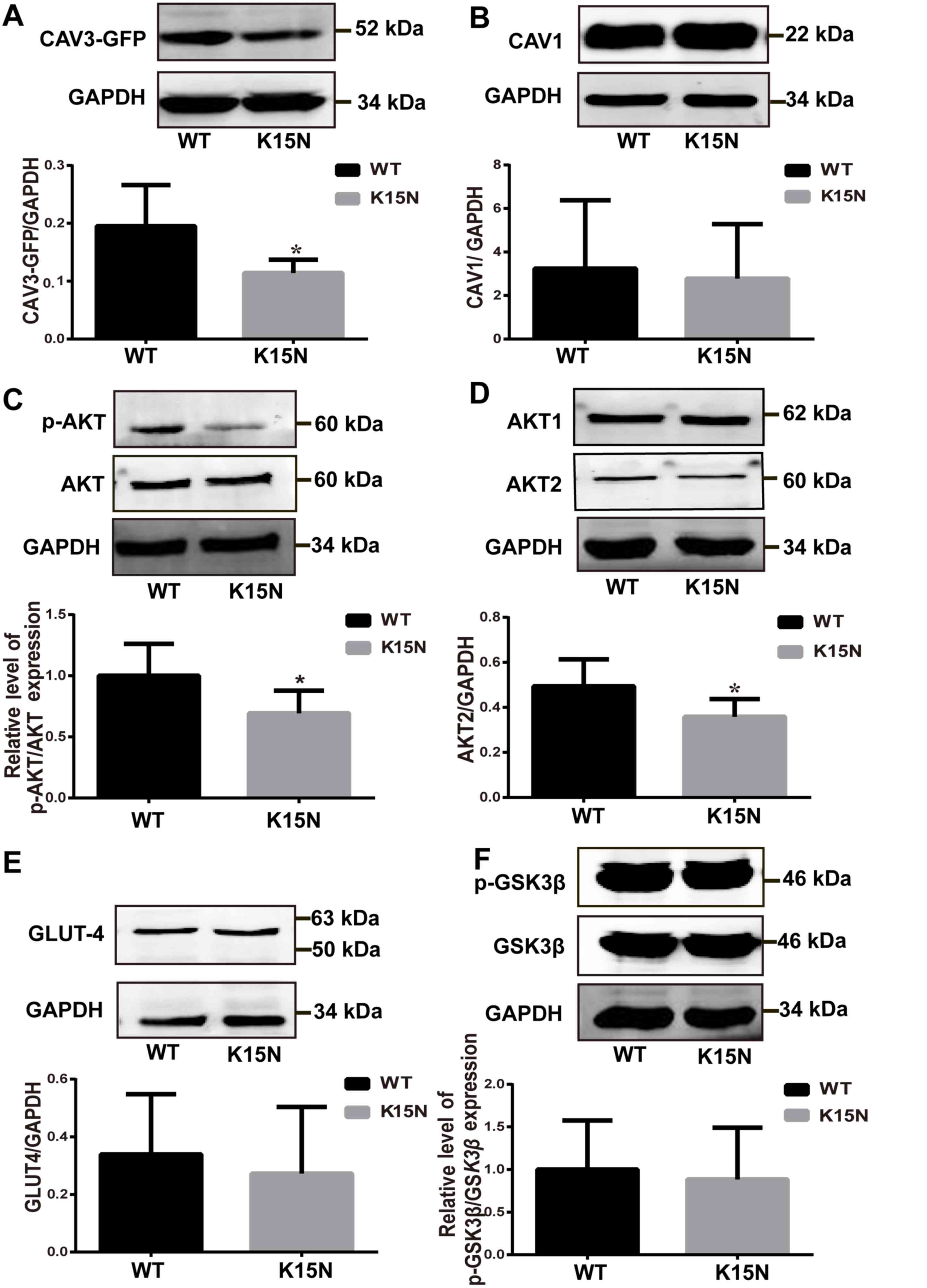 | Figure 3.K15N mutation effect on protein
expression in the AKT signaling pathway. Western blot and
densitometry analysis of (A) CAV3-eGFP, (B) CAV1, (C) p-AKT, (D)
AKT1 and AKT2, (E) GLUT-4 and (F) p-GSK3β expression in WT and K15N
cells. All data are presented as the mean ± standard deviation.
*P<0.05 vs. WT. CAV3, caveolin-3; eGFP, enhanced green
fluorescent protein; CAV-1, caveolin-1; p, phosphorylated; GLUT-4,
glucose transporter type 4; GSK3β, glycogen synthase kinase 3β; WT,
wild-type; K15N, CAV3 K15N mutation. |
K15N mutation reduces p-AKT and AKT2
levels but not total GLUT-4 or p-GSK3β expression
AKT activation requires phosphorylation and is
associated with the activation of the PI3K/AKT insulin signaling
pathway. Two AKT phosphorylation sites exist, including T308 and
S473 (22). However, optimal AKT
activity requires the regulation of the Ser473 phosphorylation site
(23–25). Therefore, S473 was used to detect AKT
phosphorylation levels. AKT S473 phosphorylation in the K15N group
was significantly decreased compared with the CAV3 WT group
(P=0.040; Fig. 3C), however the
total AKT protein content in the cell exhibited no marked
difference between the two groups.
In the present study, AKT1 and AKT2 protein
expression levels were assessed using western blot analysis and the
results demonstrate that AKT2 expression was significantly lower in
the K15N group when compared with the WT group (P=0.042; Fig. 3D), whereas AKT1 protein expression
was not significantly different between the two groups. The results
also demonstrated that the expression of downstream molecule GLUT-4
(Fig. 3E) and the relative
p-GSK3β/GSK3β ratio (Fig. 3F) did
not differ significantly between the WT and K15N group.
K15N mutation decreases the
translocation of cell membrane GLUT-4
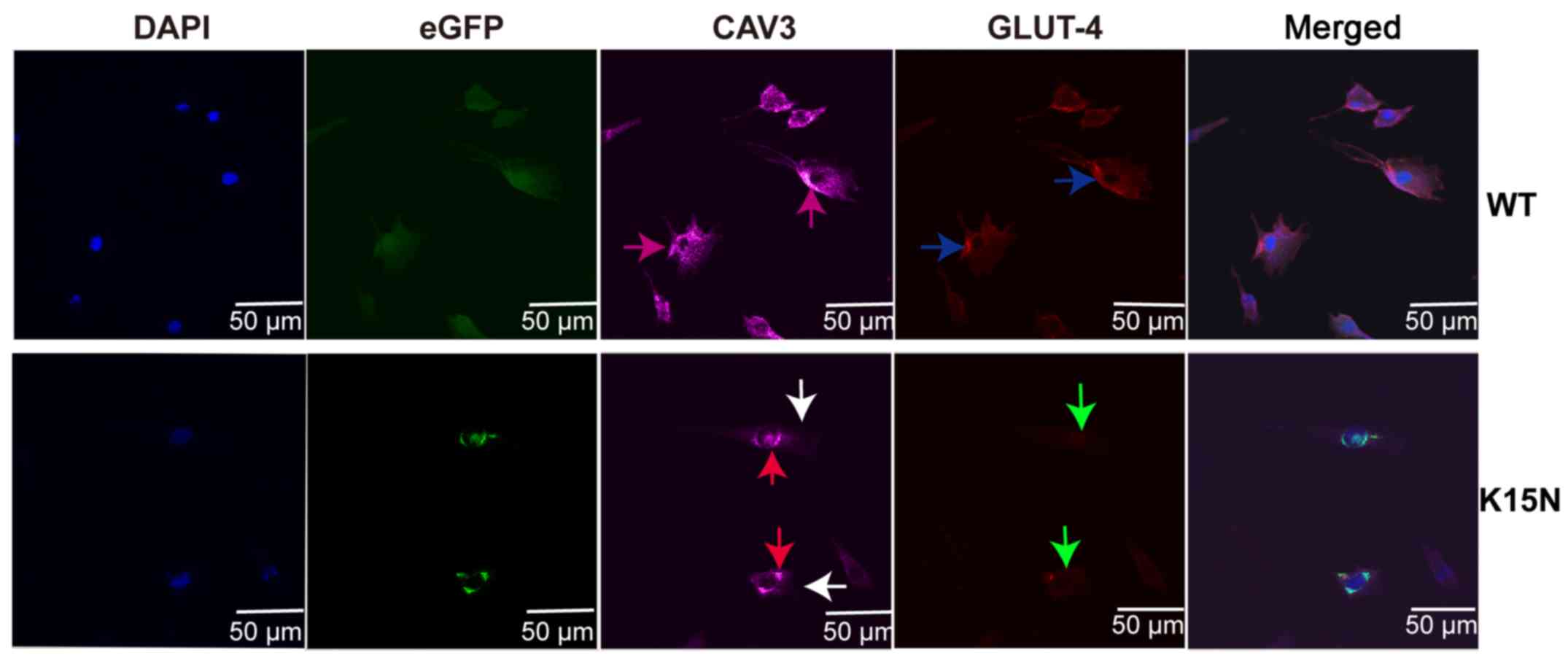
CAV3 protein was evenly localized in the cytoplasm
and on the cell membrane (purple arrows) in the WT group; however,
its expression on the cell membrane of the K15N group was decreased
(white arrow) and accumulated in the vesicles around the nucleus
(red arrow; Fig. 4). GLUT-4
immunolabeling strongly outlined the cell surface membrane in WT
cells (blue arrows), while the translocation of GLUT-4 to the cell
membrane of K15N group cells was markedly decreased as GLUT-4 was
not localized to the cell membrane (green arrow).
K15N mutation impairs glycometabolism
in muscle cells
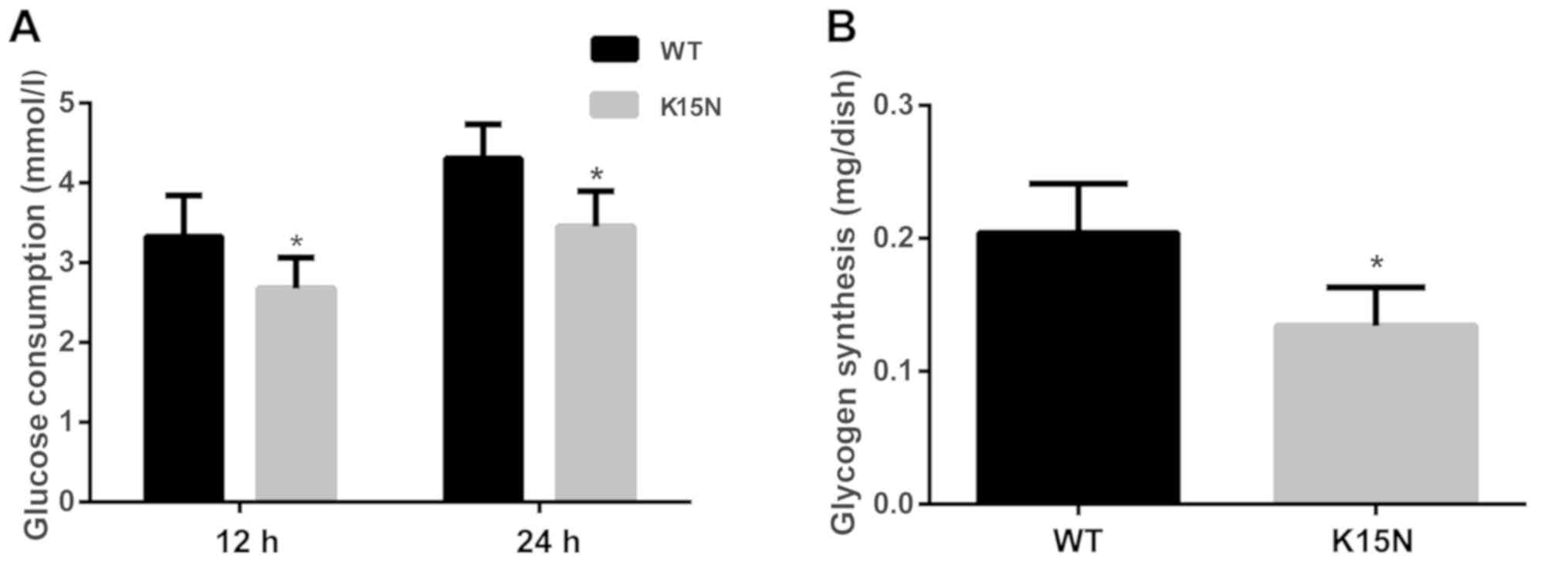
In the present study, differences in glucose uptake
between the WT group and the K15N group were observed at 12 and 24
h. The results indicate that the K15N mutation significantly
reduces glucose uptake after insulin stimulation at 12 and 24 h
(P=0.021 and P=0.003, respectively; Fig.
5A). The quantity of synthesized glycogen in the K15N group was
also significantly lower than that in the WT group (P=0.004;
Fig. 5B).
Discussion
The CAV3 protein is 151 amino acids long and is
divided into five separate domains (9). The K15N mutation is located in the
N-terminus, which is highly conserved in the CAV3 protein and may
impact protein configuration (9,26).
Mutations in this region may impair the location and maturity of
the CAV3 protein in the cell membrane (27,28).
This may induce effects on the signaling pathways, which are
associated with this protein (29).
CAV3 proteins have been demonstrated to act as
molecular scaffolds, regulating the activity of a variety of
signaling molecules and modulating their function (3,30,31).
CAV3 defects cause downstream signaling molecules (that are
regulated by CAV3) to degrade at a faster rate (12). The K15N mutation reduces the CAV3
protein on the muscle cell membranes by ~95% (32). This reduction causes the abnormal
localization of proto-oncogene tyrosine-protein kinase Src on the
Golgi (33) and leads to CAV3
protein retention (15). Consistent
with these data, western blot analysis performed in the current
study indicated that the CAV3 K15N mutation led to decreased
recombinant CAV3 protein expression in muscle cells. CAV1, another
subtype of CAV, plays an important role in maintaining insulin
signaling (4), especially in
regulating glucose uptake in skeletal muscle cells (34). Therefore the authors also detected
the expression of CAV1 protein. However, CAV1 expression was not
significantly different between the two groups. Thus, in that
experiment, the authors were able to rule out its effect on glucose
metabolism. Furthermore, confocal scanning microscopy revealed that
the K15N mutation caused CAV3 to accumulate around the nucleus and
not the cell membrane. The Golgi body is the site of protein
packaging and maturation (35). CAV3
protein retention indicates an increased likelihood that the K15N
mutation may lead to an amino acid sequence change; retention may
be due to the Golgi's inability to recognize the mutated protein,
thus allowing the CAV3 to mature into the membrane protein
(16). Subsequently, the stability
and effects of related molecules on the skeletal muscle cell
membrane may also be affected (12).
The IR/PI3K/AKT/GLUT signaling pathway is primarily
associated with insulin signaling in skeletal muscle and liver
cells (36). Additionally, this
pathway has been demonstrated to be a major mechanism in the
development of insulin resistance (37). However, IR and GLUT-4 are associated
with glucose metabolism and are localized to membrane caveolae,
with their expression being regulated by CAV3 on the cell membrane.
CAV3 can enhance the expression of IR (38–40) by
stimulating IR kinase activity, increasing the stability of IR at
the sarcolemmal membrane and reducing its degradation (12), stimulating the phosphorylation of
IRS-1 and activating the PI3K/AKT signaling pathway (15,18).
Activated AKT not only promoted the translocation of GLUT-4 to the
plasma membrane and enhanced glucose uptake, but also induced the
phosphorylation of GSK3β, which leads to glycogen synthesis via the
activation of glycogen synthase (41,42).
Therefore, any alteration of protein expression in the P13K/AKT
signaling pathway may influence insulin sensitivity.
Insulin resistance is a major factor in T2DM
development and can result in the dysfunction of the insulin
signaling pathway (43–46). Therefore, to investigate the effect
of the CAV3 K15N mutation on insulin-stimulated glucose metabolism,
the expression and activation state of insulin signaling
pathway-related molecules was assessed in the current study. There
are three subtypes of AKT: AKT1, AKT2 and AKT3. AKT1 is associated
with cell physiological growth and function, AKT2 is associated
with the insulin-mediated regulation of glucose homeostasis and
AKT3 is associated with various neurological conditions (47,48). The
CAV3 K15N mutation decreased AKT phosphorylation and AKT2 levels,
but total AKT, AKT1, GSK3β and p-GSK3β protein levels did not
differ significantly between groups. A limitation of the current
study is shown in the expression of these proteins, as some of the
samples were large and caused double banding. The results indicate
that the activation of AKT signaling may be inhibited when the CAV3
protein is decreased on the cell membrane. The overexpression of
CAV3 enhanced the tyrosine phosphorylation of IRS-1 and activated
the AKT signaling pathway (49). The
K15N mutation resulted in decreased CAV3 protein expression on the
cell membrane, which inhibited the phosphorylation of AKT and may
subsequently affect the activation of various downstream signaling
molecules. This result may be due to downregulated CAV3 expression,
causing CAV3 to attach to the cell membrane abnormally, affecting
the expression and stability of weakening the physiological effect
of IR on the PI3K/AKT signaling pathway. Various downstream
signaling pathways, including AKT phosphorylation and GLUT-4
protein translocation may also be selectively suppressed.
AKT serves a role in cell metabolism via its
association with glucose transporters and glucose uptake, and the
conversion of stored glycogen to glucose (50). Studies have demonstrated that mice
with an AKT1 gene deletion show normal glucose metabolism (51), whereas mice with an AKT2 gene
deletion or humans with an AKT2 gene mutation develop insulin
resistance and a type 2 diabetes-like phenotype (51–53).
Furthermore, in vitro studies have demonstrated that AKT2
silencing causes inhibition of insulin-induced GLUT-4 translocation
to the plasma membrane (52,54). Due to the role of AKT2 in
insulin-mediated glucose metabolism, the results of the present
study indicate that AKT2 expression decreased in cells transfected
with K12N compared with the. WT. Decreased AKT2 expression may
decrease the insulin sensitivity of skeletal muscle cells and may
be an important molecular mechanism associated with insulin
resistance in patients with T2DM.
Insulin can promote glucose transport in skeletal
muscles by stimulating the translocation of GLUT-4 from
intracellular storage vesicles to the plasma membrane (55). CAV3 promotes glucose uptake in
skeletal muscle cells by enhancing GLUT-4 translocation to the
plasma membrane (56). In
individuals that exhibit insulin resistance, GLUT-4 expression is
normal (57), but translocation to
the cell membrane is decreased (58). In the present study, the results
revealed that in CAV3 K15N cells, GLUT-4 was less concentrated on
the cell membrane due to reduced translocation, but the total
GLUT-4 expression of GLUT-4 protein did not change. These results
indicated that K15N mutations affect glucose metabolism by reducing
GLUT-4 translocation to the cell membrane and not through the
overall expression of GLUT-4 protein.
CAV3-null myotubes exhibit low levels of
insulin-stimulated glucose uptake as PI3K/AKT activation and plasma
membrane GLUT-4 translocation are reduced (56). Consistent with this effect, the
current study demonstrated that subsequent to insulin stimulation,
the CAV3 K15N mutation reduced p-AKT and AKT2 expression, GLUT-4
translocation to the membrane, glucose uptake and glycogen
synthesis. Therefore, the K15N mutation in patients with T2DM may
impair glycometabolism in skeletal muscle cells and increase blood
glucose.
In total, the authors hypothesized that the CAV3
gene K15N mutation, which was located in patients with T2DM,
decreased total CAV3 expression, caused CAV3 proteins to aggregate
in the Golgi and caused the proteins to lose their normal
functional role. This may affect the stability of IR on the cell
membrane and inhibit the AKT pathway, which is associated with AKT
phosphorylation and GLUT-4 protein translocation (12,16). The
IR/PI3K/AKT/GLUT signaling pathway may be restricted due the K15N
mutation and may result in glucose accumulation, which is exhibited
in hypoglycemia and during the development of T2DM (59).
The current study demonstrated an association
between the CAV3 K15N mutation and the pathogenesis of T2DM.
However, subsequent observations are required in other cell lines
and animal models to further validate these results. The current
study assessed the effect of CAV3 K15N mutation on impaired glucose
metabolism induced by insulin. The K15N mutation exhibited the same
effect as the P104L mutation in CAV3 (16), which demonstrated impaired glucose
metabolism in the C2C12 cell line. These kinds of patients showed
different symptoms, which may be attributed to one or more
mutations in their genes, including the CAV3 gene, and the
interactions of the genes (60,61). To
the best of our knowledge, the current study is the first to assess
the contribution of K15N mutation in T2DM.
The present study identified AKT is a key factor in
the regulation of glucose metabolism in cells. The present study
also assessed the downstream signaling factors of AKT, including
GSK3β, p-GSK3β and GLUT-4. Other downstream effectors of AKT
signaling including glucose transporter 2, glycogen synthase and
phosphoinositide 5-phosphate 4-kinase type II remain to be
investigated.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81660360).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YH and QH designed and performed the experiments,
analyzed the data, and wrote the manuscript. YD, LS, LY, JH and QH
performed the research and analyzed the data. JM, XL, HZ and JX
designed and executed the search strategies and contributed to the
critical revision of the manuscript and approved the final version.
GL designed and supervised the research. All authors have read and
approved the final draft of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Horikawa YT, Panneerselvam M, Kawaraguchi
Y, Tsutsumi YM, Ali SS, Balijepalli RC, Murray F, Head BP, Niesman
IR, Rieg T, et al: Cardiac-specific overexpression of caveolin-3
attenuates cardiac hypertrophy and increases natriuretic peptide
expression and signaling. J Am Coll Cardiol. 57:2273–2283. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Song KS, Scherer PE, Tang Z, Okamoto T, Li
S, Chafel M, Chu C, Kohtz DS and Lisanti M: Expression of
caveolin-3 in skeletal, cardiac, and smooth muscle cells.
Caveolin-3 is a component of the sarcolemma and co-fractionates
with dystrophin and dystrophin-associated glycoproteins. J Biol
Chem. 271:15160–15165. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Talukder MA, Preda M, Ryzhova L, Prudovsky
I and Pinz IM: Heterozygous caveolin-3 mice show increased
susceptibility to palmitate-induced insulin resistance.
Physiological reports. 4:e127362016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oshikawa J, Otsu K, Toya Y, Tsunematsu T,
Hankins R, Kawabe J, Minamisawa S, Umemura S, Hagiwara Y and
Ishikawa Y: Insulin resistance in skeletal muscles of
caveolin-3-null mice. Proc Natl Acad Sci USA. 101:12670–12675.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Patel HH, Murray F and Insel PA:
G-protein-coupled receptor-signaling components in membrane raft
and caveolae microdomains. Handb Exp Pharmacol. 167–184. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lei S, Li H, Xu J, Liu Y, Gao X, Wang J,
Ng KF, Lau WB, Ma XL, Rodrigues B, et al: Hyperglycemia-induced
protein kinase C β2 activation induces diastolic cardiac
dysfunction in diabetic rats by impairing caveolin-3 expression and
Akt/eNOS signaling. Diabetes. 62:2318–2328. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gervásio OL, Whitehead NP, Yeung EW,
Phillips WD and Allen DG: TRPC1 binds to caveolin-3 and is
regulated by Src kinase-role in Duchenne muscular dystrophy. J Cell
Sci. 121:2246–2255. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Su W, Zhang Y, Zhang Q, Xu J, Zhan L, Zhu
Q, Lian Q, Liu H, Xia ZY, Xia Z and Lei S: N-acetylcysteine
attenuates myocardial dysfunction and postischemic injury by
restoring caveolin-3/eNOS signaling in diabetic rats. Cardiovasc
Diabetol. 15:1462016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Woodman SE, Sotgia F, Galbiati F, Minetti
C and Lisanti MP: Caveolinopathies: Mutations in caveolin-3 cause
four distinct autosomal dominant muscle diseases. Neurology.
62:538–543. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Milone M, McEvoy KM, Sorenson EJ and Daube
JR: Myotonia associated with caveolin-3 mutation. Muscle Nerve.
45:897–900. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Woodman SE, Park DS, Cohen AW, Cheung MW,
Chandra M, Shirani J, Tang B, Jelicks LA, Kitsis RN, Christ GJ, et
al: Caveolin-3 knock-out mice develop a progressive cardiomyopathy
and show hyperactivation of the p42/44 MAPK cascade. J Biol Chem.
277:38988–38997. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Capozza F, Combs TP, Cohen AW, Cho YR,
Park SY, Schubert W, Williams TM, Brasaemle DL, Jelicks LA, Scherer
PE, et al: Caveolin-3 knockout mice show increased adiposity and
whole body insulin resistance, with ligand-induced insulin receptor
instability in skeletal muscle. Am J Physiol Cell Physiol.
288:C1317–C1331. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nader NS and Kumar S: Type 2 diabetes
mellitus in children and adolescents: Where do we stand with drug
treatment and behavioral management? Curr Diab Rep. 8:383–388.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang Q, Huang YY, Deng YF, Xian J, Lu WS
and Wei HQ: Caveolin-3 gene polymorphism in Chinese H an diabetic
patients. J Pract Med. 30:1757–1759. 2014.
|
|
15
|
Shang L, Chen T, Deng Y, Huang Y, Huang Y,
Xian J, Lu W, Yang L and Huang Q: Caveolin-3 promotes
glycometabolism, growth and proliferation in muscle cells. PLoS
One. 12:e01890042017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Deng YF, Huang YY, Lu WS, Huang YH, Xian
J, Wei HQ and Huang Q: The Caveolin-3 P104L mutation of LGMD-1C
leads to disordered glucose metabolism in muscle cells. Biochem
Biophys Res Commun. 486:218–223. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Koistinen HA, Galuska D, Chibalin AV, Yang
J, Zierath JR, Holman GD and Wallberg-Henriksson H:
5-amino-imidazole carboxamide riboside increases glucose transport
and cell-surface GLUT4 content in skeletal muscle from subjects
with type 2 diabetes. Diabetes. 52:1066–1072. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim HS, Kim HJ, Kim YS, Park SC, Harris R
and Kim CK: Caveolin, GLUT4 and insulin receptor protein content in
human arm and leg muscles. Eur J Appl Physiol. 106:173–179. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Visvanathan R, Jayathilake C and Liyanage
R: A simple microplate-based method for the determination of
alpha-amylase activity using the glucose assay kit (GOD method).
Food Chem. 211:853–859. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Luo J, Xu Q, Jiang B, Zhang R, Jia X, Li
X, Wang L, Guo C, Wu N and Shi D: Selectivity, cell permeability
and oral availability studies of novel bromophenol derivative HPN
as protein tyrosine phosphatase 1B inhibitor. Br J Pharmacol.
175:140–153. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Feng W, Mao G, Li Q, Wang W, Chen Y, Zhao
T, Li F, Zou Y, Wu H, Yang L and Wu X: Effects of chromium malate
on glycometabolism, glycometabolism-related enzyme levels and lipid
metabolism in type 2 diabetic rats: A dose-response and curative
effects study. J Diabetes Investig. 6:396–407. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ben Jemaa A, Bouraoui Y, Sallami S, Banasr
A, Nouira Y, Horchani A and Oueslati R: PSMA-PSA clones controlled
by full Akt phosphorylation (T308+,S473+) recapitulate molecular
features of human prostate cancer. Tunis Med. 93:556–564.
2015.PubMed/NCBI
|
|
23
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of Akt/PKB by the
rictor-mTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guertin DA, Stevens DM, Thoreen CC, Burds
AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ and Sabatini DM:
Ablation in mice of the mTORC components raptor, rictor, or mLST8
reveals that mTORC2 is required for signaling to Akt-FOXO and
PKCalpha, but not S6K1. Dev Cell. 11:859–871. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chu N, Salguero AL, Liu AZ, Chen Z,
Dempsey DR, Ficarro SB, Alexander WM, Marto JA, Li Y, Amzel LM, et
al: Akt kinase activation mechanisms revealed using protein
semisynthesis. Cell. 174:897–907.e14. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Spisni E, Tomasi V, Cestaro A and Tosatto
SC: Structural insights into the function of human caveolin 1.
Biochem Biophys Res Commun. 338:1383–1390. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim JH, Schlebach JP, Lu Z, Peng D,
Reasoner KC and Sanders CR: A pH-mediated topological switch within
the N-terminal domain of human caveolin-3. Biophys J.
110:2475–2485. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vaidyanathan R, Van Ert H, Haq KT, Morotti
S, Esch S, McCune EC, Grandi E and Eckhardt LL: Inward rectifier
potassium channels (Kir2.x) and caveolin-3 domain-specific
interaction: Implications for purkinje cell-dependent ventricular
arrhythmias. Circ Arrhythm Electrophysiol. 11:e0058002018.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fischer D, Schroers A, Blümcke I, Urbach
H, Zerres K, Mortier W, Vorgerd M and Schröder R: Consequences of a
novel caveolin-3 mutation in a large German family. Ann Neurol.
53:233–241. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Couet J, Li S, Okamoto T, Ikezu T and
Lisanti MP: Identification of peptide and protein ligands for the
caveolin-scaffolding domain. Implications for the interaction of
caveolin with caveolae-associated proteins. J Biol Chem.
272:6525–6533. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Couet J, Sargiacomo M and Lisanti MP:
Interaction of a receptor tyrosine kinase, EGF-R, with caveolins.
Caveolin binding negatively regulates tyrosine and serine/threonine
kinase activities. J Biol Chem. 272:30429–30438. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Minetti C, Sotgia F, Bruno C, Scartezzini
P, Broda P, Bado M, Masetti E, Mazzocco M, Egeo A, Donati MA, et
al: Mutations in the caveolin-3 gene cause autosomal dominant
limb-girdle muscular dystrophy. Nat Genet. 18:365–368. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hernández-Deviez DJ, Martin S, Laval SH,
Lo HP, Cooper ST, North KN, Bushby K and Parton RG: Aberrant
dysferlin trafficking in cells lacking caveolin or expressing
dystrophy mutants of caveolin-3. Hum Mol Genet. 15:129–142. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Oh YS, Cho KA, Ryu SJ, Khil LY, Jun HS,
Yoon JW and Park SC: Regulation of insulin response in skeletal
muscle cell by caveolin status. J Cell Biochem. 99:747–758. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang X and Wang Y: Glycosylation quality
control by the Golgi structure. J Mol Biol. 428:3183–3193. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gao Y, Zhang M, Wu T, Xu M, Cai H and
Zhang Z: Effects of D-pinitol on insulin resistance through the
PI3K/Akt signaling pathway in type 2 diabetes mellitus rats. J
Agric Food Chem. 63:6019–6026. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang M, Ren Y, Lin Z, Tang C, Jia Y, Lai
Y, Zhou T, Wu S, Liu H, Yang G and Li L: Krüppel-like factor 14
increases insulin sensitivity through activation of PI3K/Akt signal
pathway. Cell Signal. 27:2201–2208. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tan Z, Zhou LJ, Mu PW, Liu SP, Chen SJ, Fu
XD and Wang TH: Caveolin-3 is involved in the protection of
resveratrol against high-fat-diet-induced insulin resistance by
promoting GLUT4 translocation to the plasma membrane in skeletal
muscle of ovariectomized rats. J Nutr Biochem. 23:1716–1724. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ferrannini E, Bjorkman O, Reichard GA Jr,
Pilo A, Olsson M, Wahren J and DeFronzo RA: The disposal of an oral
glucose load in healthy subjects. A quantitative study. Diabetes.
34:580–588. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gazzerro E, Sotgia F, Bruno C, Lisanti MP
and Minetti C: Caveolinopathies: From the biology of caveolin-3 to
human diseases. Eur J Hum Genet. 18:137–145. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Manning BD and Cantley LC: AKT/PKB
signaling: Navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee J and Kim MS: The role of GSK3 in
glucose homeostasis and the development of insulin resistance.
Diabetes Res Clin Pract. 77 (Suppl 1):S49–S57. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Biddinger SB and Kahn CR: From mice to
men: Insights into the insulin resistance syndromes. Annu Rev
Physiol. 68:123–158. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Boura-Halfon S and Zick Y: Phosphorylation
of IRS proteins, insulin action, and insulin resistance. Am J
Physiol Endocrinol Metab. 296:E581–E591. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Saltiel AR and Kahn CR: Insulin signalling
and the regulation of glucose and lipid metabolism. Nature.
414:799–806. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gual P, Le Marchand-Brustel Y and Tanti
JF: Positive and negative regulation of insulin signaling through
IRS-1 phosphorylation. Biochimie. 87:99–109. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dummler B and Hemmings BA: Physiological
roles of PKB/Akt isoforms in development and disease. Biochem Soc
Trans. 35:231–235. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cohen MM Jr: The AKT genes and their roles
in various disorders. Am J Med Genet A 161A. 2931–2937. 2013.
View Article : Google Scholar
|
|
49
|
Hadj Sassi A, Monteil J, Sauvant P and
Atgié C: Overexpression of caveolin-3-enhanced protein synthesis
rather than proteolysis inhibition in C2C12 myoblasts: Relationship
with myostatin activity. J Physiol Biochem. 68:683–690. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yudushkin I: Getting the Akt together:
Guiding intracellular Akt activity by PI3K. Biomolecules.
9:E672019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cho H, Thorvaldsen JL, Chu Q, Feng F and
Birnbaum MJ: Akt1/PKBalpha is required for normal growth but
dispensable for maintenance of glucose homeostasis in mice. J Biol
Chem. 276:38349–38352. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Garofalo RS, Orena SJ, Rafidi K, Torchia
AJ, Stock JL, Hildebrandt AL, Coskran T, Black SC, Brees DJ, Wicks
JR, et al: Severe diabetes, age-dependent loss of adipose tissue,
and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin
Invest. 112:197–208. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
George S, Rochford JJ, Wolfrum C, Gray SL,
Schinner S, Wilson JC, Soos MA, Murgatroyd PR, Williams RM, Acerini
CL, et al: A family with severe insulin resistance and diabetes due
to a mutation in AKT2. Science. 304:1325–1328. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Katome T, Obata T, Matsushima R, Masuyama
N, Cantley LC, Gotoh Y, Kishi K, Shiota H and Ebina Y: Use of RNA
interference-mediated gene silencing and adenoviral overexpression
to elucidate the roles of AKT/protein kinase B isoforms in insulin
actions. J Biol Chem. 278:28312–28323. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Huang S and Czech MP: The GLUT4 glucose
transporter. Cell Metab. 5:237–252. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fecchi K, Volonte D, Hezel MP, Schmeck K
and Galbiati F: Spatial and temporal regulation of GLUT4
translocation by flotillin-1 and caveolin-3 in skeletal muscle
cells. FASEB J. 20:705–707. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Pedersen O, Bak JF, Andersen PH, Lund S,
Moller DE, Flier JS and Kahn BB: Evidence against altered
expression of GLUT1 or GLUT4 in skeletal muscle of patients with
obesity or NIDDM. Diabetes. 39:865–870. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ryder JW, Yang J, Galuska D, Rincón J,
Björnholm M, Krook A, Lund S, Pedersen O, Wallberg-Henriksson H,
Zierath JR and Holman GD: Use of a novel impermeable biotinylated
photolabeling reagent to assess insulin- and hypoxia-stimulated
cell surface GLUT4 content in skeletal muscle from type 2 diabetic
patients. Diabetes. 49:647–654. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Konno S, Alexander B, Zade J and Choudhury
M: Possible hypoglycemic action of SX-fraction targeting insulin
signal transduction pathway. Int J Gen Med. 6:181–187. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Dewulf M, Köster DV, Sinha B, Viaris de
Lesegno C, Chambon V, Bigot A, Bensalah M, Negroni E, Tardif N,
Podkalicka J, et al: Dystrophy-associated caveolin-3 mutations
reveal that caveolae couple IL6/STAT3 signaling with mechanosensing
in human muscle cells. Nat Commun. 10:19742019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Pál E, Zima J, Hadzsiev K, Ito YA, Hartley
T; Care4Rare Canada Consortium, ; Boycott KM and Melegh B: A novel
pathogenic variant in TNPO3 in a Hungarian family with limb-girdle
muscular dystrophy 1F. Eur J Med Genet. 62:1036622019. View Article : Google Scholar : PubMed/NCBI
|