Introduction
The most effective strategy to reduce acute
myocardial ischemic injury and subsequent mortality is to promptly
recover coronary reflow using thrombolytic therapy or percutaneous
intervention (1). However,
reperfusion can induce myocardial ischemia reperfusion injury
(MIRI). The inflammatory response, oxidative stress and cell
apoptosis are considered to be critical factors associated with
mediating the effects of MIRI (2–4).
Targeting these factors is important in the prevention and
reduction of MIRI.
Adropin, first described by Kumar et al
(5) in 2008, is a secreted protein
and an endogenous biologically active substance encoded for by an
energy homeostasis-associated gene. Lovren et al (6) demonstrated that adropin is expressed in
the endothelial cells of the umbilical veins and coronary arteries.
The aforementioned study also revealed that adropin may exhibit
nonmetabolic properties, which includes the regulation of
endothelial function through the upregulation of endothelial nitric
oxide synthase (eNOS) via the PI3K-Akt and ERK1/2, which are the
two major components of the reperfusion injury salvage kinase
(RISK) pathway. The RISK pathway represents one of the most
important survival mechanisms against ischemic reperfusion injury.
Apart from the RISK pathway, the survivor activating factor
enhancement (SAFE) pathway also serves a role in ischemic
postconditioning. The major components of the SAFE pathway are
TNF-α and Janus kinase (JAK), which phosphorylates the
transcription factor STAT3. Additionally, adropin has been revealed
to improve murine limb perfusion and elevate capillary density
following the induction of hindlimb ischemia (6). Clinical research has demonstrated that
adropin is associated with a variety of metabolic risk factors.
Butler et al (7) demonstrated
that plasma adropin levels are negatively associated with obesity
and insulin resistance. Celik et al (8) revealed that serum adropin levels were
negatively associated with cardiac X syndrome due to coronary
microvascular perfusion dysfunction and that low serum adropin
levels were an independent risk factor of X syndrome. Adropin has
been revealed to be negatively correlated with inflammatory
biomarker-C reactive protein and it has been demonstrated that
patients with severe atherosclerosis exhibit lower adropin levels
(9). These results indicated that
adropin may influence the anti-inflammatory response and reduce
atherosclerosis (9). Yang et
al (10) demonstrated that
adropin reduces endothelial cell permeability and modulates
ischemia-induced blood-brain barrier injury. However, to the best
of our knowledge, the role of adropin in myocardial reperfusion
injury has not yet been assessed.
In the current study, a hypoxia/reoxygenation model
was established in neonatal rat cardiomyoblast cells (H9c2) to
simulate ischemia/reperfusion (SI/R) injury. The effect of adropin
on SI/R injury and the mechanisms that govern this effect were
subsequently assessed.
Materials and methods
Cell culture
H9c2 cells were obtained from the Type Culture
Collection of the Chinese Academy of Sciences. Cells were passaged
up to 4 times and were cultured in DMEM (GE Healthcare Life
Sciences) containing 10% (v/v) heat-inactivated FBS (Gibco; Thermo
Fisher Scientific, Inc.), 100 IU/ml penicillin (GE Healthcare Life
Sciences) and 100 µg/ml streptomycin (GE Healthcare Life Sciences),
under a 5% CO2 atmosphere at 37°C.
H9c2 cells subjected to
hypoxia/reoxygenation induced injury
Hypoxia was induced as described previously
(11). H9c2 cells were cultured to
70–80% confluency, fresh DMEM without FBS was subsequently added
and the cells were transferred to a triple gas incubator with
either hypoxic (5% CO2, 1% O2 and 94%
N2) or SI/R (hypoxia: 5% CO2, 1%
O2 and 94% N2, followed by reoxygenation: 5%
CO2, 21% O2 and 74% N2) settings.
A hypoxia/reoxygenation model was established and cells were
divided into 11 groups. All groups except the control group were
treated with hypoxic conditions for 12 h and reoxygenation for 24
h. Postconditioning of cardiomyocytes was achieved as follows: At
the end of 12 h of hypoxia, the cells were initially received
different doses of adropin and then returned to the reoxygenated
condition for another 24 h. The groups were classified as follows:
i) Control group, normoxic conditions (37°C, 5% CO2, 21%
O2, 71% N2); ii) SI/R group; iii) SI/R + low
dose adropin (10 ng/ml; Phoenix Pharmaceuticals, Inc.), in which
adropin was added prior to reoxygenation (adropin-L); iv) SI/R +
moderate dose adropin group (25 ng/ml; adropin-M); v) SI/R + high
dose adropin group (50 ng/ml; adropin-H); vi) LY294002 group, 40
µmol/l PI3K specific inhibitor LY294002 (Sigma-Aldrich; Merck KGaA)
was added to the medium prior to hypoxia as described previously
(12); vii) adropin + LY294002
group, in which 40 µmol/l LY294002 and 25 ng/ml adropin were added
to the medium prior to hypoxia (12)
and reoxygenation, respectively; viii) PD98059 group, in which 25
µmol/l ERK1/2-specific inhibitor PD98059 (Sigma-Aldrich; Merck
KGaA) was added to the medium (12)
prior to hypoxia; ix) adropin + PD98059 group, in which 25 µmol/l
PD98059 and 25 ng/ml adropin were added to the medium (12) prior to hypoxia and reoxygenation,
respectively; x) AG490 group, in which 100 µmol/l JAK2 inhibitor
AG490 (Sigma-Aldrich; Merck KGaA) was added to the medium prior to
hypoxia as described previously (13); xi) adropin + AG490 group, in which
100 µmol/l AG490 and 25 ng/ml adropin were added to the medium
(13) prior to hypoxia and
reoxygenation, respectively.
MTT measurement of cell viability
A total of 1×105 H9c2 cells/ml were
seeded into a 96-well culture plate and incubated at 5%
CO2 and 37°C for 24 h. Cell viability was determined
using an MTT assay. At 12 h following reoxygenation, 20 µl MTT
solution was added into each well (5 mg/ml) and plates were
incubated for 4 h at 37°C. A microplate reader was used to measure
the absorbance at a wavelength of 490 nm.
ELISA assay and colorimetry
The expression of creatine kinase MB (CK-MB; cat.
no. H197), tumor necrosis factor α (TNF-α; cat. no. H052) and
interleukin (IL)-10 (cat. no. H009) were measured using ELISA assay
kits (Nanjing Jiancheng Bioengineering Institute). Malondialdehyde
(MDA; cat. no. A003-4) and superoxide dismutase (SOD; cat. no.
A001-1) concentrations were determined using colorimetry kits
according to manufacturer's protocols (Nanjing Jiancheng
Bioengineering Institute). The experiment was performed at least
three times and CK-MB level was expressed as IU/l. TNF-α and IL-10
levels were expressed as pg/ml. The MDA level and SOD were
expressed as nmol/mg protein and as U/mg protein, respectively.
Apoptosis analysis
Early cell apoptosis was measured using flow
cytometry. The analysis of phosphatidylserine on the outer
apoptotic cell membranes was performed using annexin-V-fluorescein
and propidium iodide (Annexin-V-FLUOS Staining kit; Roche
Diagnostics). Collected cells were rinsed with ice-cold PBS and
resuspended in 250 µl of binding buffer and ~1–5×105
cells were analyzed in each of the samples. A total of 100 µl
annexin-V-FLUOS labeling solution was added to the cells, which
were then incubated for 15 min at 25°C. The cells were analyzed
using FlowJo software (version 10.4.1; BD FACScanto II; Becton,
Dickinson and Company).
Measurement of caspase-3 activity
Caspase-3 activity was measured using a colorimetric
activity assay kit (Ac-DEVD-pNA; Beyotime Institute of
Biotechnology) according to manufacturer's protocol. In brief,
cells were lysed in ice-cold lysis buffer, placed on ice for 15
min, then centrifuged at 4°C for 15 min at 16,000 × g and
supernatant was subsequently incubated with caspase-3 substrate on
a 96-well plate. Protein concentration was determined using
Bradford protein assay kit (cat. no. P0006; Beyotime Institute of
Biotechnology). Caspase-3 activity was determined using a
microplate reader at a wavelength of 405 nm.
Western blot analysis
H9c2 cells were washed with PBS, enzymatically
dissociated with the use of trypsin (HyClone; GE Healthcare Life
Sciences), and prepared in lysis buffer with protease inhibitor
cocktail (cat. no. P0013B; Beyotime Institute of Biotechnology).
Protein quantification was measured by using a BCA protein assay
kit (cat. no. P0012; Beyotime Institute of Biotechnology). Equal
quantities of protein (30 µg/lane) from whole cell lysates of
cultured H9c2 cells were separated by 10% SDS-PAGE and transferred
to a PVDF membrane. Following blocking with 5% BSA for 1 h at room
temperature for binding non-specific sites, membranes were
incubated with primary antibodies overnight at 4°C. The following
primary antibodies were used: Phosphorylated (p)-Akt polyclonal
antibody (1:1,000; cat. no. YP0864) and Akt polyclonal antibody
(1:1,000; cat. no. YT0173) were purchased from ImmunoWay
Biotechnology Company. p-ERK 1/2 monoclonal antibody (1:1,000; cat.
no. sc-136521), ERK 1/2 monoclonal antibody (1:1,000; cat. no.
sc-514302), p-STAT3 antibody (1:1000; cat. no. sc-7993) and STAT3
antibody (1:1,000; cat. no. sc-8019) were purchased from Santa Cruz
Biotechnology, Inc. P-GSK3β antibody (1:1,000; cat. no. ab131097),
GSK3β antibody (1:5,000; cat. no. ab32391), Bcl-2 antibody
(1:1,000; cat. no. ab59348) and Bax antibody (1:1,000; cat. no.
ab32503) were purchased from Abcam. Following incubation with
horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG
(1:5,000; cat. no. ZB-5301; OriGene Technologies, Inc.) or
HRP-conjugated goat anti-mouse IgG (1:5,000; cat. no. ZB-2305;
OriGene Technologies, Inc.) at 37°C for 1 h, the signals were
detected with Pierce™ ECL Western Blotting Substrate kit (cat. no.
32209, Pierce; Thermo Fisher Scientific, Inc.) and bands were
subsequently quantified using Quantity One software (version 4.6.2;
Bio-Rad Laboratories, Inc.).
Statistical analysis
Data are presented as the mean ± standard deviation.
Comparisons between groups were performed using one-way ANOVA with
Student-Newman-Keuls correction for multiple comparisons.
Statistical analyses were performed using SPSS version 13.0 (SPSS
Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
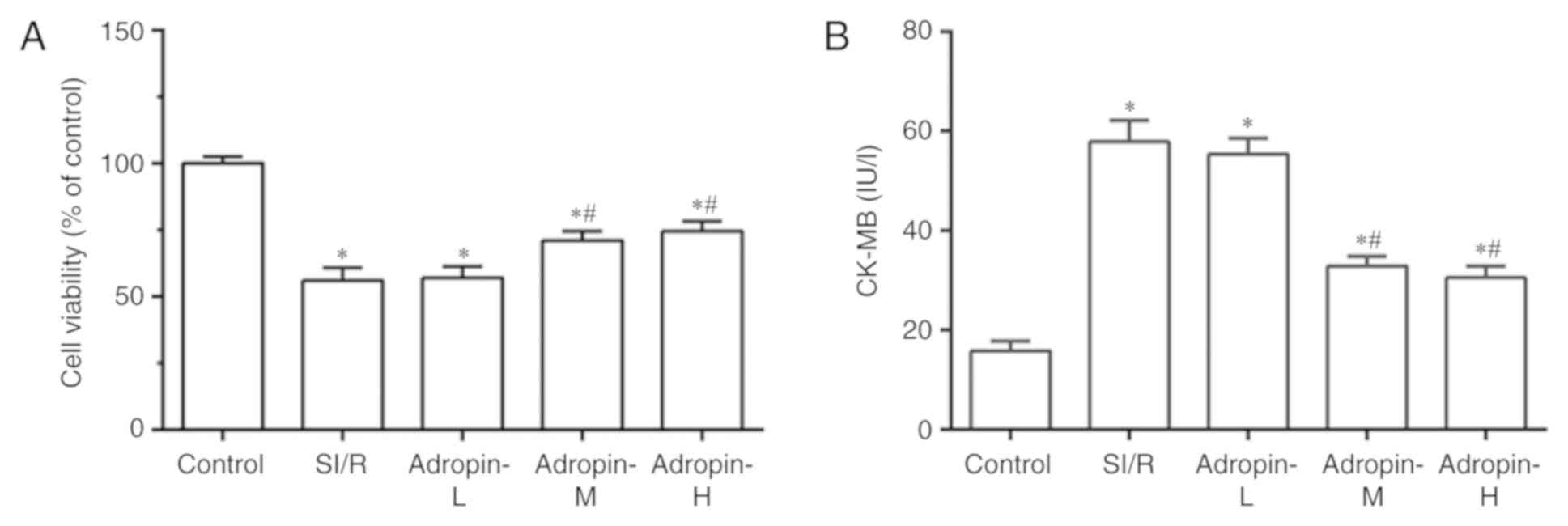
Effect of adropin dose on cell
viability and CK-MB levels
Cell viability was examined using a MTT assay and
CK-MB levels were measured to assess cardiomyocyte injury. As
presented in Fig. 1A and B, SI/R
group cell viability was significantly reduced (P<0.001) and
CK-MB levels significantly increased (P<0.001) compared with the
control group. Cell viability was significantly higher and CK-MB
levels were significantly lower in the adropin-M and adropin-H
groups when compared with the SI/R group. In addition, no
significant difference in cell viability and CK-MB levels was
observed between the adropin-M and adropin-H groups. The adropin-L
group did not exhibit any significant effect on cell viability or
CK-MB expression when compared with the SI/R group, indicating that
moderate and high adropin levels can reduce SI/R injury. The
subsequent experiments were performed using moderate-dose adropin
as the adropin group.
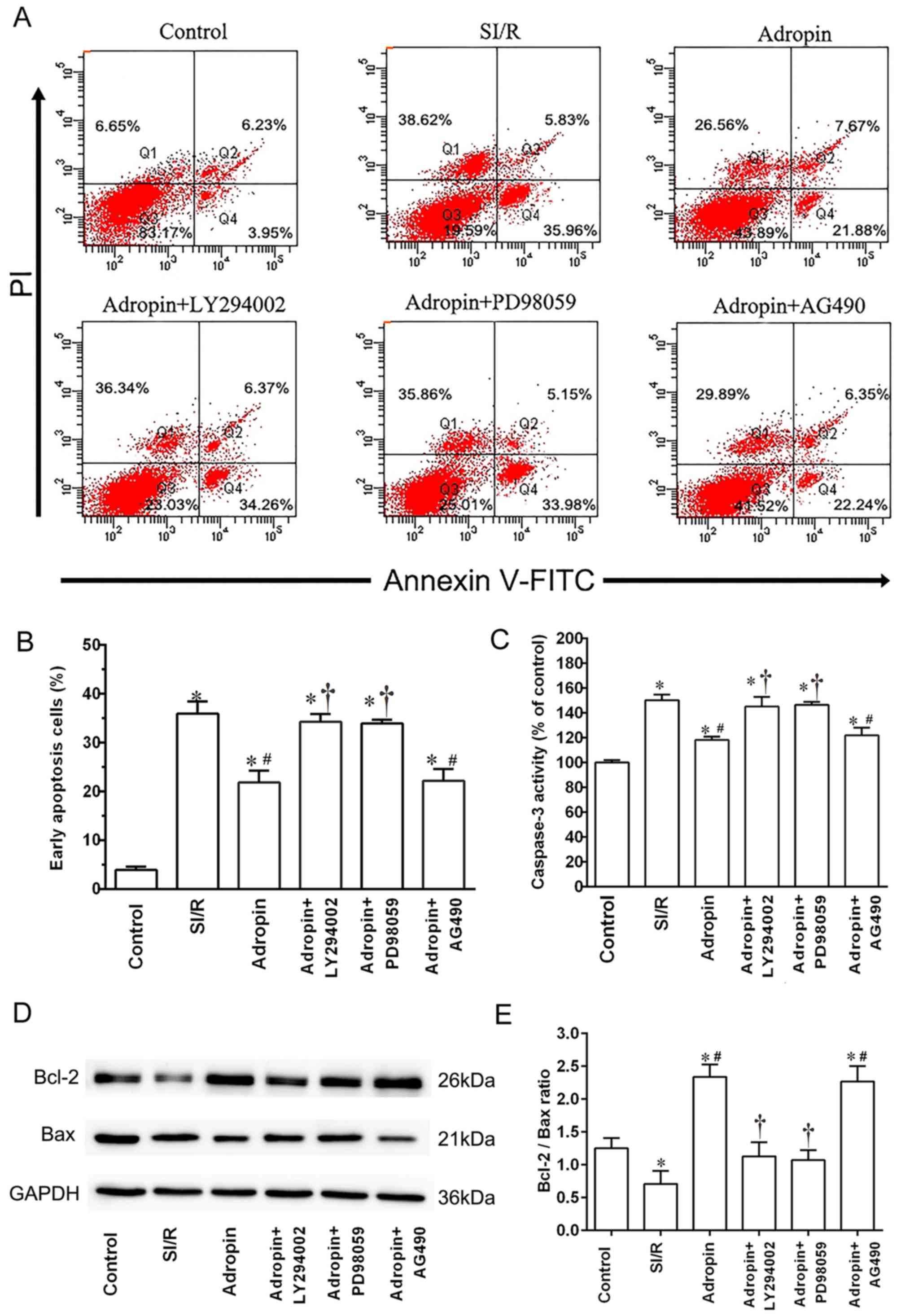
Effect of adropin on myocardial
apoptosis
Flow cytometry was used to assess myocardial
apoptosis and caspase-3 activity subsequent to reoxygenation
(Fig. 2). The SI/R group exhibited a
higher early apoptosis rate (P<0.001) and higher caspase-3
activity (P<0.001) when compared with the control group
(Fig. 2B and C, respectively).
Moderate-dose adropin exhibited a significantly lower early
apoptosis rate (P<0.001) and caspase-3 activity (P<0.001)
compared with the SI/R group. Additionally, LY294002 and PD98059
significantly reversed the protective effects of adropin on
apoptosis rate (P<0.001) and significantly increased caspase-3
activity (P<0.001) compared with the adropin group (Fig. 2A-C). However, AG490 exhibited no
significant effect on early apoptosis rate or caspase-3 activity
when compared with the adropin group (Fig. 2A-C).
Western blot analysis was used to detect the effect
of adropin and the aforementioned inhibitors on the Bcl-2/Bax
ratio. As presented in Fig. 2D and
E, the SI/R group had a significantly lower Bcl-2/Bax ratio
compared with the control group (P<0.05). When compared with the
SI/R group, the adropin group exhibited a significantly higher
Bcl-2/Bax ratio (P<0.05). Additionally, the adropin + LY294002
and adropin + PD98059 groups exhibited significantly lower
Bcl-2/Bax ratios (P<0.05) compared with the adropin-only group.
However, no significant differences were determined in the
Bcl-2/Bax ratio between the adropin and adropin + AG490 group
(Fig. 2D and E).
Effects of different doses of adropin
on the inflammatory response
The inflammatory response was assessed using TNF-α
and IL-10 expression measurements (Fig.
3A and B, respectively). TNF-α levels significantly increased
(P<0.001) and IL-10 levels decreased (P<0.001) in the SI/R
group compared with the control group. The adropin-M and adropin-H
groups exhibited significantly reduced TNF-α expression
(P<0.001) and significantly increased IL-10 expression
(P<0.001) when compared with the SI/R group. In addition, no
significant difference was determined in TNF-α and IL-10 expression
levels between the adropin-M and adropin-H groups (P>0.05). The
adropin-L group did not affect TNF-α or IL-10 levels compared with
the control group, suggesting that moderate and high concentrations
of adropin can protect the heart by alleviating the inflammatory
response.
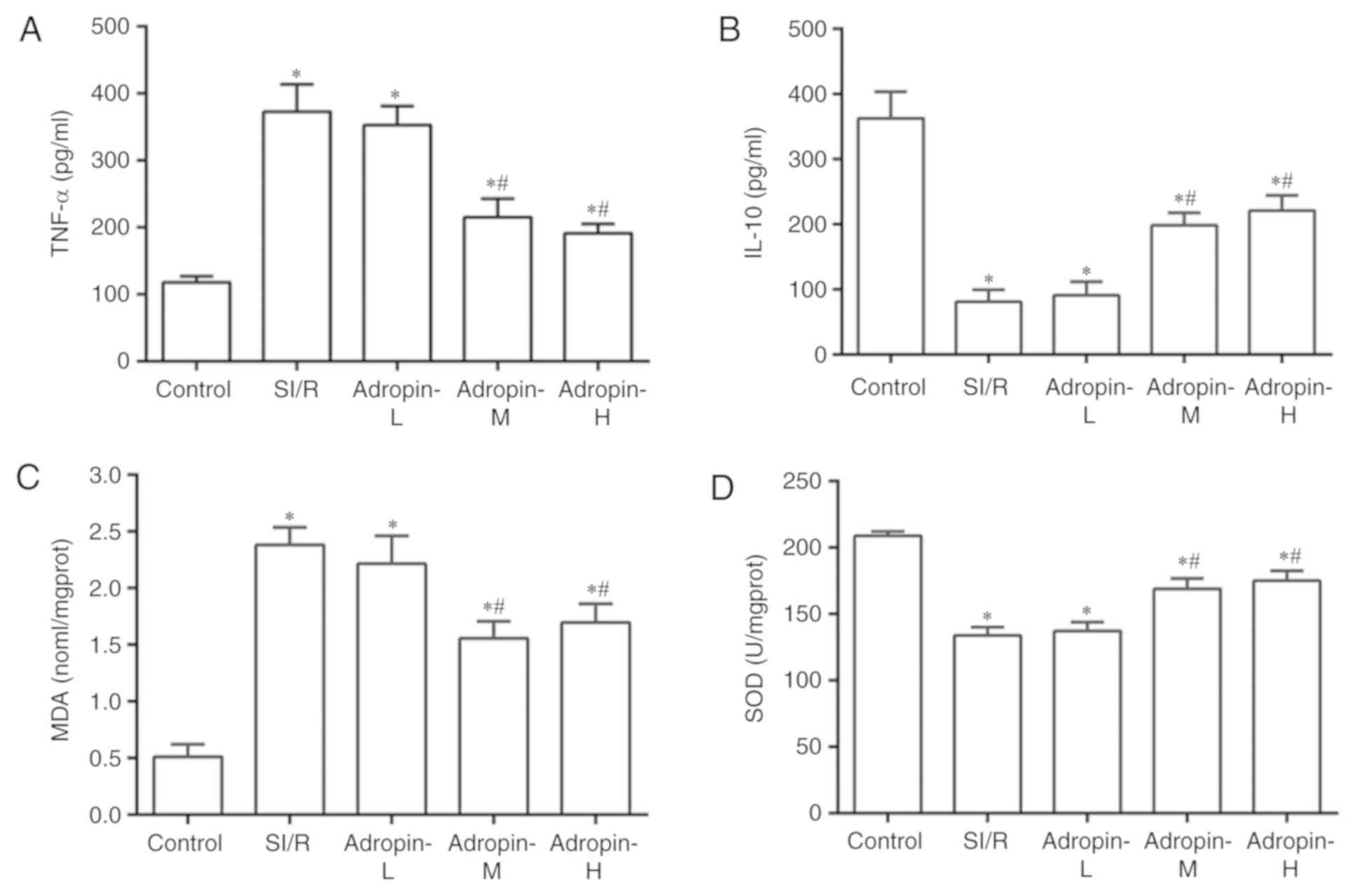 | Figure 3.Effects of adropin on the
inflammatory response and oxidative stress. Effect of adropin on
(A) TNF-α, (B) IL-10, (C) MDA and (D) SOD of H9c2 cells subjected
to SI/R. *P<0.001 vs. control and #P<0.001 vs.
SI/R. Results are representative of three independent experiments.
TNF-α, tumor necrosis factor α; IL-10, interleukin 10; MDA,
malondialdehyde; SOD, superoxide dismutase; SI/R, simulated
ischemia/reperfusion; L, low; M, medium; H, high. |
Effects of different doses of adropin
on oxidative stress
Oxidative stress was examined by measuring MDA
levels and SOD activity (Fig. 3C and
D, respectively). MDA levels significantly increased
(P<0.001) and SOD activity was significantly reduced in the SI/R
group (P<0.001) compared with the control group. The adropin-M
and adropin-H groups exhibited reduced MDA levels (P<0.001) and
exhibited higher SOD activity (P<0.001) compared with the SI/R
group. The results indicated that adropin may inhibit lipid
peroxide production and increase scavenging superoxide radical
activity. No significant difference in MDA levels and SOD activity
were determined between the adropin-M and adropin-H groups.
Adropin-L did not reduce MDA levels or increase SOD activity when
compared with the SI/R group, demonstrating the dose-dependent role
of adropin in the antioxidative effect.
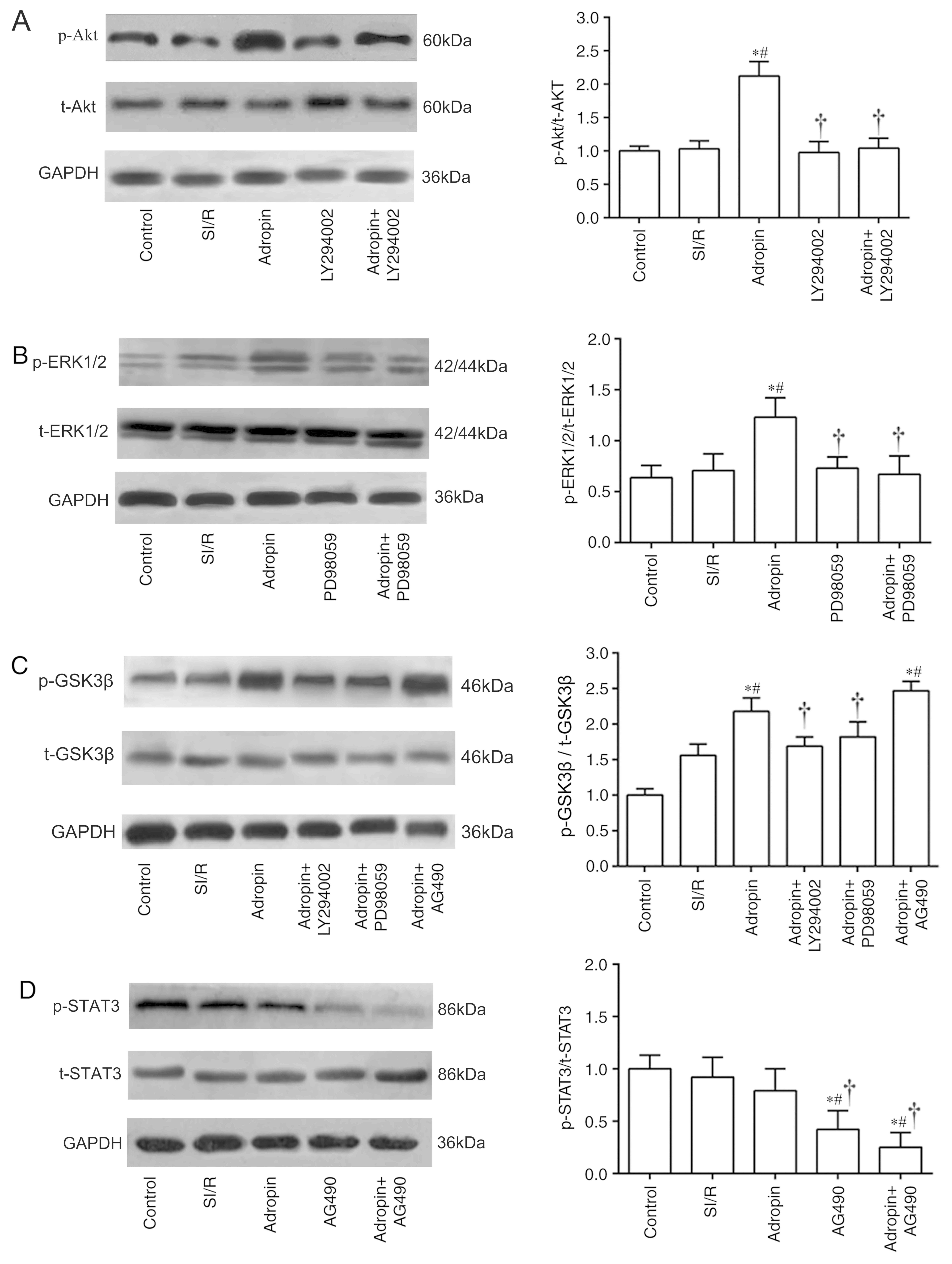
Reperfusion injury salvage kinase
(RISK) pathway is associated with the reduction of SI/R injury by
adropin
The results of the present study demonstrated that
adropin inhibited myocardial injury induced by SI/R in a
dose-dependent manner. The Adropin-M group (the minimum optimal
concentration) was used as the Adropin group in subsequent
experiments to further assess the molecular mechanisms associated
with the reduction of adropin in SI/R injury.
As presented in Fig.
4A, the adropin group induced a significant elevation in
p-Akt/t-Akt ratio (P<0.05) compared with the SI/R group.
LY294002 group exhibited a significantly decreased p-Akt/t-Akt
ratio when compared with the adropin group (P<0.05). In
addition, the adropin group had a significantly higher
p-ERK1/2/t-ERK1/2 ratio (P<0.05) compared with the SI/R group.
PD98059 exhibited a significantly decreased p-ERK1/2/t-ERK1/2 ratio
compared with the adropin group (Fig.
4B). Furthermore, adropin significantly increased
p-GSK3β/t-GSK3β ratio compared with the SI/R group (P<0.05),
which was partially but significantly reversed by additive
treatments with LY294002 (P<0.05) or PD98059 (P<0.05).
However, the adropin and adropin + AG490 groups demonstrated no
significant difference in the p-GSK3β/t-GSK3β ratio (Fig. 4C). Compared with the control, the
AG490 and adropin + AG490 groups significantly inhibited the
phosphorylation levels of STAT3 (P<0.05). Notably, adropin and
control groups exhibited no difference in the p-STAT3/t-STAT ratio
(Fig. 4D).
Discussion
Adropin is a newly identified endogenous bioactive
substance that serves an important role in energy metabolism.
Lovren et al (6) demonstrated
that adropin may directly affect endothelial cells and may possess
nonmetabolic properties, including the protection of endothelial
function through the RISK pathway. Adropin upregulates eNOS and
increases the production of NO through the PI3K-Akt and ERK1/2
pathways. Adropin also serves a role in improving murine limb
perfusion and elevating capillary density after ischemia (6). Exogenous adropin reduces insulin
resistance and metabolic disorders, protects endothelial cells and
attenuates organ ischemia (6,10). These
results indicate that adropin may also be associated with ischemia
reperfusion injury and may serve a cardioprotective role in
MIRI.
Apoptosis is an important factor in the pathogenesis
of MIRI (14,15). Mitochondria serve a central role in
apoptosis regulation and control cytochrome C release through
channels formed by Bcl-2 gene family expression, which is a key
mechanism that regulates apoptosis (14). The inhibition of myocardial apoptosis
can prevent myocardial cell loss and delay the occurrence of heart
failure (16,17). In the present study, the results
indicated that adropin treatment after hypoxia induction can
inhibit hypoxia/reoxygenation-induced injury in H9c2 cells. Adropin
reduced the proportion of early apoptosis in myocardial cells,
decreased the activity of caspase-3, reduced the expression of Bax
gene and increased Bcl-2 gene expression. These results
demonstrated that adropin can reduce SI/R injury by regulating the
mitochondrial apoptosis pathway.
A number of inflammatory factors including TNF-α,
IL-1, IL-6 and IL-8 are released by myocardium subjected to
ischemia-reperfusion (18,19). Oxidative stress also serves an
important role in myocardial injury located in the infarcted and
reperfused myocardium (20,21). Myocardial cells generate numerous
reactive oxygen species during the ischemia-reperfusion process and
increase TNF-α synthesis, which can lead to an increase in the
apoptosis cascade reaction, the interactions between inflammatory
and endothelial cells and intracellular calcium overload (22). Various novel antioxidants have been
associated with renal protection through the antioxidative and
antiapoptotic pathways (23,24). A previous study (9) has demonstrated that adropin is
negatively correlated with the inflammatory marker C reactive
protein. In patients with severe coronary atherosclerosis, adropin
serum level is low (9), which
indicates that adropin possesses a potential anti-inflammatory
effect. In the current study, moderate and high concentrations of
adropin were indicated to reduce the inflammatory response and
oxidative stress during SI/R injury. Additionally, adropin was
revealed to inhibit SI/R-induced myocardial injury by reducing
early myocardial apoptosis, inflammatory response and oxidative
stress, and increasing myocardial cell viability.
In 2007, Yellon et al (25) proposed a new cardioprotective
strategy to reduce MIRI at the early stages of reperfusion by
targeting the RISK-mitochondrial permeability transition pathway
(mPTP). This study revealed that ischemic or pharmacological
postconditioning prior to reperfusion can activate RISK or inhibit
mPTP opening to limit infarct size and reduce MIRI (25). Ischemic and pharmacological
postconditioning invoke the activation of signal transduction
cascades by autacoids triggers and eventually inhibit the opening
of mPTP (26). However,
pharmacological postconditioning performed prior to continuous
reperfusion is operable in clinical practice and can avoid
mechanical manipulation and associated complications (26). The activation of the RISK signaling
pathway (PI3K/Akt and ERK1/2) may serve a role in cardioprotection
in myocardial reperfusion and therefore, this pathway may become an
important drug target (27). The
activation of PI3K and its downstream target (Akt) is also
associated with myocardial reperfusion injury (28,29). In
the ischemic myocardium, the phosphorylation of Akt can inhibit
myocardial apoptosis and promote the cell survival pathway
(30). Additionally, ERK1/2 is an
important kinase of the RISK pathway and its activation in
myocardial ischemia/reperfusion is beneficial to reduce apoptosis
and to help recover cardiac function (31).
In addition to the RISK pathway, the survivor
activating factor enhancement (SAFE) pathway has been revealed to
be an additional pro-survival signaling pathway associated with the
early reperfusion period and is composed of TNF-α and STAT-3
(32). mPTP is the downstream
effector of the SAFE and RISK pathways (32). mPTP may be the common final effector
of cardioprotective effects exhibited by pre and postconditioning
(27). Furthermore, complex
crosstalk between RISK and SAFE pathways may exist.
The current study assessed whether the RISK and SAFE
pathways are associated with the role of adropin in the reduction
of SI/R injury in cardiomyocytes. The results demonstrated that a
moderate concentration of adropin significantly increased the
phosphorylation of Akt and ERK1/2 and these results are consistent
with Lovren et al (6). It was
also revealed that adropin can promote the phosphorylation of GSK3β
(a prosurvival signaling pathway downstream target protein). PI3K
specific inhibitor LY294002 or ERK1/2 inhibitor PD98059 also
significantly inhibited the cardioprotective effects of adropin,
indicating that these effects may be dependent on the PI3K/Akt and
ERK1/2 pathway. The adropin treatment did not significantly
increase the phosphorylation of STAT3, which is the most important
target of the SAFE pathway (33).
STAT3 is also the substrate of JAK2 kinase. The JAK2 kinase
specific inhibitor, AG490, did not significantly inhibit the
protective role of adropin in SI/R injury.
In conclusion, the results of the present study
demonstrate that adropin reduces SI/R injury in H9c2 myocardial
cells through the RISK pathway (PI3K/Akt and ERK1/2) by activating
the downstream target GSK3β to regulate the mitochondrial
apoptosis. However, the SAFE pathway (JAK-STAT3) was not indicated
to be associated with the exhibited myocardial protection. The
current study may provide a potential therapeutic target for
ischemia reperfusion injury and a theoretical basis for the
clinical use of adropin.
Although H9c2 cells have been widely used in the
study of cardiovascular disease, these studies may not accurately
represent the in vivo reaction of normal myocardial cells to
drug treatments. In the current study, the effects of adropin were
only assessed in relation to a few inflammatory factors. However,
other inflammatory factors such as leukocyte adhesion, aggregation
and inflammatory stimulation signals and their receptors have not
been involved. Reactive oxygen species (ROS) levels were not
directly assessed and ROS scavenger was also not used. Therefore,
the mechanisms underlying the changes in SOD and MDA levels
observed in the current study following treatment with adropin
remain to be determined.
Acknowledgements
Not applicable.
Funding
The current study was mainly supported by The
National Natural Science Foundation of China (grant no. 81500352)
and partially by the Natural Science Foundation of Fujian Province
of China (grant no. 2016J05186), the Program for New Century
Excellent Talents in Fujian Province University (grant no.
2015B021), the Medical Elite Cultivation Program of Fujian (grant
no. 2015-ZQN-ZD-12), the Youth Foundation of Fujian Provincial
Health and Family Planning Commission of China (grant no.
2015-1-40) and the Science Foundation for Distinguished Young
Scholars of Fujian Province (grant no. 2013J06015).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LW and JF performed data analysis, wrote the
manuscript and contributed to the critical revision of the
manuscript. LW, XY and CX conducted the experiments and statistical
analysis. LC performed data analysis and contributed to the
critical revision of the manuscript. All authors are the guarantors
of this work and, as such, had full access to all the data in the
study and take responsibility for the integrity of the data and the
accuracy of the data analysis. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chandrasekar B, Smith JB and Freeman GL:
Ischemia-reperfusion of rat myocardium activates nuclear
factor-KappaB and induces neutrophil infiltration via
lipopolysaccharide-induced CXC chemokine. Circulation.
103:2296–2302. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kaminski KA, Bonda TA, Korecki J and
Musial WJ: Oxidative stress and neutrophil activation-the two
keystones of ischemia/reperfusion injury. Int J Cardiol. 86:41–59.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eefting F, Rensing B, Wigman J, Pannekoek
WJ, Liu WM, Cramer MJ, Lips DJ and Doevendans PA: Role of apoptosis
in reperfusion injury. Cardiovasc Res. 61:414–426. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kumar KG, Trevaskis JL, Lam DD, Sutton GM,
Koza RA, Chouljenko VN, Kousoulas KG, Rogers PM, Kesterson RA,
Thearle M, et al: Identification of adropin as a secreted factor
linking dietary macronutrient intake with energy homeostasis and
lipid metabolism. Cell Metab. 8:468–481. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lovren F, Pan Y, Quan A, Singh KK, Shukla
PC, Gupta M, Al-Omran M, Teoh H and Verma S: Adropin is a novel
regulator of endothelial function. Circulation 122 (11 Suppl).
S185–S192. 2010. View Article : Google Scholar
|
|
7
|
Butler AA, Tam CS, Stanhope KL, Wolfe BM,
Ali MR, O'Keeffe M, St-Onge MP, Ravussin E and Havel PJ: Low
circulating adropin concentrations with obesity and aging correlate
with risk factors for metabolic disease and increase after gastric
bypass surgery in humans. J Clin Endocrinol Metab. 97:3783–3791.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Celik A, Balin M, Kobat MA, Erdem K,
Baydas A, Bulut M, Altas Y and Aydin S and Aydin S: Deficiency of a
new protein associated with cardiac syndrome X; called adropin.
Cardiovasc Ther. 31:174–178. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu L, Fang J, Chen L, Zhao Z, Luo Y, Lin C
and Fan L: Low serum adropin is associated with coronary
atherosclerosis in type 2 diabetic and non-diabetic patients. Clin
Chem Lab Med. 52:751–758. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang C, DeMars KM, Hawkins KE and
Candelario-Jalil E: Adropin reduces paracellular permeability of
rat brain endothelial cells exposed to ischemia-like conditions.
Peptides. 81:29–37. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tanaka M, Ito H, Adachi S, Akimoto H,
Nishikawa T, Kasajima T, Marumo F and Hiroe M: Hypoxia induces
apoptosis with enhanced expression of Fas antigen messenger RNA in
cultured neonatal rat cardiomyocytes. Circ Res. 75:426–433. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Singla DK, Singla RD and McDonald DE:
Factors released from embryonic stem cells inhibit apoptosis in
H9c2 cells through PI3K/Akt but not ERK pathway. Am J Physiol Heart
Circ Physiol. 295:H907–H913. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ogata Y, Takahashi M, Ueno S, Takeuchi K,
Okada T, Mano H, Ookawara S, Ozawa K, Berk BC, Ikeda U, et al:
Antiapoptotic effect of endothelin-1 in rat cardiomyocytes in
vitro. Hypertension. 41:1156–1163. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Scarabelli TM, Knight R, Stephanou A,
Townsend P, Chen-Scarabelli C, Lawrence K, Gottlieb R, Latchman D
and Narula J: Clinical implications of apoptosis in ischemic
myocardium. Curr Probl Cardiol. 31:181–264. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pchejetski D, Kunduzova O, Dayon A, Calise
D, Seguelas MH, Leducq N, Seif I, Parini A and Cuvillier O:
Oxidative stress-dependent sphingosine kinase-1 inhibition mediates
monoamine oxidase A-associated cardiac cell apoptosis. Circ Res.
100:41–49. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fu J, Huang H, Liu J, Pi R, Chen J and Liu
P: Tanshinone IIA protects cardiac myocytes against oxidative
stress-triggered damage and apoptosis. Eur J Pharmacol.
568:213–221. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Song JQ, Teng X, Cai Y, Tang CS and Qi YF:
Activation of Akt/GSK-3beta signaling pathway is involved in
intermedin(1–53) protection against myocardial apoptosis induced by
ischemia/reperfusion. Apoptosis. 14:1299–1307. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Frangogiannis NG, Smith CW and Entman ML:
The inflammatory response in myocardial infarction. Cardiovasc Res.
53:31–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Marx N, Neumann FJ, Ott I, Gawaz M, Koch
W, Pinkau T and Schömig A: Induction of cytokine expression in
leukocytes in acute myocardial infarction. J Am Coll Cardiol.
30:165–170. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kumar D and Jugdutt BI: Apoptosis and
oxidants in the heart. J Lab Clin Med. 142:288–297. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kumar D, Lou H and Singal PK: Oxidative
stress and apoptosis in heart dysfunction. Herz. 27:662–668. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao ZQ: Oxidative stress-elicited
myocardial apoptosis during reperfusion. Curr Opin Pharmacol.
4:159–165. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liang R, Zhao Q, Jian G, Cheng D, Wang N,
Zhang G and Wang F: Tanshinone IIA attenuates contrast-induced
nephropathy via Nrf2 activation in rats. Cell Physiol Biochem.
46:2616–2623. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kong Y, Yin J, Cheng D, Lu Z, Wang N, Wang
F and Liang M: Antithrombin III attenuates AKI following acute
severe pancreatitis. Shock. 49:572–579. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ovize M, Baxter GF, Di Lisa F, Ferdinandy
P, Garcia-Dorado D, Hausenloy DJ, Heusch G, Vinten-Johansen J,
Yellon DM and Schulz R; Working Group of Cellular Biology of Heart
of European Society of Cardiology, : Postconditioning and
protection from reperfusion injury: Where do we stand? Position
paper from the working group of cellular biology of the heart of
the European Society of Cardiology. Cardiovasc Res. 87:406–423.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hausenloy DJ and Yellon DM: New directions
for protecting the heart against ischaemia-reperfusion injury:
Targeting the reperfusion injury salvage kinase (RISK)-pathway.
Cardiovasc Res. 61:448–460. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu M, Feng J, Lucchinetti E, Fischer G,
Xu L, Pedrazzini T, Schaub MC and Zaugg M: Ischemic
postconditioning protects remodeled myocardium via the PI3K-PKB/Akt
reperfusion injury salvage kinase pathway. Cardiovasc Res.
72:152–162. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Armstrong SC: Protein kinase activation
and myocardial ischemia/reperfusion injury. Cardiovasc Res.
61:427–436. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mullonkal CJ and Toledo-Pereyra LH: Akt in
ischemia and reperfusion. J Invest Surg. 20:195–203. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jeong JJ, Ha YM, Jin YC, Lee EJ, Kim JS,
Kim HJ, Seo HG, Lee JH, Kang SS, Kim YS and Chang KC: Rutin from
Lonicera japonica inhibits myocardial ischemia/reperfusion-induced
apoptosis in vivo and protects H9c2 cells against hydrogen
peroxide-mediated injury via ERK1/2 and PI3K/Akt signals in vitro.
Food Chem Toxicol. 47:1569–1576. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lacerda L, Somers S, Opie LH and Lecour S:
Ischaemic postconditioning protects against reperfusion injury via
the SAFE pathway. Cardiovasc Res. 84:201–208. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lecour S: Activation of the protective
survivor activating factor enhancement (SAFE) pathway against
reperfusion injury: Does it go beyond the RISK pathway? J Mol Cell
Cardiol. 47:32–40. 2009. View Article : Google Scholar : PubMed/NCBI
|