|
1
|
Domper Arnal MJ, Ferrández Arenas Á and
Lanas Arbeloa Á: Esophageal cancer: Risk factors, screening and
endoscopic treatment in Western and Eastern countries. World J
Gastroenterol. 21:7933–7943. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brown CS, Gwilliam N, Kyrillos A, Lutfi W,
Lapin B, Kim KW, Krantz SB, Howington JA, Yao K and Ujiki MB:
Predictors of pathologic upstaging in early esophageal
adenocarcinoma: Results from the national cancer database. Am J
Surg. 216:124–130. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen M, Huang J, Zhu Z, Zhang J and Li K:
Systematic review and meta-analysis of tumor biomarkers in
predicting prognosis in esophageal cancer. BMC Cancer. 13:5392013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Darlavoix T, Seelentag W, Yan P, Bachmann
A and Bosman FT: Altered expression of CD44 and DKK1 in the
progression of Barrett's esophagus to esophageal adenocarcinoma.
Virchows Arch. 454:629–637. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Smith CM, Watson DI, Michael MZ and Hussey
DJ: MicroRNAs, development of Barrett's esophagus, and progression
to esophageal adenocarcinoma. World J Gastroenterol. 16:531–537.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Leidner RS, Ravi L, Leahy P, Chen Y,
Bednarchik B, Streppel M, Canto M, Wang JS, Maitra A, Willis J, et
al: The MicroRNAs, MiR-31 and MiR-375, as candidate markers in
Barrett's esophageal carcinogenesis. Genes Chromosomes Cancer.
51:473–479. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41:D991–D995. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Z, Cheng Y, Abraham JM, Yan R, Liu X,
Chen W, Ibrahim S, Schroth GP, Ke X, He Y and Meltzer SJ: RNA
sequencing of esophageal adenocarcinomas identifies novel fusion
transcripts, including NPC1-MELK, arising from a complex
chromosomal rearrangement. Cancer. 123:3916–3924. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene Ontology: Tool for the unification of biology The gene
ontology consortium. Nat Genetics. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang DW, Sherman BT, Tan Q, Collins JR,
Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC and Lempicki
RA: The DAVID gene functional classification tool: A novel
biological module-centric algorithm to functionally analyze large
gene lists. Genome Biol. 8:R1832007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Langfelder P and Horvath S: Fast R
functions for robust correlations and hierarchical clustering. J
Stat Softw. 46:i112012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin H and Zelterman D: Modeling survival
data: Extending the cox model. J Am Stat Assoc. 44:85–86. 2002.
|
|
17
|
Wang Q, Ma C and Kemmner W: Wdr66 is a
novel marker for risk stratification and involved in
epithelial-mesenchymal transition of esophageal squamous cell
carcinoma. BMC Cancer. 13:1372013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Thomas A, Klein MS, Stevens AP, Reinders
Y, Hellerbrand C, Dettmer K, Gronwald W, Oefner PJ and Reinders J:
Changes in the hepatic mitochondrial and membrane proteome in mice
fed a non-alcoholic steatohepatitis inducing diet. J Proteomics.
80:107–122. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zehmer JK, Bartz R, Liu P and Anderson RG:
Identification of a novel N-terminal hydrophobic sequence that
targets proteins to lipid droplets. J Cell Sci. 121:1852–1860.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu CC, MacCoss MJ, Mardones G, Finnigan C,
Mogelsvang S, Yates JR 3rd and Howell KE: Organellar proteomics
reveals golgi arginine dimethylation. Mol Biol Cell. 15:2907–2919.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
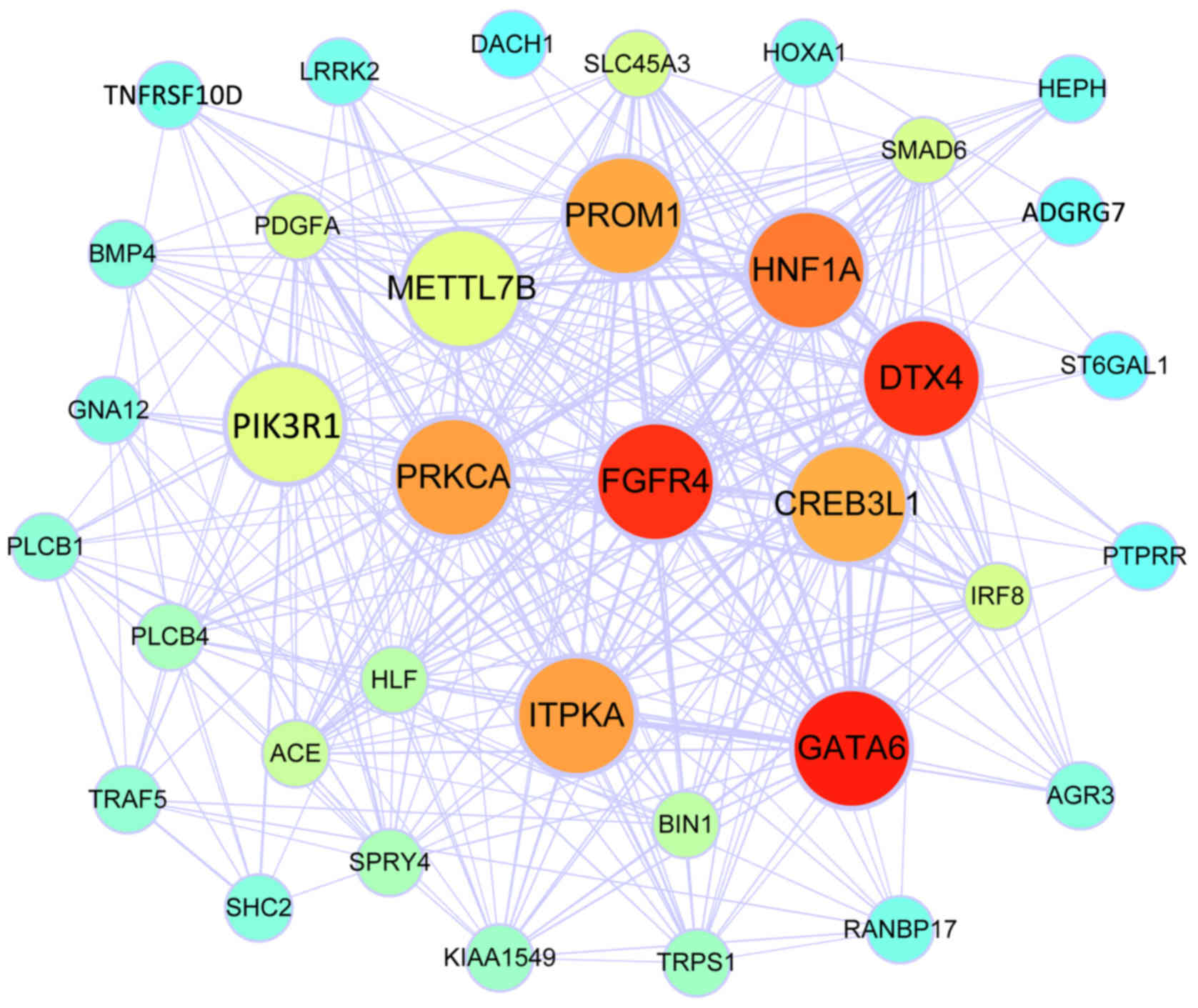
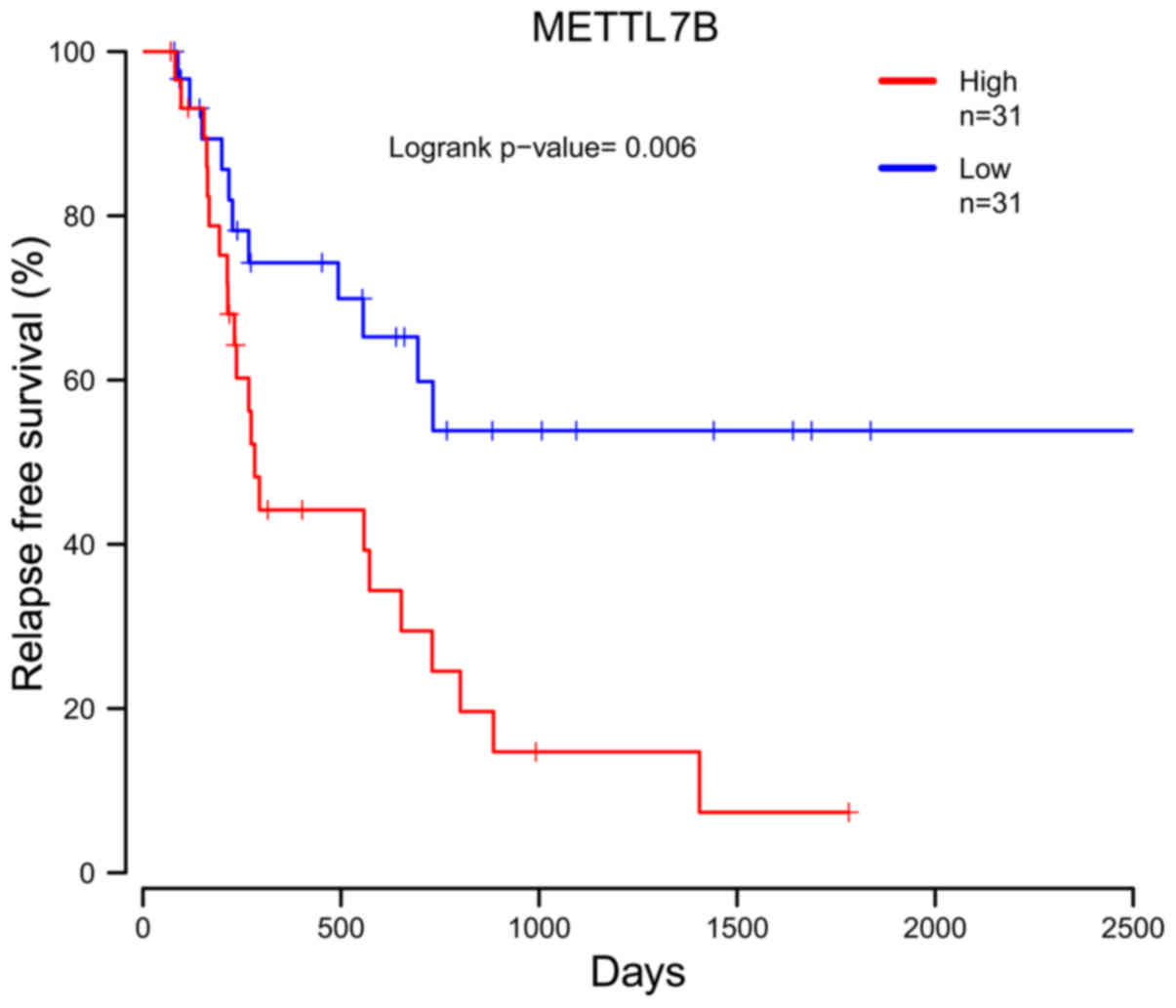
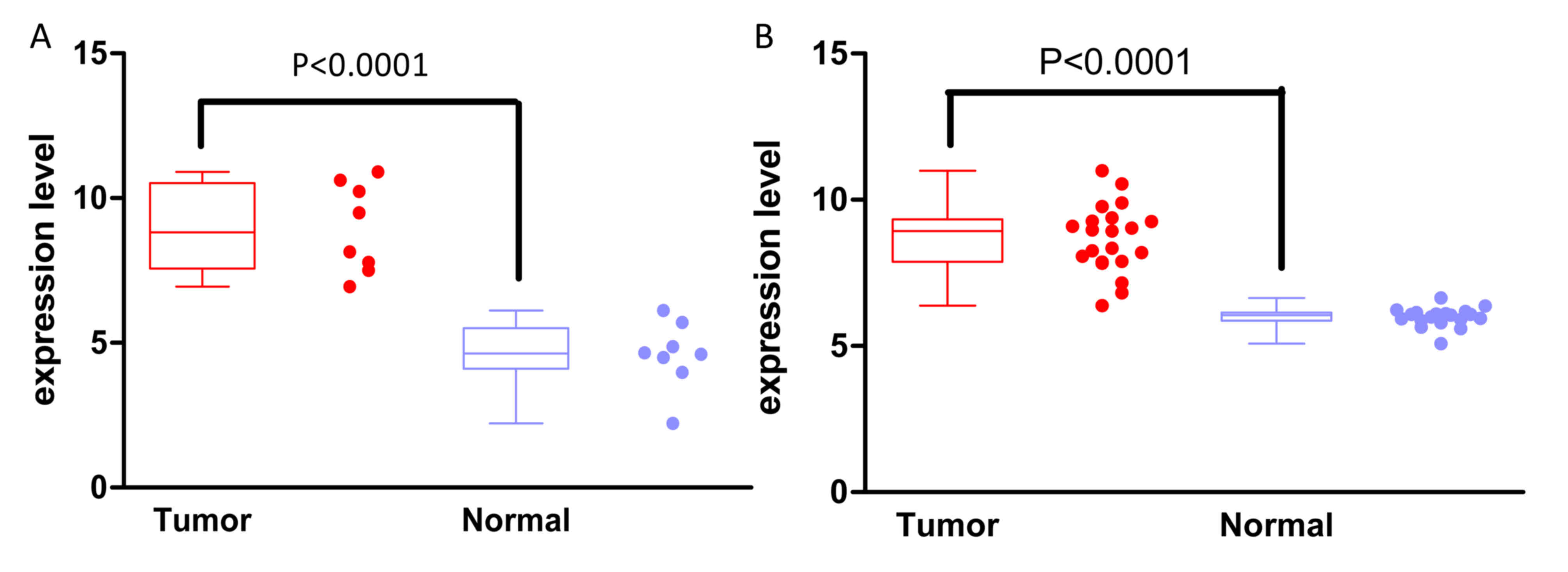
21
|
McKinnon CM and Mellor H: The tumor
suppressor RhoBTB1 controls Golgi integrity and breast cancer cell
invasion through METTL7B. BMC Cancer. 17:1452017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fan W, Sun L, Zhou JQ, Zhang C, Qin S,
Tang Y, Liu Y, Lin SS and Yuan ST: Marsdenia tenacissima extract
induces G0/G1 cell cycle arrest in human esophageal carcinoma cells
by inhibiting mitogen-activated protein kinase (MAPK) signaling
pathway. Chin J Nat Med. 13:428–437. 2015.PubMed/NCBI
|
|
23
|
Onwuegbusi BA, Rees JR, Lao-Sirieix P and
Fitzgerald RC: Selective loss of TGF beta smad-dependent signalling
prevents cell cycle arrest and promotes invasion in oesophageal
adenocarcinoma cell lines. Plos One. 2:e1772007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Connor CA, Adriaens M, Pierini R, Johnson
IT and Belshaw NJ: Procyanidin induces apoptosis of esophageal
adenocarcinoma cells via JNK activation of c-Jun. Nutr Cancer.
66:335–341. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sutter AP, Maaser K, Barthel B and
Scherübl H: Ligands of the peripheral benzodiazepine receptor
induce apoptosis and cell cycle arrest in oesophageal cancer cells:
Involvement of the p38MAPK signalling pathway. Br J Cancer.
89:564–572. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hu JL, Xiao L, Li ZY, Wang Q, Chang Y and
Jin Y: Upregulation of HO-1 is accompanied by activation of p38MAPK
and mTOR in human oesophageal squamous carcinoma cells. Cell Biol
Int. 37:584–592. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Menon J, Doebele RC, Gomes S, Bevilacqua
E, Reindl KM and Rosner MR: A Novel interplay between Rap1 and PKA
regulates induction of angiogenesis in prostate cancer. Plos One.
7:e498932012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Banerjee R, Russo N, Liu M, Van Tubergen E
and D'Silva NJ: Rap1 and its regulatory proteins: The tumor
suppressor, oncogene, tumor suppressor gene axis in head and neck
cancer. Small GTPases. 3:192–197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang YL, Wang RC, Cheng K, Ring BZ and Su
L: Roles of Rap1 signaling in tumor cell migration and invasion.
Cancer Biol Med. 14:90–99. 2017. View Article : Google Scholar : PubMed/NCBI
|