Introduction
Diabetes is a metabolic disorder of carbohydrates,
proteins and lipids that contributes to development of cardiac
fibrosis. It is predicted that the number of people with diabetes
in the world could reach up to 592 million by the year 2035
(1). Thus, there is a pressing need
to develop therapeutic interventions to attenuate diabetes and its
associated cardiac fibrosis. Unfortunately, the molecular
mechanisms responsible for cardiac fibrosis in type 2 diabetes
mellitus (T2DM) are not well understood.
The literature suggests that accumulation of
advanced glycation end products (AGEs), that crosslink with
extracellular matrix (ECM) proteins, transduce fibrogenic signals
through generation of reactive oxygen species (ROS) and activation
of the receptor for AGEs (RAGE). In addition, activation of
transforming growth factor β (TGFβ)/SMAD signaling may activate
fibroblasts to induce deposition of structural ECM proteins. Thus,
activation of several distinct but overlapping pathways promotes
diabetes-associated cardiac fibrosis (2).
Experimental induction of T2DM in animal models is
essential to understand the oxidant stress and fibrogenic signals
in T2DM (3). In this context,
Streptozotocin (STZ), a preferential toxic compound of β cells, has
been used for a long time in rodents to induce DM2 and to observe
complications of the human form of the disease (4,5).
High-fat, high-carbohydrate diet (HFCD) has been used to induce
insulin resistance in combination with STZ, to reduce the
compensatory capacity of β-cells facing insulin hypersecretion,
resulting in hyperglycemia (6).
Therefore, this combined model of HFCD-fed/STZ-treated rats is one
of the best that simulates the progression of the natural disease
and the typical metabolic characteristics of individuals at higher
risk of developing T2DM.
Animal models of diabetes have revealed that
aminoguanidine (AG), a known inducible nitric oxide synthase (iNOS)
inhibitor, prevents diabetes-associated cardiovascular
deterioration, nephropathy, retinopathy and neuropathy (7–9). In
addition, it has been demonstrated that AG inhibits the formation
of AGEs by interacting with and quenching dicarbonyl compounds
(10), protecting against myocardial
contractile dysfunction (11). Thus,
elucidating and dissecting the involvement of AG in
diabetes-associated cardiac fibrosis could be useful in preventing
or treating ROS- and AGE-related pathologies on the verge of an
epidemic such as T2DM. Our aim was to investigate whether AG
supplementation may reduce cardiac fibrosis and the gene expression
profile of oxidative stress in diabetes-associated cardiac
fibrosis.
Materials and methods
Sources of antibodies and qPCR
primers
The sources of commercially available antibodies can
be found in Table SI. The collagen
type I antibody (1/5,000) was generated and provided by Dr Schuppan
(University of Mainz, Mainz, Germany) (12,13). The
qPCR primers used are shown in Table
SII.
Cell culture and treatments
Cell culture experiments were performed in primary
rat myofibroblasts at high glucose (>17.5 mM, considering
glucose concentration in culture medium) and high insulin
concentrations, induced hyperglycemia/hyperinsulinemia conditions
similar to diabetes (14,15). Primary heart cultures were obtained
from neonatal rats, and myofibroblasts were isolated according to
Boateng et al (16). Three
days after cell isolation, rat myofibroblasts were seeded on
six-well plates (300,000 cells/well) in DMEM/F12 supplemented with
10% FBS, 0–50 nM insulin (4512-01 CellPrime® r Insulin;
EMD Millipore Corporation), 0–17.5 mM glucose (Thermo Fisher
Scientific, Inc.), fungizone, penicillin and streptomycin for 48 h.
The medium was replaced with serum-free medium before treatment
with 0–1 mM AG (Acros Organics). The untreated cells were
maintained in DMEM/F-12 supplemented with 10% FBS. According to the
manufacturer's description, DMEM/F-12 medium contains 3.15 g/l
(17.5 mM) of glucose, plus traces of glucose originating from the
serum. Concentrations of glucose above 10 mM are analogous to a
diabetic condition within the cell culture system (17).
Time-course (15 min to 24 h) and dose-response
(0.5–1.0 mM) experiments were carried out to determine the final
concentration of AG and the best time-point for collagen type I
induction (24 h). In some experiments, either 0.12 µM (5 µg/ml) of
rat RAGE-neutralizing antibody (AF1616-SP) or normal goat IgG
control (AB-108-C; R&D Systems) was added to the cells 1 h
prior to incubation with AG, according to the recommended
concentration for blockade of receptor-ligand interaction from the
manufacturer. The optimal concentration for collagen type I
induction was determined experimentally.
Animals
This study was carried out on 45 male Wistar rats
with weights ranging between 100 and 150 g. Rats were anesthetized
with ketamine/xylazine (i.p. 100 mg/kg and 10 mg/kg b.w.) followed
by a lethal 3× anesthesia dose, according to The Guidelines of The
American Veterinary Medical Association. Confirmation of death was
performed by observation of cardiac arrest for 10 min or longer.
This was variable among animals until lack of pulse, breathing and
response to firm toe pinch was observed. The verification of death
was supplemented by rigor mortis verification. Rats were obtained
from the animal facility of the University Center of Health Science
(CUCS), Guadalajara University. All animal studies and humane
endpoints set out for this study were in accordance with the rules
of Ethical and Technical Specifications for Care and Management
outlined in The National Institutes of Health's Guide, and approved
by Jalisco State Agency for the Care and Use of Laboratory Animals
(approval no. 16/UG-JAL/2008). During the study, the animals were
kept in polypropylene cages with ad libitum access to water
in order to gain weight until reaching 200–250 g, when their
fasting blood glucose levels were monitored prior to diabetic
induction.
Induction of experimental diabetes in
rats
Rats were fed for diabetic induction with an HFCD
composed of 20% sucrose, 10% coconut oil and 1% cholesterol per
kilogram for 2 weeks. Two-thirds of the rats fed with HFCD (n=30)
received daily intraperitoneal injections of freshly prepared
solution with 20 mg/kg of STZ (Sigma-Aldrich Co.) dissolved in 0.1
M sodium citrate buffer (pH 4.5) during the following 5 days, while
age-matched controls received buffer-only injections and HFCD diet.
On the first day after injection, the fasting blood glucose was
measured from rat tail-veins by using a blood glucometer. Rats with
fasting blood glucose levels >200 mg/dl were deemed diabetic.
One-third (n=15) of the animals continued to be fed with HFCD diet
(control group).
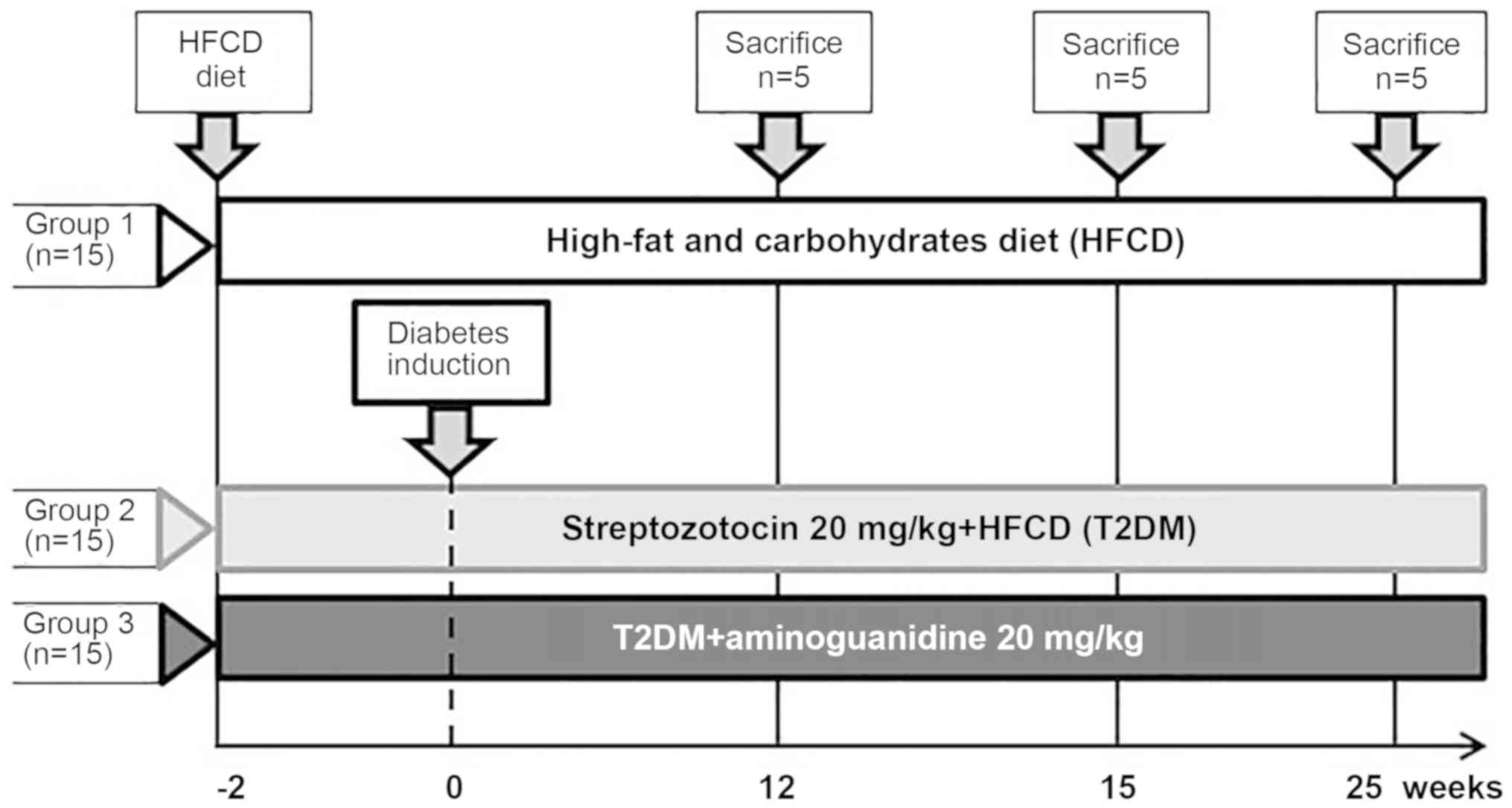
Groups and treatment
The rats not receiving diabetic induction were used
as negative controls (non-diabetic group). The diabetic rats were
randomly allotted to non-treated diabetic (HFCD+STZ) and
diabetic-treated (HFCD+STZ+AG) groups (n=15 rats/group). The
HFCD+STZ+AG group was treated with daily intraperitoneal
administration of AG (20 mg/kg) throughout the experiment (Fig. 1). Methodologically, a third control
group without diabetes but with AG treatment was not included since
two control groups, negative control (HFCD-fed) and positive
control (STZ-injected), were already used in the study (18,19). At
15, 20 and 25 weeks after treatment, all animals were sacrificed
prior to blood extraction. Blood was taken directly from the
cardiac chamber and the heart was removed for molecular and
histological analysis. Blood samples were collected and stored at
−80°C, and myocardial tissue samples were collected and
subsequently formalin-fixed for further analysis.
Histological analysis
Hearts were excised, washed with saline solution and
placed in 10% formalin. Then, they were cut transversely, close to
the apex, to visualize the left ventricle and right ventricle, and
several sections of heart (5-µm thick) were prepared. Cardiac
histology was assessed using hematoxylin and eosin (H&E)
staining in paraffin-embedded sections using standard commercial
methods. Fibrosis was assessed using both Masson's trichrome and
Sirius red/Fast green staining in paraffin-embedded sections using
established methodology (20). The
cardiac histology was evaluated by a pathologist who was blinded
from the experimental conditions. The number of microscopic fields
with fibrosis was recorded, and five affected fields were
photographed. Image analysis (n=5 rats/week) was performed using a
digital microscope and AxioVision v.7 software.
Western blotting
Cells lysate samples were processed using sodium
dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) 5–10%
gels and nitrocellulose membranes. Actin served as an endogenous
control of protein expression. Membranes were incubated with the
primary antibodies shown in Table
SI and corresponding peroxidase-coupled secondary antibodies
from EMD Millipore Corporation. Images were captured with an
Amersham™ Imager 600, and results were analyzed using ImageQuant™TL
analysis software v8.1 (GE Healthcare Bio-Sciences) after enhanced
chemiluminescence using Pierce™ ECL substrate (Thermo Fisher
Scientific, Inc.).
α-Smooth muscle actin
immunohistochemistry
Immunohistochemical staining on paraffin-embedded
heart tissue was performed for each experimental time-point using a
primary antibody against αSMA (Millipore anti-alpha-actin (smooth
muscle) clone E184, rabbit monoclonal; dilution: 1 : 500), followed
by a secondary biotinylated anti-rabbit IgG, and developed using
the Histostain® Plus detection system (Invitrogen;
Thermo Fisher Scientific, Inc.).
cDNA synthesis and reverse
transcription-quantitative PCR (qPCR)
Total RNA was isolated from 100 mg of tissue using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.),
quantified by determination at OD260 and stored at
−80°C. Two micrograms of total RNA was reversed-transcribed using
an oligo(dT) primer and M-MLV reverse transcriptase (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. The cDNA generated was stored at −20°C.
Real-time PCR was developed with diluted cDNAs using
pre-developed and validated TaqMan® gene expression
assays by Applied Biosystems; Thermo Fisher Scientific, Inc.:
Nos2 ID: Rn02132634_s1; Nox4 ID: Rn00585380_m1;
Col1a1 ID: Rn01463848_m1; Ucp2 ID: Rn01754856_m1;
Nrf2 ID: Rn00477784_m1; β-actin ID: Rn00667869_m1,
according to the manufacturer's protocol. The β-actin qPCR
gene efficiency had been validated previously. Amplification by
real-time PCR was performed on the StepOne™ Real-Time PCR System
(Applied Biosystems; Thermo Fisher Scientific, Inc.). Each qPCR
analysis included duplicate wells, and appropriate control
reactions were performed for all samples. The expression level of
each gene of interest was calculated using the 2−∆∆Cq
method (21). Gene amplification was
normalized against β-actin expression in each sample, and
gene expression levels are shown as relative expression units by
comparing them to the control group as an internal calibrator.
Total RNA from cultured primary rat myofibroblasts
was isolated using TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.). Total RNA was reverse-transcribed using an
Ecodry® Premix Kit (Clontech), and qPCR was performed in
a Roche Light Cycler 480 with FastStart SYBR-Green Master (Roche
Diagnostics). Expression of the target gene was calculated by the
cycle-threshold method, and results are expressed as the relative
fold-change with respect to the appropriate control group. Values
were normalized to the housekeeping gene β-actin. Each qPCR
reaction was run in triplicate.
Statistical analysis
Data are expressed as mean ± standard deviation of
the mean (SD). Statistical comparisons within groups were performed
by two-way ANOVA followed by Tukey's multiple comparison test. Gene
expression comparisons were performed by Kruskal-Wallis test
followed by Mann-Whitney U test post hoc test. Data are shown in
all figures, and P<0.05 was used to indicate a statistically
significant difference.
Results
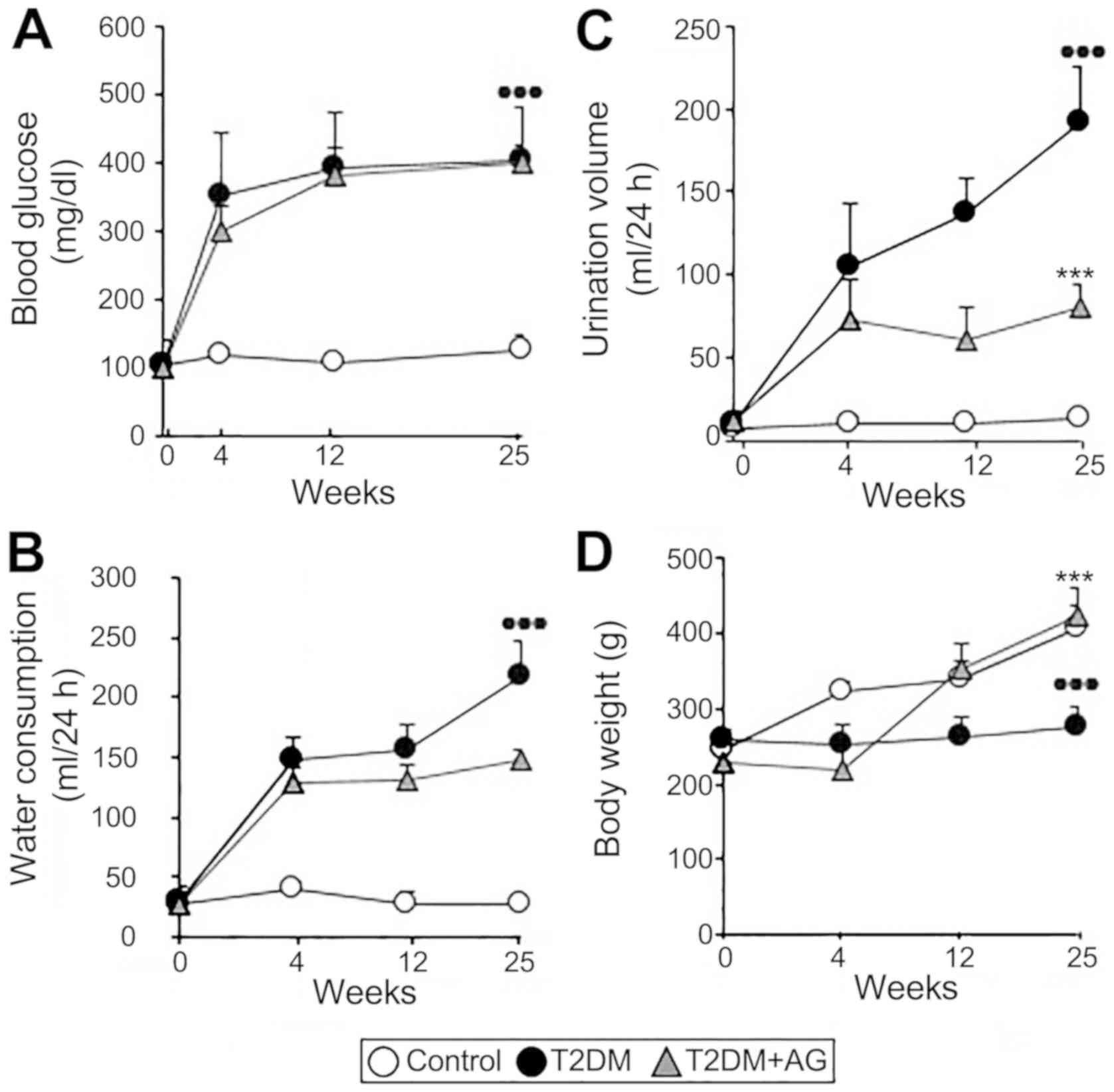
T2DM rats show increased blood
glucose, urinary volume and water consumption with modest body
weight gain
To begin our experimental investigations, we
confirmed that our T2DM model shows representative symptoms of
human T2DM such as hyperglycemia, polydipsia and polyuria. The
HFCD, concomitantly with STZ, induced insulin resistance resulting
in hyperglycemia maintained throughout the study (Fig. 2A). This was accompanied by increased
water consumption (Fig. 2B) and
urinary volume (Fig. 2C) as well as
modest body weight gain (Fig. 2D).
Partial but not full pancreatic islet β-cell destruction-a central
feature in T2DM (22) -was observed
in induced-T2DM rats (Fig. S1).
Importantly, AG had no hypoglycemic effect as shown by unchanged
fasting blood sugar concentration and water consumption but
improved urinary volume and weight in AG-treated diabetic rats.
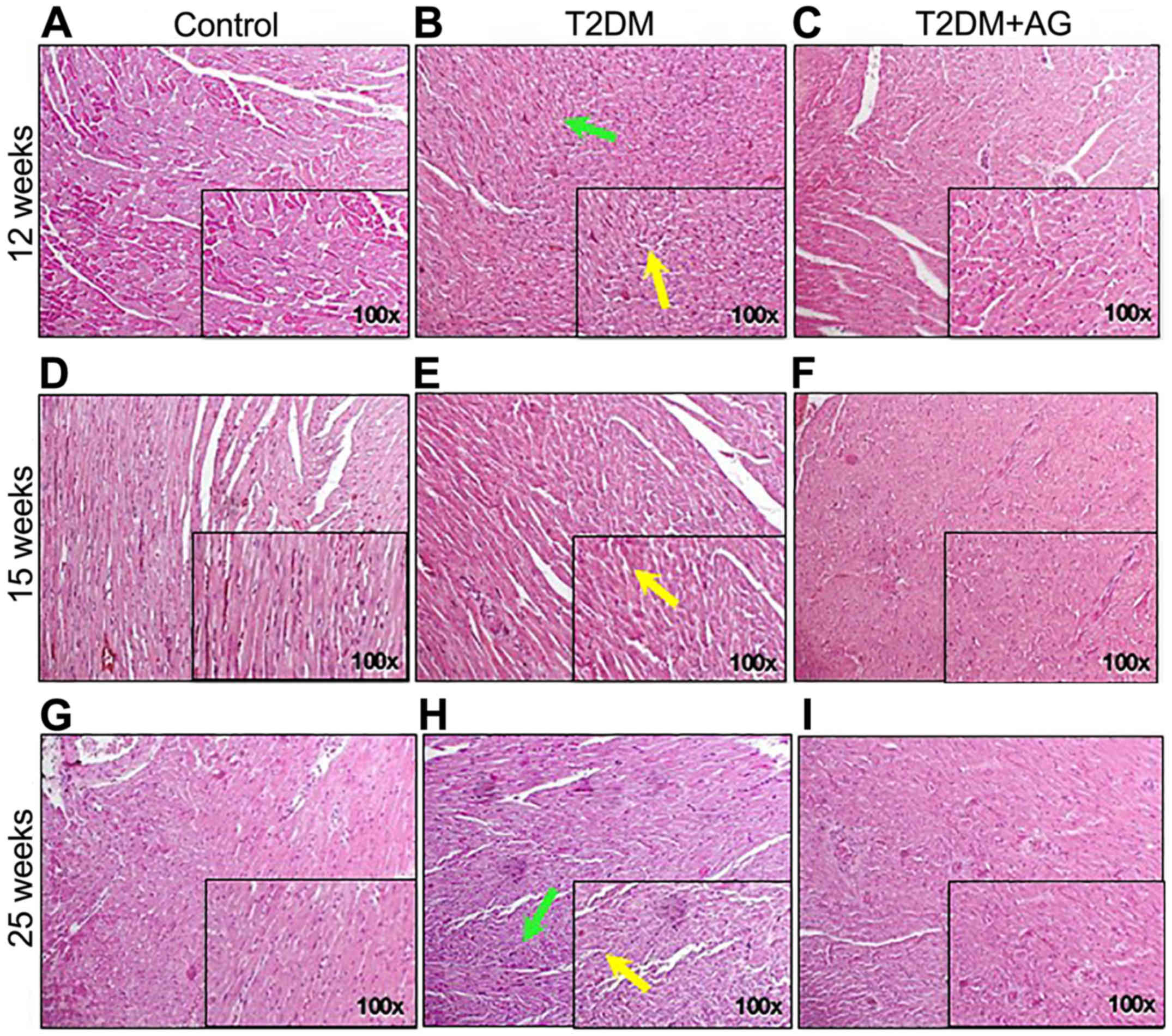
AG-treated rats showed improvement of
myocardial architecture compared to non-treated diabetic rats
To analyze the prevention of diabetes-associated
cardiovascular damage by AG, rats were sacrificed in weeks 12, 15
and 25, and their cardiac tissues were subjected to histological
analysis. After 15 weeks, T2DM rats already had irregular
myofibrillar orientation as shown by H&E staining (Fig. 3E and H). By contrast, AG-treated
diabetic rats showed improvement of myocardial architecture
compared to non-treated diabetic rats (Fig. 3F and I).
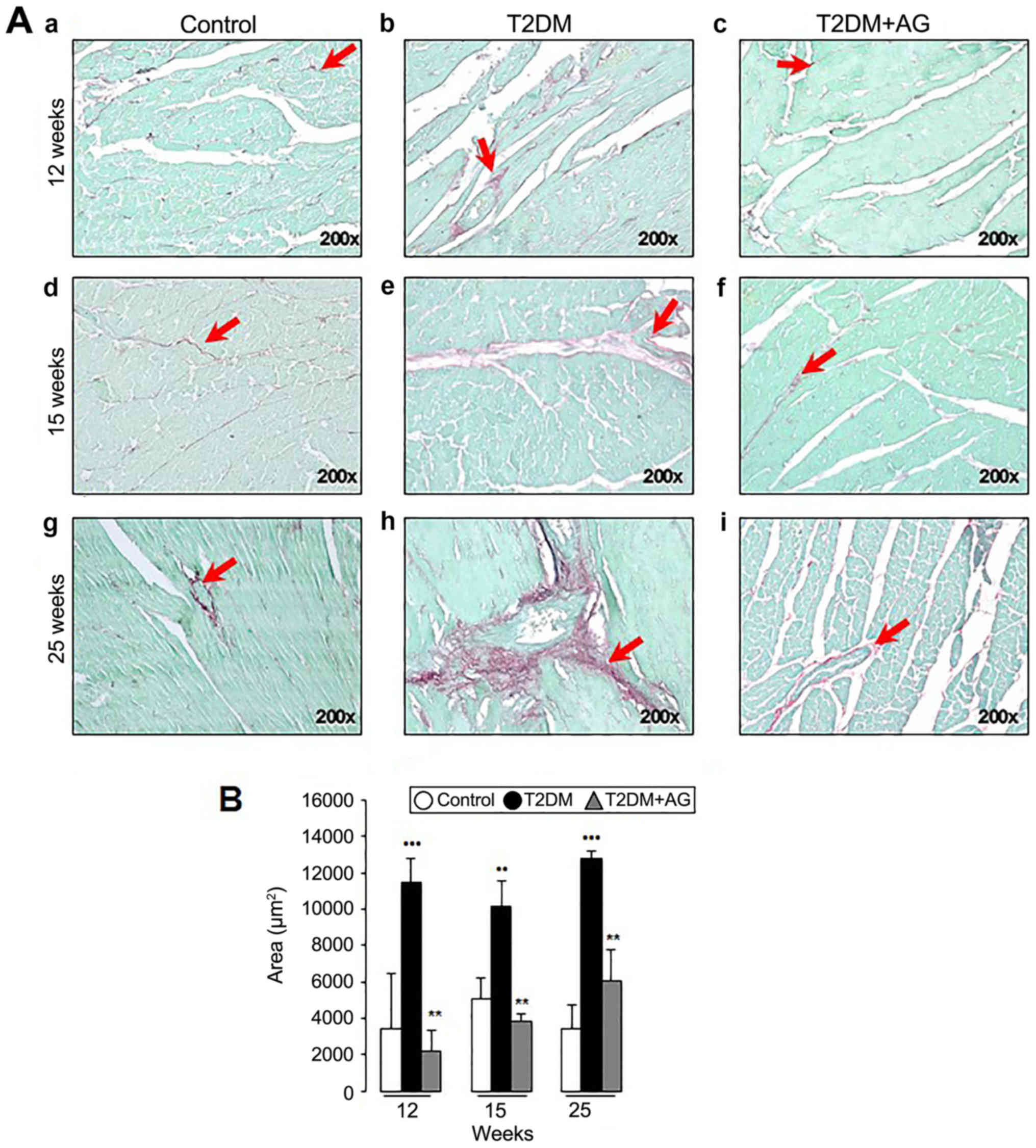
AG-treated rats exhibit less fibrosis
and α-smooth muscle actin (αSMA) protein expression than T2DM
rats
To further evaluate the potential antifibrotic role
of AG in vivo, hearts were processed for Sirius red/Fast
green, Masson's trichrome staining and αSMA immunohistochemistry
(IHC). We observed progressive interstitial myocardial fibrosis in
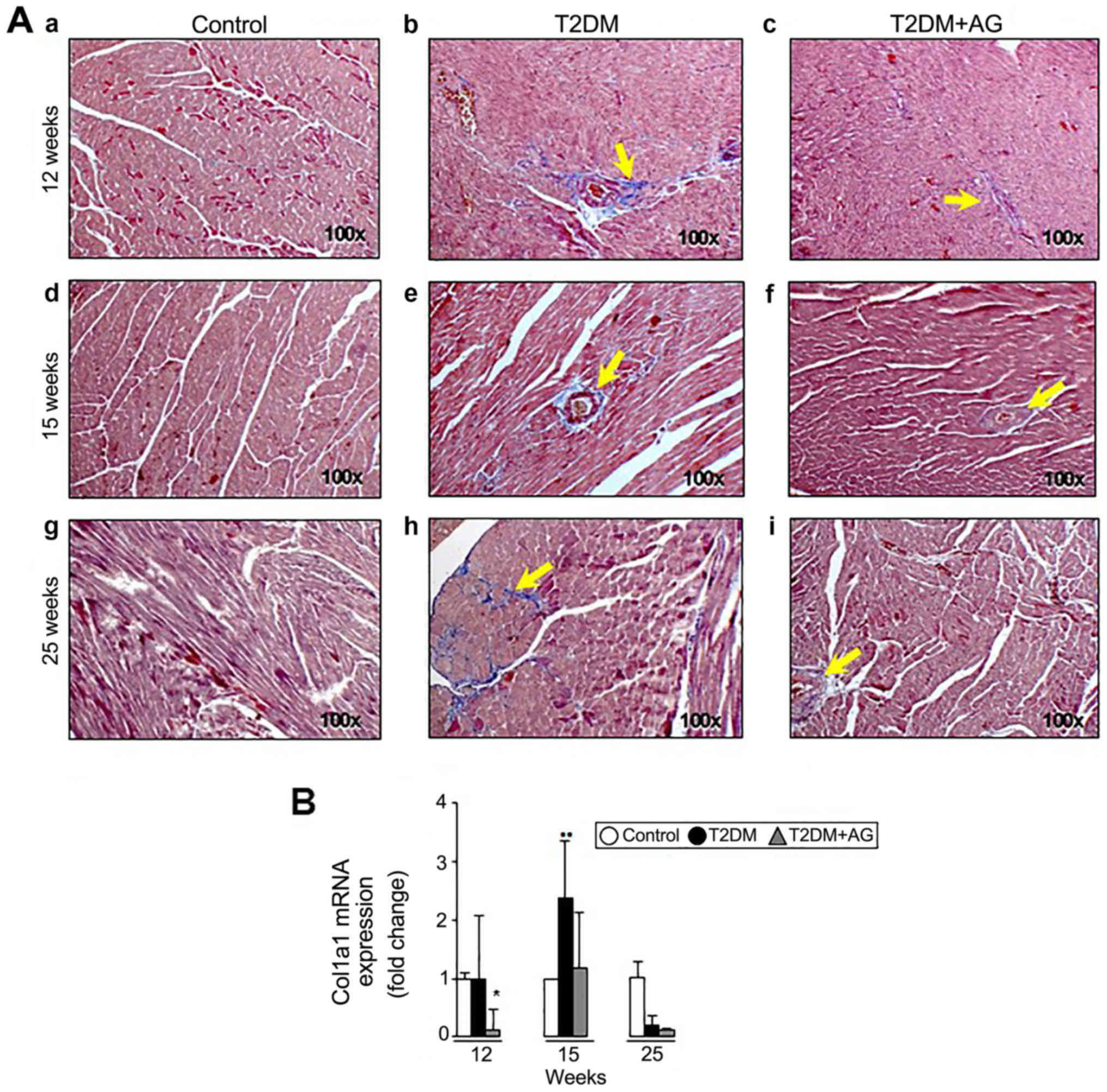
T2DM rats by 12 weeks after induction (Fig. 4A-e and A-h) while AG-treated diabetic
rats exhibited less fibrosis (Fig. 4A-f
and A-i). Likewise, Sirius red/Fast green quantification
(Fig. 4B) and Masson's trichrome
staining (Fig. 5A) revealed less
fibrosis compared to the T2DM group. Consistently, AG significantly
downregulated Col1a1 mRNA expression 8.3-fold at week 12,
although no changes were observed at week 25 in T2DM+AG compared to
T2DM rats (Fig. 5B).
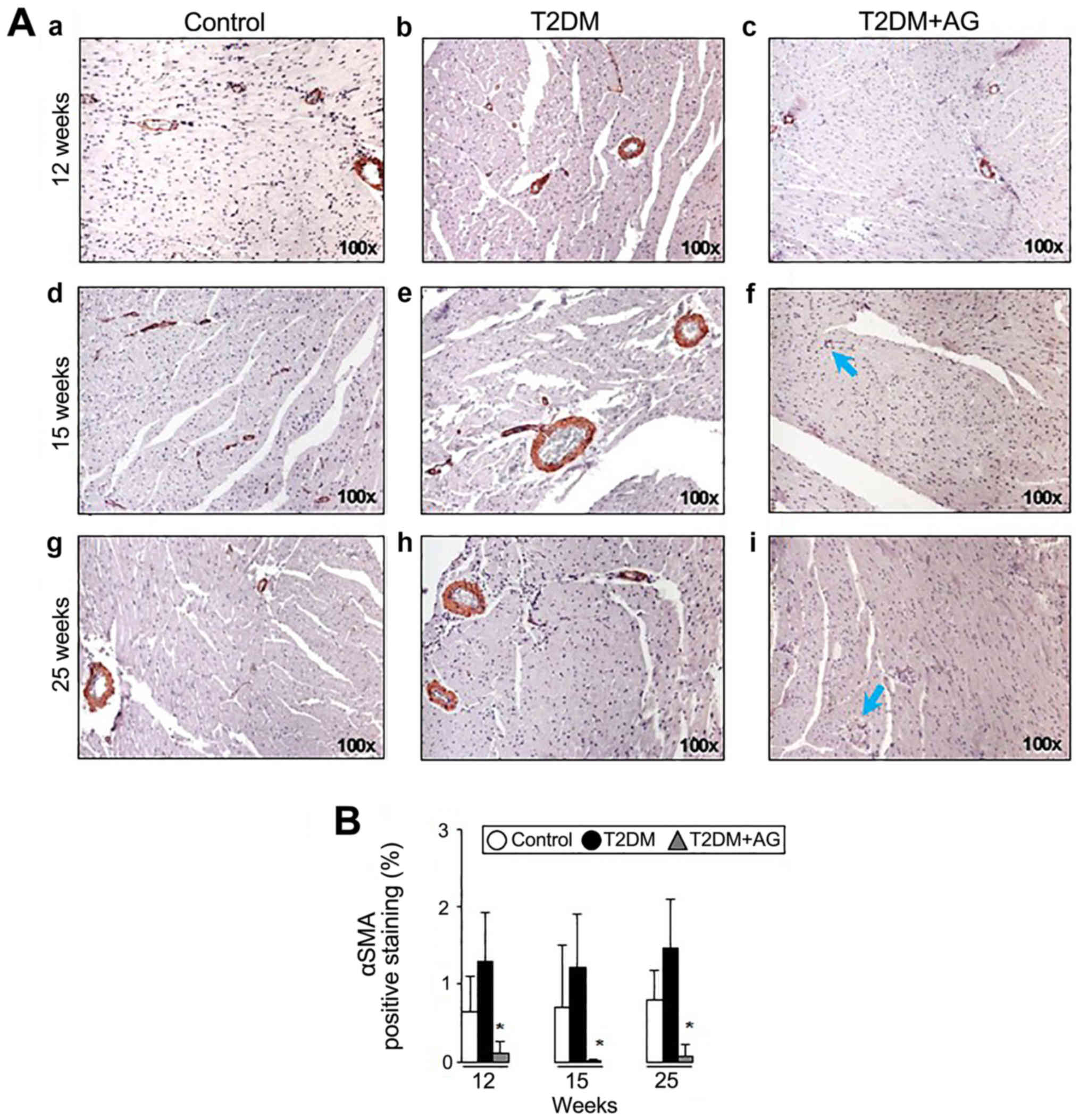
Considering the expression of αSMA in myofibroblasts
as a marker in fibrosis (23), we
next studied the role of AG in αSMA protein expression. AG-treated
rats exhibited less αSMA expression (Fig. 6A-f and A-i) compared to non-treated
T2DM rats (Fig. 6A-e and A-h).
Likewise, αSMA IHC quantification showed lower αSMA expression in
T2DM+AG compared to T2DM rats (Fig.
6B).
Collectively, these results suggest that AG has an
important antifibrotic role in T2DM rats by reducing fibrosis
deposition and αSMA induction in diabetes-associated cardiac
fibrosis.
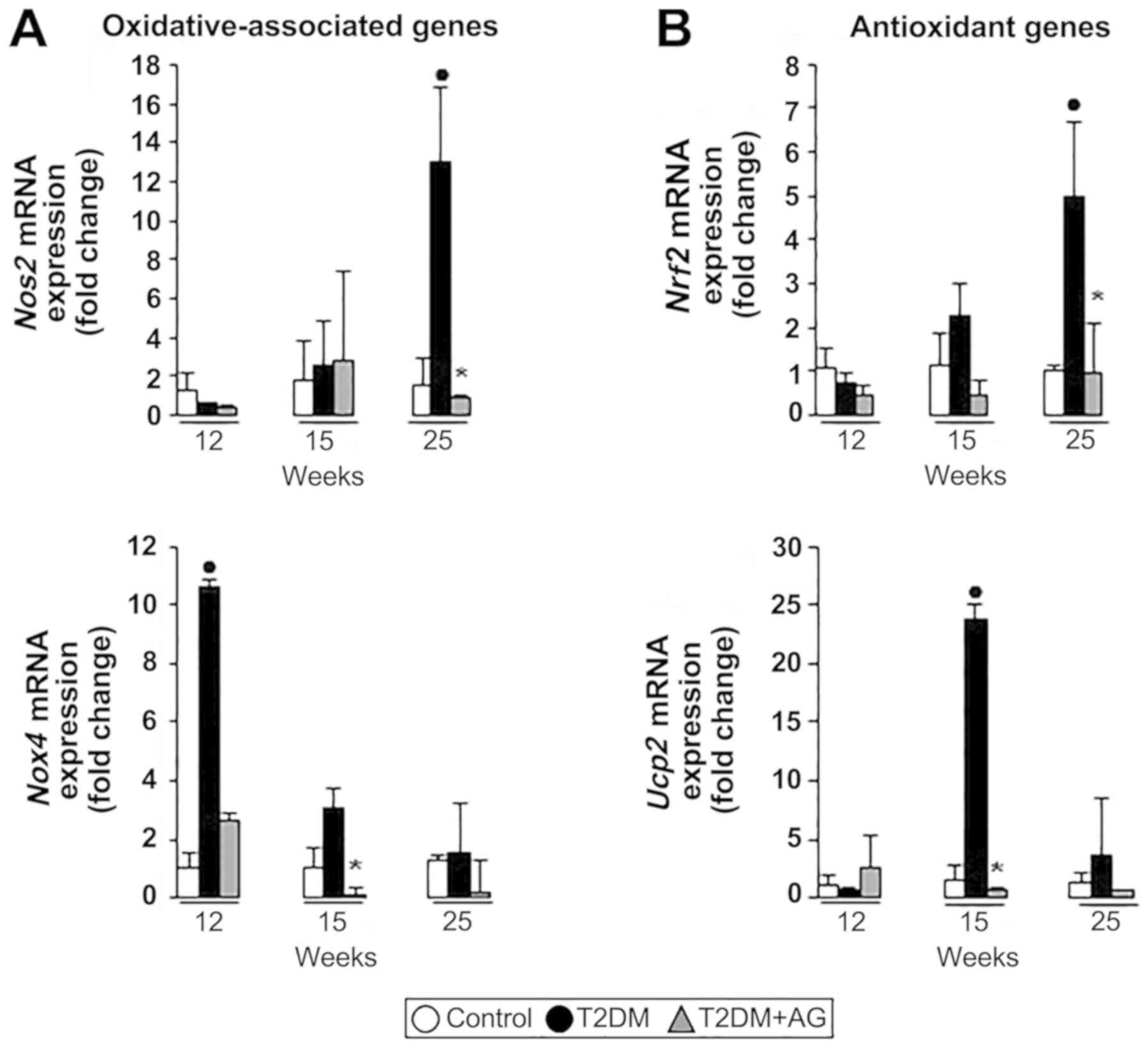
AG treatment downregulates
oxidation-associated gene expression in cardiac tissue
Since oxidant stress-sensitive proteins are known to
regulate cardiac collagen type I (24), we looked into the mRNA profile of
oxidant and antioxidant genes in rats and in primary myofibroblasts
treated with AG. We found that Nos2 (endothelial nitric
oxide synthase) and Nox4 (NADPH oxidase 4) mRNA expression
was upregulated in diabetic rats. By contrast, AG-treated rats had
less Nos2 and Nox4 mRNA expression in cardiac tissue,
in a time-dependent manner (Fig.
7A). Likewise, Nrf2 (nuclear factor (erythroid-derived
2)-like 2) and Ucp2 (uncoupling protein 2) mRNAs, well-known
antioxidant-associated genes capable of modulating ROS (25,26),
were upregulated under oxidative conditions in T2DM rats and
reduced upon AG treatment (Fig.
7B).
Taken together, these results suggest an antioxidant
effect of AG in diabetic-associated cardiac fibrosis.
AG reduces oxidation-associated
collagen type I expression through ERK1/2 and SMAD2/3 signaling in
myofibroblasts
Considering that AG has ROS-detoxification activity
(27–29), and that ROS lead to ECM accumulation
in diabetes (30), we investigated
the role of AG in AGE signaling in primary myofibroblasts. Primary
myofibroblasts from neonatal rats were exposed for 48 h to extended
high insulin (0–50 nM) and high glucose (0–17.5 mM) levels to
induce the impairment of insulin signaling and to desensitize
glucose transport (HIG conditions) (31). Cells were further treated with AG
(0–1 mM) for 24 h to analyze the effect of AG on the activation of
kinase(s), such as the JAK/STAT pathway (32), the MEK1/2 pathway (33,34),
SMAD2/3 (35) and AKT signaling
(36), implicated in
oxidative-associated fibrogenesis.
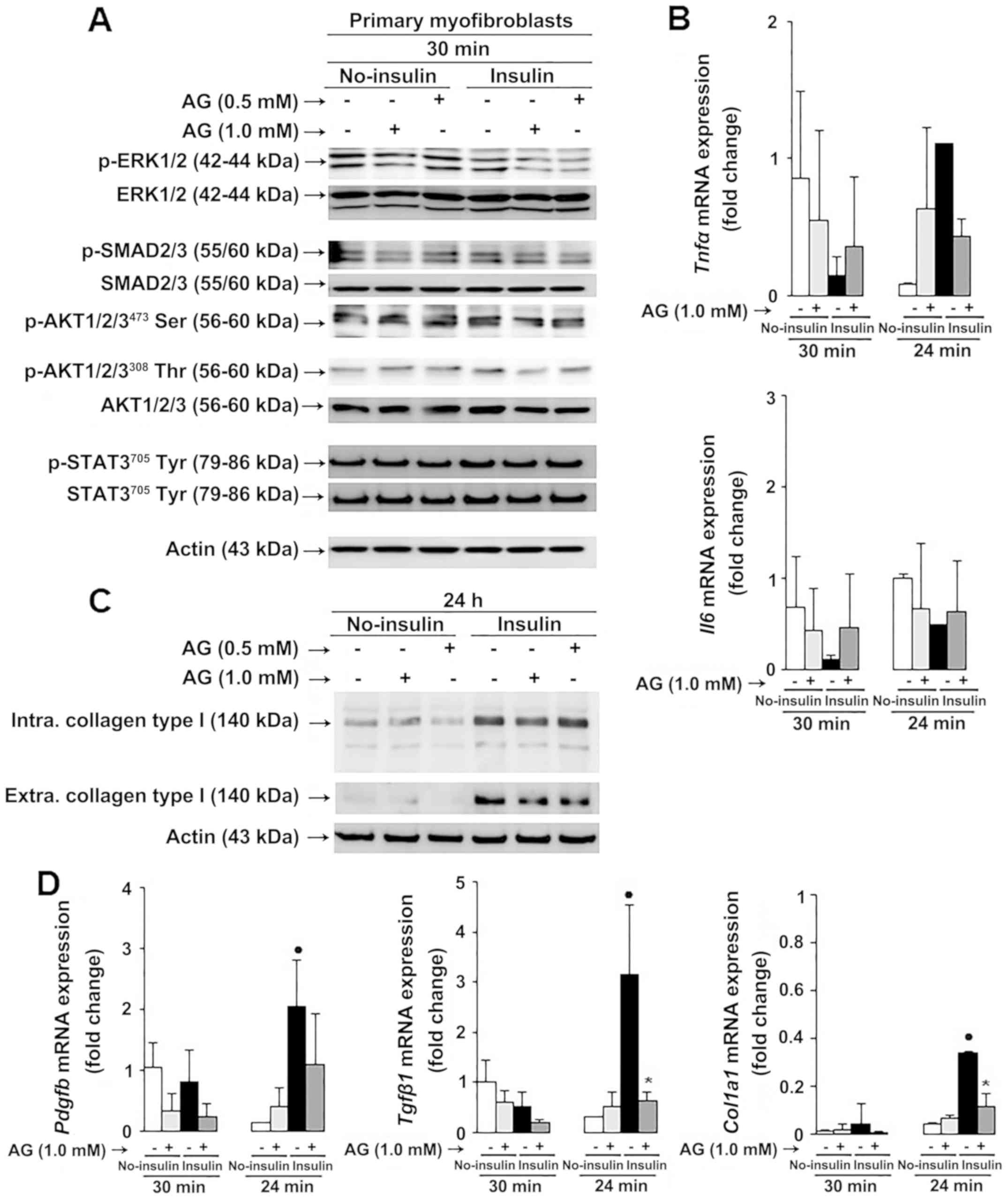
AG reduced phosphorylation of ERK1/2 and SMAD2/3 but
not AKT1/2/3473Ser, AKT1/2/3308Thr or
STAT3705Tyr in primary rat myofibroblasts (Fig. 8A). Moreover, we looked into oxidant
stress-sensitive pro-inflammatory mediators such as interleukin-6
(IL-6), and tumor necrosis factor α (TNFα), known to regulate
collagen type I expression, and observed similar Tnfα aSnd
Il6 mRNA expression in AG-treated compared to untreated
myofibroblasts in diabetic conditional medium (Fig. 8B). Similarly, no changes in Nos2,
Nox4 and Ucp2 mRNA expression were observed in
AG-treated compared to untreated myofibroblasts (Fig. S2).
Based on the in vivo findings, and because
collagen is one of the main targets of reducing sugars and
dicarbonyl compounds in diabetes (37,38), we
further investigated the effect of AG on collagen type I expression
in primary rat myofibroblasts. Intra- and extracellular collagen
type I protein expression (Fig. 8C)
and Pdgfb, Tgfβ1 and Col1a1 mRNA expression (Fig. 8D) were downregulated in AG-treated
compared to untreated myofibroblasts in diabetic conditional
medium. Therefore, our results indicate that AG reduces collagen
type I deposition in myofibroblasts through convergent
ROS-sensitive ERK1/2 and SMAD2/3 signaling.
AG reduces collagen type I expression
via RAGE in myofibroblasts
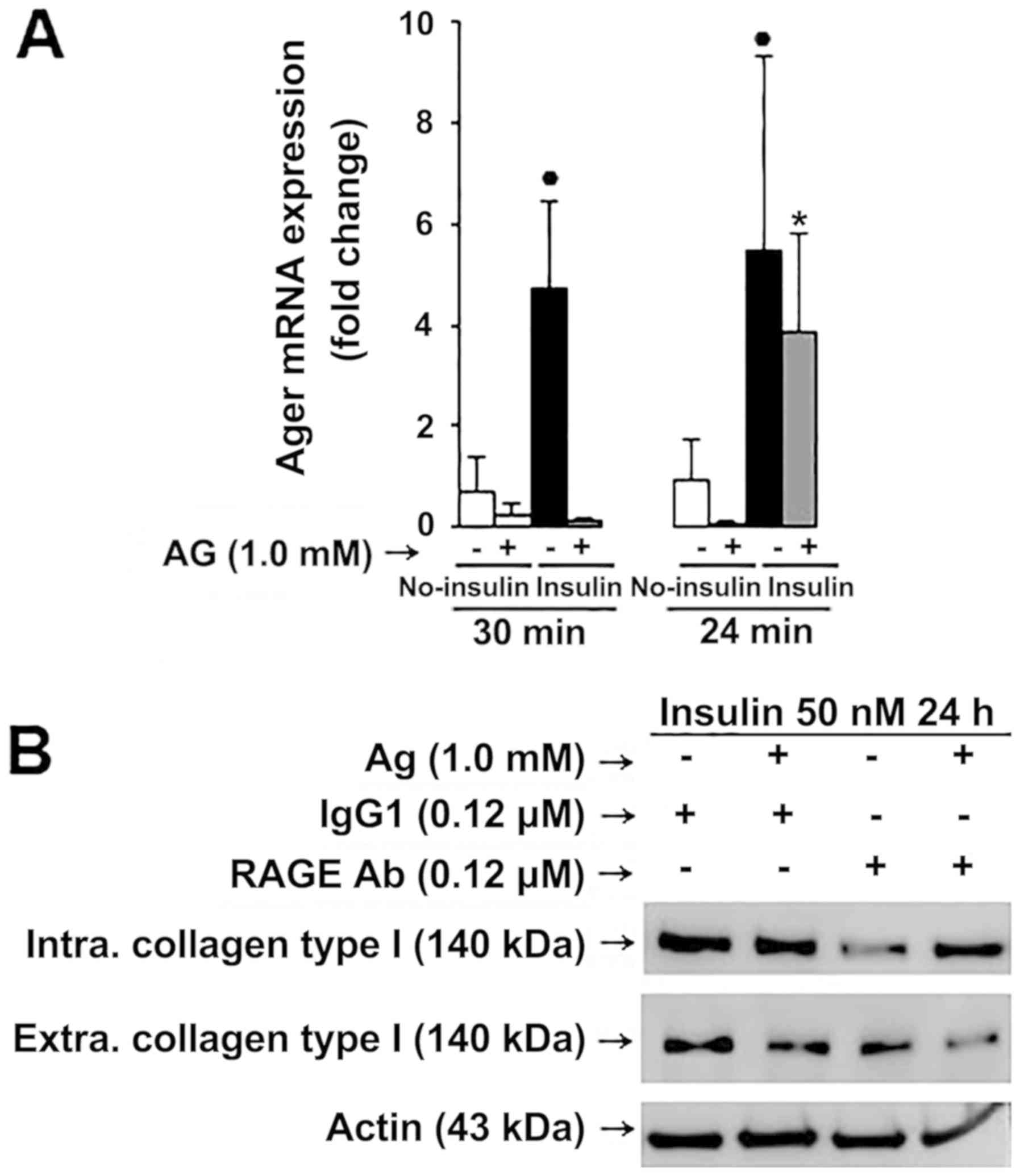
Since Ager mRNA was significantly after HIG
conditions (Fig. 9A), the effect of
AG and the importance of RAGE for collagen type I expression were
explored using an antibody blockade strategy. Treatment with a
RAGE-neutralizing antibody before AG treatment resulted in a
significant decrease of total collagen type I expression in
myofibroblasts (Fig. 9B), suggesting
that AGE/RAGE activation is requisite for increased collagen type I
expression in myofibroblasts (Fig.
10).
Discussion
Until now, many kinds of animal models have been
used for diabetes research, but neither surgical nor chemical
models simulate human T2DM (3,39–42). STZ
induces cardiac fibrosis, contractile dysregulation and
mitochondrial damage (43), all
AGE-related disorders (44). In
fact, lower doses of STZ (≤50 mg/kg) in conjunction with a diet
rich in fat (40% of total dietary kcal) have been successfully
employed to induce T2DM (44,45). In
the present study, we reduced the dose of STZ to 20 mg/kg (46), and extended the time reported from 18
weeks to 25 weeks (47), to
replicate chronic features of T2DM and evoke cardiac fibrosis.
In our model, diabetic rats showed slight weight
gain, polydipsia and polyuria without tendency to cause ketosis and
without gradual recovery to normoglycemia. In addition, the use of
insulin therapy to sustain life was not necessary, suggesting that
STZ doses plus HFCD only reduced the pancreatic β cell mass, but
did not eliminate them. Importantly, a symptom for the onset of
T2DM is a sudden loss of weight due to frequent urination and high
blood glucose that is no longer being taken up, and when some form
of treatment is applied to allow the uptake of glucose, patients
often see an increase in body weight (45). This was indeed observed in the
STZ-induced diabetic rats, indicating the presence of T2DM.
However, insulin resistance and its associated multiorgan
dysfunction assessment were not performed, which might be a
limitation of this study.
Recent investigations on diabetes have demonstrated
that impaired mechanisms dependent on UCP2 (46), Nrf2 (47,48),
Nox4 (49–52) and endothelial nitric oxide synthase
(eNOS) (53–55) participate in collagen type I
deposition, therefore reducing myocardial compliance (56,57).
However, it is still unclear how these molecular signals
participate in the diabetes-associated fibrogenic response.
For this reason, we used AG as a therapeutic agent
for cardiac fibrosis associated with T2DM. AG is a water-soluble
molecule that scavenges toxic 1,2-dicarbonyl compounds such as
methylglyoxal, and attenuates oxidative stress by inhibiting the
formation of AGEs and nitric oxide synthase (58).
The treatment with AG reduced cardiac Nox4
and Nos2 mRNA expression compared to untreated diabetic
rats. By contrast, the unchanged Nos2, Nox4 and Ucp2
mRNA expression in primary myofibroblasts but not in the diabetic
rats suggests a dual effect of AG in the diabetic heart:
Antioxidant in non-myofibroblasts, and antifibrotic in
myofibroblasts. This is supported by the downregulation of collagen
type I protein, Pdgfb, Tgfβ1 and Col1a1 mRNA
expression and ERK1/2-SMAD2/3 signaling in AG-treated
myofibroblasts. It is also possible that AG in vivo has a
positive effect on renal AGE-associated damage (59,60),
since AG treatment improved urinary output. This effect may not be
related to changes in blood pressure but to reduction of
AGE-mediated tubulointerstitial injury and endothelium-dependent
relaxation (61). Nevertheless, more
in vivo studies are needed to dissect the organ-specific
molecular signals activated by AG in diabetes-associated
fibrosis.
In vitro, hyperglycemia is involved in the
activation of cardiac fibroblasts, promoting a fibrogenic phenotype
and release of cytokines and growth factors with potential to
generate ROS (62–64); however, the significance of these
findings in vivo is unclear, and robust evidence is required
to demonstrate that diabetes-associated myocardial fibrosis is due
to hyperglycemia. Although some metabolic conditions that occur in
diabetes cannot be resembled in vitro, myofibroblasts were
cultured in hyperglycemic medium, with and without insulin, to
analyze the AG effect in the diabetic niche (65,66).
The appearance of hyperglycemia in T2DM is preceded
by a phase of hyperinsulinemia, which is able to overcome tissue
resistance and transport glucose into cells. This is followed by a
stage of hyperglycemia that may overlap with hyperinsulinemia for
some time. Thus, experimental conditions of high glucose and high
glucose plus high insulin were designed to recapitulate T2DM
(15,67), and to induce insulin resistance in
myofibroblasts. Thus, AKT signaling was upregulated because insulin
conveys its metabolic action through this pathway. However, western
blot results suggested that AG, via AKT-independent signaling,
reduces collagen type I likely through ERK1/2 and SMAD2/3 in
myofibroblasts.
Importantly, the in vitro generation of final
products of glycosylation was not performed, rendering an important
limitation to the study. Nevertheless, the alteration of Pdgfb,
Tgfβ1 and Col1a1 transcript levels and ERK1/2 and
SMAD2/3 phosphorylation suggests the induction of diabetic
conditions and a restoration of the pro-fibrotic program in
myofibroblasts by AG.
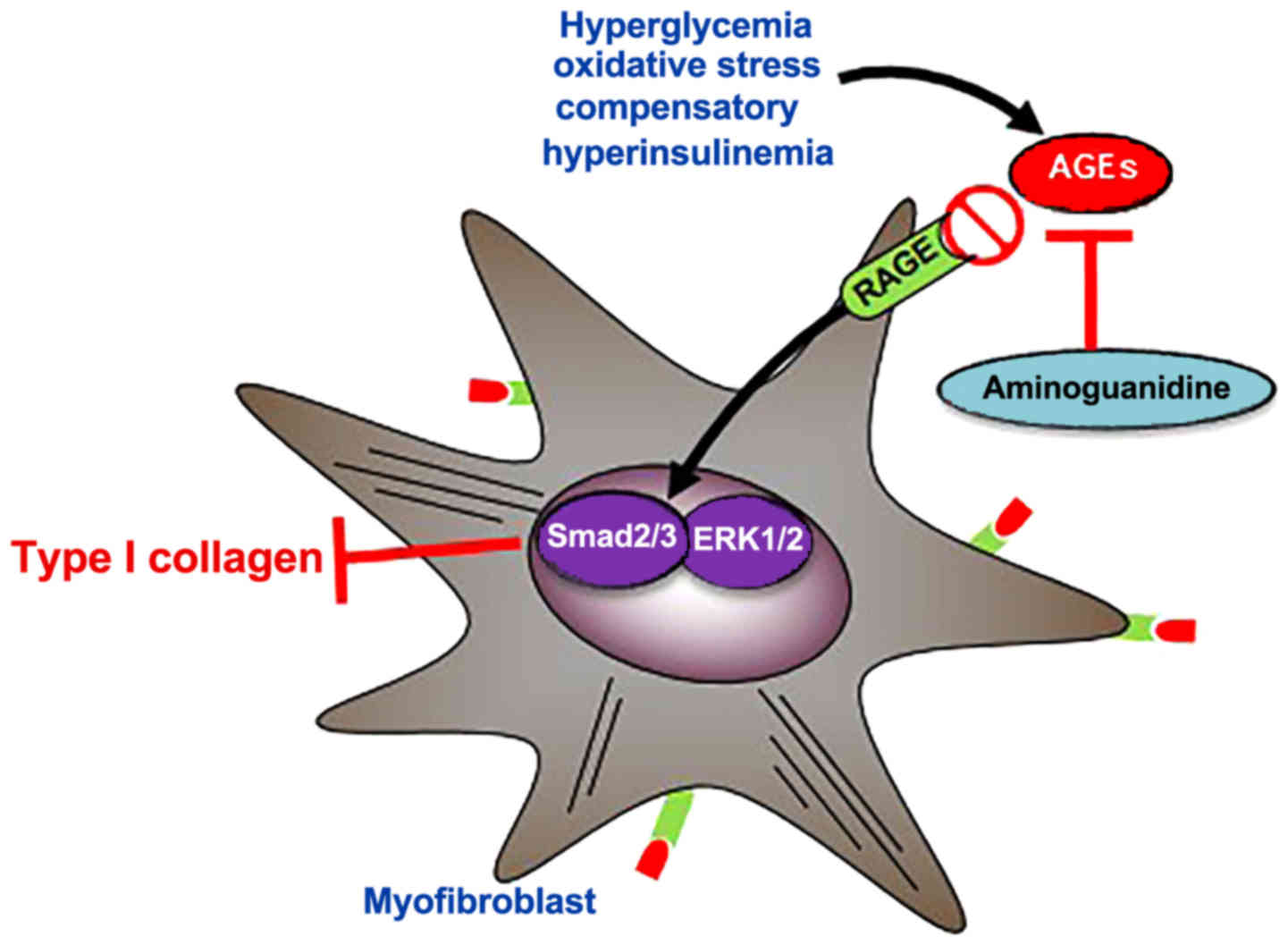
Here, we propose that AG attenuates the fibrogenic
response in T2DM rats, considering that AG reduces AGEs, advanced
oxidation protein products (AOPP) (68), and inhibits iNOS activity (69,70),
thus reducing oxidative-associated cardiac fibrosis. This was
observed in vivo by αSMA IHC, Sirius red/Fast green and
Masson's trichrome staining, and confirmed by the in vitro
experiments on myofibroblasts. Therefore, our study provides strong
support for the association between diabetes and myocardial
fibrosis. Many studies have documented the underlying possible
mechanism of AG in diabetes associated cardiac fibrosis, however,
the effect of AG on collagen type I deposition through
SMAD2/3-ERK1/2 via AGE/RAGE signaling represents the novelty of
this study. These can be explained due to the glycation and
AOPP-inhibiting properties of AG widely described in the scientific
literature (68,71).
To provide further evidence for the contribution of
SMAD2/3-ERK1/2 signaling, the use of specific MAP kinase inhibitors
and/or siRNAs, might reverse the effects of AG-mediated
cardioprotection. However, this was not assessed in vitro,
which we acknowledge is another constraint to our research.
Since AG mitigates oxidative stress (72) and AGE levels (73) and reduces ROS formation (55,74), we
analyzed oxidant and antioxidant-associated genes capable of
modulating ROS, and oxidant stress-sensitive proteins known to
regulate collagen type I expression. Our results suggest that AG
reduces collagen type I expression and myofibroblast activation by
either extenuating oxidative stress or by reducing the level of
AGE-associated oxidative modifications such as RAGE binding
(75). For instance, RAGE
activation, depending on the cellular context and interacting
ligand, can trigger different signaling pathways, which modulate
oxidative stress that contributes to cellular dysfunction
associated with diabetes (76). In
this study, we demonstrated that AG exerts an antifibrotic effect
likely through AGE/RAGE signaling, which is not associated to
hypoglycemia. This is of special relevance since RAGE signaling
contributes to oxidative stress-associated cardiac fibrosis
(77).
In conclusion, STZ plus HFCD promotes T2DM and
evokes pro-oxidative and pro-fibrotic responses attenuated in the
presence of AG. Our results suggest that AG reduces
oxidative-associated cardiac fibrosis by reducing the expression of
pERK1/2, pSMAD2/3 and collagen type I, likely through AGE/RAGE
signaling.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Li Jieli
(Department of Physiology and Biophysics, College of Medicine,
University of Illinois) for her excellent assistance in the
isolation of primary rat myofibroblasts.
Funding
This work was supported in part by PROINPEP 2014
(AR), awarded to support Ph.D. pharmacology program students, and
by US Public Health Service (grant nos. R01 DK069286, R56 DK069286
and R56 DK069286-06S1) from the National Institute of Diabetes and
Digestive and Kidney Diseases (NN), and US Public Health Service
Grants (grant nos. P20 AA017067, P20 AA017067-01S1, P20
AA017067-03S1 and U01 AA021887-03) from the National Institute on
Alcohol Abuse and Alcoholism (NN).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
FM was responsible for the study supervision,
statistical analysis, drafting and editing of the manuscript, and
data acquisition and interpretation. CCB, CLCN, AMGL, AOVA and AGMD
contributed to data acquisition and editing of the manuscript. MCIC
was responsible for the study design and concept, data acquisition
and editing of the manuscript. NN interpreted the data, was
responsible for the financial support for the study, drafting and
editing the manuscript and the critical revision for important
intellectual content. ARRS obtained financial support for the
study, drafted and edited the manuscript, contributed to the study
design and conception, critically revised the manuscript for
important intellectual content and gave final approval of the
version for submission. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal studies and humane endpoints set out for
this study were in accordance with the rules of Ethical and
Technical Specifications for Care and Management outlined in The
National Institutes of Health's Guide, and approved by Jalisco
State Agency for the Care and Use of Laboratory Animals (approval
no. 16/UG-JAL/2008).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AOPP
|
advanced oxidation protein
products
|
|
αSMA
|
α-smooth muscle actin
|
|
AG
|
aminoguanidine
|
|
AGEs
|
advanced glycation end products
|
|
COL1A1
|
collagen type 1 α 1
|
|
eNOS
|
endothelial nitric oxide synthase
|
|
ECM
|
extracellular matrix
|
|
HFCD
|
high-fat, high-carbohydrate diet
|
|
IL-6
|
interleukin-6
|
|
MAPK/ERK
|
mitogen-activated protein
kinase/extracellular signal-regulated kinase
|
|
NADPH
|
nicotinamide adenine dinucleotide
phosphate
|
|
Nox4
|
NADPH oxidase 4
|
|
NO
|
nitric oxide
|
|
Nrf2
|
nuclear factor (erythroid-derived
2)-like 2
|
|
PDGF
|
platelet-derived growth factor
|
|
RAGE
|
receptor for advanced glycation end
products
|
|
ROS
|
reactive oxygen species
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
STZ
|
streptozotocin
|
|
TGFβ
|
transforming growth factor β
|
|
TNFα
|
tumor necrosis factor α
|
|
T2DM
|
type 2 diabetes mellitus
|
|
UCPs
|
uncoupling proteins
|
References
|
1
|
Patel D, Kumar R, Laloo D and Hemalatha S:
Diabetes mellitus: An overview on its pharmacological aspects and
reported medicinal plants having antidiabetic activity. Asian Pac J
Trop Biomed. 2:411–420. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kayama Y, Raaz U, Jagger A, Adam M,
Schellinger IN, Sakamoto M, Suzuki H, Toyama K, Spin JM and Tsao
PS: Diabetic cardiovascular disease induced by oxidative stress.
Int J Mol Sci. 16:25234–25263. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kumar S, Singh R, Vasudeva N and Sharma S:
Acute and chronic animal models for the evaluation of anti-diabetic
agents. Cardiovasc Diabetol. 11:92012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang HJ, Jin YX, Shen W, Neng J, Wu T, Li
YJ and Fu ZW: Low dose streptozotocin (STZ) combined with high
energy intake can effectively induce type 2 diabetes through
altering the related gene expression. Asia Pac J Clin Nutr. 16
(Suppl 1):S412–S417. 2007.
|
|
5
|
Malfitano C, de Souza Junior AL, Carbonaro
M, Bolsoni-Lopes A, Figueroa D, de Souza LE, Silva KA,
Consolim-Colombo F, Curi R and Irigoyen MC: Glucose and fatty acid
metabolism in infarcted heart from streptozotocin-induced diabetic
rats after 2 weeks of tissue remodeling. Cardiovasc Diabetol.
14:1492015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nugent DA, Smith DM and Jones HB: A review
of islet of Langerhans degeneration in rodent models of type 2
diabetes. Toxicol Pathol. 36:529–551. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Serban AI, Stanca L, Geicu OI, Munteanu
MC, Costache M and Dinischiotu A: Extracellular matrix is modulated
in advanced glycation end products milieu via a RAGE receptor
dependent pathway boosted by transforming growth factor-β1 RAGE. J
Diabetes. 7:114–124. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Du AJ, Ren B, Gao XW, Yang L, Fu Y and
Zhao XD: Effects of aminoguanidine on retinal apoptosis in mice
with oxygen-induced retinopathy. Int J Ophthalmol. 6:436–441.
2013.PubMed/NCBI
|
|
9
|
Serhiyenko VA and Serhiyenko AA: Diabetic
cardiac autonomic neuropathy: Do we have any treatment
perspectives? World J Diabetes. 6:245–258. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thornalley PJ: Use of aminoguanidine
(Pimagedine) to prevent the formation of advanced glycation
endproducts. Arch Biochem Biophys. 419:31–40. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Soliman M: Preservation of myocardial
contractile function by aminoguanidine, a nitric oxide synthase
inhibitors, in a rat model of hemorrhagic shock. Pak J Med Sci.
29:1415–1419. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Niki T, Rombouts K, De Bleser P, De Smet
K, Rogiers V, Schuppan D, Yoshida M, Gabbiani G and Geerts A: A
histone deacetylase inhibitor, trichostatin A, suppresses
myofibroblastic differentiation of rat hepatic stellate cells in
primary culture. Hepatology. 29:858–867. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rombouts K, Niki T, Greenwel P,
Vandermonde A, Wielant A, Hellemans K, De Bleser P, Yoshida M,
Schuppan D, Rojkind M and Geerts A: Trichostatin A, a histone
deacetylase inhibitor, suppresses collagen synthesis and prevents
TGF-beta(1)-induced fibrogenesis in skin fibroblasts. Exp Cell Res.
278:184–197. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Grabiec K, Gajewska M, Milewska M,
Blaszczyk M and Grzelkowska-Kowalczyk K: The influence of high
glucose and high insulin on mechanisms controlling cell cycle
progression and arrest in mouse C2C12 myoblasts: The comparison
with IGF-I effect. J Endocrinol Invest. 37:233–245. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang CC, Gurevich I and Draznin B: Insulin
affects vascular smooth muscle cell phenotype and migration via
distinct signaling pathways. Diabetes. 52:2562–2569. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Boateng SY, Hartman TJ, Ahluwalia N,
Vidula H, Desai TA and Russell B: Inhibition of fibroblast
proliferation in cardiac myocyte cultures by surface
microtopography. Am J Physiol Cell Physiol. 285:C171–C182. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li M and Hagerman AE: Effect of
(−)-epigallocatechin-3-gallate on glucose-induced human serum
albumin glycation. Free Radic Res. 49:946–953. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Johnson PD and Besselsen DG: Practical
aspects of experimental design in animal research. ILAR J.
43:202–206. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Festing MF and Altman DG: Guidelines for
the design and statistical analysis of experiments using laboratory
animals. ILAR J. 43:244–258. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lattouf R, Younes R, Lutomski D, Naaman N,
Godeau G, Senni K and Changotade S: Picrosirius red staining: A
useful tool to appraise collagen networks in normal and
pathological tissues. J Histochem Cytochem. 62:751–758. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Halban PA, Polonsky KS, Bowden DW, Hawkins
MA, Ling C, Mather KJ, Powers AC, Rhodes CJ, Sussel L and Weir GC:
β-cell failure in type 2 diabetes: Postulated mechanisms and
prospects for prevention and treatment. J Clin Endocrinol Metab.
99:1983–1992. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rao KB, Malathi N, Narashiman S and Rajan
ST: Evaluation of myofibroblasts by expression of alpha smooth
muscle actin: A marker in fibrosis, dysplasia and carcinoma. J Clin
Diagn Res. 8:ZC14–ZC17. 2014.
|
|
24
|
Lijnen PJ, van Pelt JF and Fagard RH:
Stimulation of reactive oxygen species and collagen synthesis by
angiotensin II in cardiac fibroblasts. Cardiovasc Ther. 30:e1–e8.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ma ZA, Zhao Z and Turk J: Mitochondrial
dysfunction and β-cell failure in type 2 diabetes mellitus. Exp
Diabetes Res. 2012:7035382012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tian XY, Wong WT, Xu A, Lu Y, Zhang Y,
Wang L, Cheang WS, Wang Y, Yao X and Huang Y: Uncoupling protein-2
protects endothelial function in diet-induced obese mice. Circ Res.
110:1211–1216. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Parthasarathy A, Gopi V, Devi K M S,
Balaji N and Vellaichamy E: Aminoguanidine inhibits ventricular
fibrosis and remodeling process in isoproterenol-induced
hypertrophied rat hearts by suppressing ROS and MMPs. Life Sci.
118:15–26. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chowdhury P, Soulsby ME and Scott JL:
Effects of aminoguanidine on tissue oxidative stress induced by
hindlimb unloading in rats. Ann Clin Lab Sci. 39:64–70.
2009.PubMed/NCBI
|
|
29
|
Cigremis Y, Parlakpinar H, Polat A, Colak
C, Ozturk F, Sahna E, Ermis N and Acet A: Beneficial role of
aminoguanidine on acute cardiomyopathy related to
doxorubicin-treatment. Mol Cell Biochem. 285:149–154. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wegner M, Neddermann D,
Piorunska-Stolzmann M and Jagodzinski PP: Role of epigenetic
mechanisms in the development of chronic complications of diabetes.
Diabetes Res Clin Pract. 105:164–175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bertacca A, Ciccarone A, Cecchetti P,
Vianello B, Laurenza I, Maffei M, Chiellini C, Del Prato S and
Benzi L: Continually high insulin levels impair Akt phosphorylation
and glucose transport in human myoblasts. Metabolism. 54:1687–163.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lu TC, Wang ZH, Feng X, Chuang PY, Fang W,
Shen Y, Levy DE, Xiong H, Chen N and He JC: Knockdown of Stat3
activity in vivo prevents diabetic glomerulopathy. Kidney Int.
76:63–71. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yahiaoui L, Gvozdic D, Danialou G, Mack M
and Petrof BJ: CC family chemokines directly regulate myoblast
responses to skeletal muscle injury. J Physiol. 586:3991–4004.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tang M, Zhang W, Lin H, Jiang H, Dai H and
Zhang Y: High glucose promotes the production of collagen types I
and III by cardiac fibroblasts through a pathway dependent on
extracellular-signal-regulated kinase 1/2. Mol Cell Biochem.
301:109–114. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dong Y, Lakhia R, Thomas SS, Dong Y, Wang
XH, Silva KA and Zhang L: Interactions between p-Akt and Smad3 in
injured muscles initiate myogenesis or fibrogenesis. Am J Physiol
Endocrinol Metab. 305:E367–E375. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schiaffino S and Mammucari C: Regulation
of skeletal muscle growth by the IGF1-Akt/PKB pathway: Insights
from genetic models. Skelet Muscle. 1:42011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sakai M, Oimomi M and Kasuga M:
Experimental studies on the role of fructose in the development of
diabetic complications. Kobe J Med Sci. 48:125–136. 2002.PubMed/NCBI
|
|
38
|
Wang XL, Lau WB, Yuan YX, Wang YJ, Yi W,
Christopher TA, Lopez BL, Liu HR and Ma XL: Methylglyoxal increases
cardiomyocyte ischemia-reperfusion injury via glycative inhibition
of thioredoxin activity. Am J Physiol Endocrinol Metab.
299:E207–E214. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ding WY, Liu L, Wang ZH, Tang MX, Ti Y,
Han L, Zhang L, Zhang Y, Zhong M and Zhang W: FP-receptor gene
silencing ameliorates myocardial fibrosis and protects from
diabetic cardiomyopathy. J Mol Med (Berl). 92:629–640. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yu W, Wu J, Cai F, Xiang J, Zha W, Fan D,
Guo S, Ming Z and Liu C: Curcumin alleviates diabetic
cardiomyopathy in experimental diabetic rats. PLoS One.
7:e520132012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rajesh M, Mukhopadhyay P, Bátkai S, Patel
V, Saito K, Matsumoto S, Kashiwaya Y, Horváth B, Mukhopadhyay B,
Becker L, et al: Cannabidiol attenuates cardiac dysfunction,
oxidative stress, fibrosis, and inflammatory and cell death
signaling pathways in diabetic cardiomyopathy. J Am Coll Cardiol.
56:2115–2125. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
King AJ: The use of animal models in
diabetes research. Br J Pharmacol. 166:877–894. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang Y, Babcock SA, Hu N, Maris JR, Wang
H and Ren J: Mitochondrial aldehyde dehydrogenase (ALDH2) protects
against streptozotocin-induced diabetic cardiomyopathy: Role of
GSK3β and mitochondrial function. BMC Med. 10:402012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Asbun J and Villarreal FJ: The
pathogenesis of myocardial fibrosis in the setting of diabetic
cardiomyopathy. J Am Coll Cardiol. 47:693–700. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Vijan S: In the clinic. Type 2 diabetes.
Ann Intern Med. 152:ITC31–ITC15, ITC316. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ježek P, Olejár T, Smolková K, Jezek J,
Dlasková A, Plecitá-Hlavatá L, Zelenka J, Špaček T, Engstová H,
Pajuelo Reguera D and Jabůrek M: Antioxidant and regulatory role of
mitochondrial uncoupling protein UCP2 in pancreatic beta-cells.
Physiol Res. 63 (Suppl 1):S73–S91. 2014.PubMed/NCBI
|
|
47
|
Baldelli S, Aquilano K and Ciriolo MR:
Punctum on two different transcription factors regulated by PGC-α:
Nuclear factor erythroid-derived 2-like 2 and nuclear respiratory
factor 2. Biochim Biophys Acta. 1830:4137–4146. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li B, Liu S, Miao L and Cai L: Prevention
of diabetic complications by activation of Nrf2: Diabetic
cardiomyopathy and nephropathy. Exp Diabetes Res. 2012:2165122012.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lasségue B and Griendling KK: NADPH
oxidases: Functions and pathologies in the vasculature.
Arterioscler Thromb Vasc Biol. 30:653–661. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen F, Haigh S, Barman S and Fulton DJ:
From form to function: The role of Nox4 in the cardiovascular
system. Front Physiol. 3:4122012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pendyala S and Natarajan V: Redox
regulation of Nox proteins. Respir Physiol Neurobiol. 174:265–271.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Brewer AC, Murray TV, Arno M, Zhang M,
Anilkumar NP, Mann GE and Shah AM: Nox4 regulates Nrf2 and
glutathione redox in cardiomyocytes in vivo. Free Radic Biol Med.
51:205–215. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Dusting GJ and Triggle C: Are we over
oxidized? Oxidative stress, cardiovascular disease, and the future
of intervention studies with antioxidants. Vasc Health Risk Manag.
1:93–97. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kazakov A, Hall R, Jagoda P, Bachelier K,
Müller-Best P, Semenov A, Lammert F, Böhm M and Laufs U: Inhibition
of endothelial nitric oxide synthase induces and enhances
myocardial fibrosis. Cardiovasc Res. 100:211–221. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Oak JH, Youn JY and Cai H: Aminoguanidine
inhibits aortic hydrogen peroxide production, VSMC NOX activity and
hypercontractility in diabetic mice. Cardiovasc Diabetol. 8:652009.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Li W, Cui M, Wei Y, Kong X, Tang L and Xu
D: Inhibition of the expression of TGF-β1 and CTGF in human
mesangial cells by exendin-4, a glucagon-like peptide-1 receptor
agonist. Cell Physiol Biochem. 30:749–757. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yokoi H, Kasahara M, Mori K, Kuwabara T,
Toda N, Yamada R, Namoto S, Yamamoto T, Seki N, Souma N, et al:
Peritoneal fibrosis and high transport are induced in mildly
pre-injured peritoneum by 3,4-dideoxyglucosone-3-ene in mice. Perit
Dial Int. 33:143–154. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Dhar A, Dhar I, Desai KM and Wu L:
Methylglyoxal scavengers attenuate endothelial dysfunction induced
by methylglyoxal and high concentrations of glucose. Br J
Pharmacol. 161:1843–1856. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Eika B, Levin RM and Longhurst PA:
Modulation of urinary bladder function by sex hormones in
streptozotocin-diabetic rats. J Urol. 152:537–543. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Youssef S, Nguyen DT, Soulis T,
Panagiotopoulos S, Jerums G and Cooper ME: Effect of diabetes and
aminoguanidine therapy on renal advanced glycation end-product
binding. Kidney Int. 55:907–916. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wilkinson-Berka JL, Kelly DJ, Koerner SM,
Jaworski K, Davis B, Thallas V and Cooper ME: ALT-946 and
aminoguanidine, inhibitors of advanced glycation, improve severe
nephropathy in the diabetic transgenic (mREN-2)27 rat. Diabetes.
51:3283–3289. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Han DC, Isono M, Hoffman BB and Ziyadeh
FN: High glucose stimulates proliferation and collagen type I
synthesis in renal cortical fibroblasts: Mediation by autocrine
activation of TGF-beta. J Am Soc Nephrol. 10:1891–1899.
1999.PubMed/NCBI
|
|
63
|
Aguilar H, Fricovsky E, Ihm S, Schimke M,
Maya-Ramos L, Aroonsakool N, Ceballos G, Dillmann W, Villarreal F
and Ramirez-Sanchez I: Role for high-glucose-induced protein
O-GlcNAcylation in stimulating cardiac fibroblast collagen
synthesis. Am J Physiol Cell Physiol. 306:C794–C804. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Fiaschi T, Magherini F, Gamberi T,
Lucchese G, Faggian G, Modesti A and Modesti PA: Hyperglycemia and
angiotensin II cooperate to enhance collagen I deposition by
cardiac fibroblasts through a ROS-STAT3-dependent mechanism.
Biochim Biophys Acta. 1843:2603–2610. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kojima H, Fujimiya M, Matsumura K,
Nakahara T, Hara M and Chan L: Extrapancreatic insulin-producing
cells in multiple organs in diabetes. Proc Natl Acad Sci USA.
101:2458–2463. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Phadnis SM, Ghaskadbi SM, Hardikar AA and
Bhonde RR: Mesenchymal stem cells derived from bone marrow of
diabetic patients portrait unique markers influenced by the
diabetic microenvironment. Rev Diabet Stud. 6:260–270. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wang CC, Goalstone ML and Draznin B:
Molecular mechanisms of insulin resistance that impact
cardiovascular biology. Diabetes. 53:2735–2740. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Grzebyk E and Piwowar A: Inhibitory
actions of selected natural substances on formation of advanced
glycation endproducts and advanced oxidation protein products. BMC
Complement Altern Med. 16:3812016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Islas-Carbajal MC, Covarrubias A, Grijalva
G, Alvarez-Rodriguez A, Armendáriz-Borunda J and Rincón-Sánchez AR:
Nitric oxide synthases inhibition results in renal failure
improvement in cirrhotic rats. Liver Int. 25:131–140. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Corbett JA, Tilton RG, Chang K, Hasan KS,
Ido Y, Wang JL, Sweetland MA, Lancaster JR Jr, Williamson JR and
McDaniel ML: Aminoguanidine, a novel inhibitor of nitric oxide
formation, prevents diabetic vascular dysfunction. Diabetes.
41:552–556. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ara C, Karabulut AB, Kirimlioglu H, Yilmaz
M, Kirimliglu V and Yilmaz S: Protective effect of aminoguanidine
against oxidative stress in an experimental peritoneal adhesion
model in rats. Cell Biochem Funct. 24:443–448. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Stadler K, Jenei V, Somogyi A and Jakus J:
Beneficial effects of aminoguanidine on the cardiovascular system
of diabetic rats. Diabetes Metab Res Rev. 21:189–196. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Richardson MA, Furlani RE, Podell BK,
Ackart DF, Haugen JD, Melander RJ, Melander C and Basaraba RJ:
Inhibition and breaking of advanced glycation end-products (AGEs)
with bis-2-aminoimidazole derivatives. Tetrahedron Lett.
56:3406–3409. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Giardino I, Fard AK, Hatchell DL and
Brownlee M: Aminoguanidine inhibits reactive oxygen species
formation, lipid peroxidation, and oxidant-induced apoptosis.
Diabetes. 47:1114–1120. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Oldfield MD, Bach LA, Forbes JM,
Nikolic-Paterson D, McRobert A, Thallas V, Atkins RC, Osicka T,
Jerums G and Cooper ME: Advanced glycation end products cause
epithelial-myofibroblast transdifferentiation via the receptor for
advanced glycation end products (RAGE). J Clin Invest.
108:1853–1863. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Khazaei M, Karimi J, Sheikh N, Goodarzi
MT, Saidijam M, Khodadadi I and Moridi H: Effects of resveratrol on
receptor for advanced glycation end products (RAGE) expression and
oxidative stress in the liver of rats with type 2 diabetes.
Phytother Res. 30:66–71. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Tan Y, Ichikawa T, Li J, Si Q, Yang H,
Chen X, Goldblatt CS, Meyer CJ, Li X, Cai L and Cui T: Diabetic
downregulation of Nrf2 activity via ERK contributes to oxidative
stress-induced insulin resistance in cardiac cells in vitro and in
vivo. Diabetes. 60:625–633. 2011. View Article : Google Scholar : PubMed/NCBI
|