Introduction
Alzheimer's disease (AD) is a progressive
neurodegenerative disorder that accounts for 60–70% of patients
with dementia (1). While the
neuropathological mechanisms of AD remain mostly unknown, they are
associated with the accumulation of neurofibrillary tangles and
senile plaques that accelerate oxidative stress and decrease
cholinergic activity in the brain, which in turn impair memory and
cognitive function (2,3). Thus, restoration of cholinergic
function in the brain has been suggested to be a standard strategy
for delaying the progression of the disease (3,4). Several
acetylcholinesterase (AChE) inhibitors have been approved to manage
the symptoms of mild AD and are associated with various adverse
effects, including nausea, diarrhea, anorexia, vomiting, and
hepatic toxicity (3,5). Moreover, no medication has been
approved to treat patients with memory deficits. Therefore,
traditional herbs may be a valuable resource for treating memory
impairment with fewer side effects.
In East Asia, including Korea, root of ginseng
(Panax ginseng Meyer) has been prescribed for centuries to
tonify ‘qi’. It is also used to treat wide range of
diseases. Although the bioactive compounds found in this root are
also distributed in other parts of ginseng (i.e., berry, stem,
leaf, and flower), the berry has often been regarded as a ‘useless
by-product’ and discarded during the process of ginseng root
production. However, accumulated evidence suggests that ginseng
berry contains high levels of ginsenosides (6,7) and has
more potent pharmacological activities than the root (6,8,9). In particular, it has been reported that
the ginseng berry has beneficial effects in decreasing blood
glucose (9), sensitizing insulin
signaling (10), inhibiting
adipogenesis (11), reducing blood
coagulation (12), enhancing blood
circulation (6,8), ameliorating cisplatin-induced
nephrotoxicity (13) and
acetaminophen-induced hepatotoxicity (14), and inhibiting dextran sodium
sulfate-induced colitis (15).
Despite its potential beneficial effects on diverse diseases, the
effects of ginseng berry against amnesia remain poorly
understood.
Thus, the present study aimed to examine the in
vitro neuroprotective effects of ginseng berry aqueous extract
(GBE) against oxidative stress and to explore the in vivo
anti-amnesic effects of GBE in a murine model of scopolamine
(SCP)-induced memory impairment. Tacrine
(9-amino-1,2,3,4-tetrahydroacridine hydrochloride) is a prototypic
cholinesterase inhibitor that increases cholinergic transmission at
synapses (3). Tacrine has been shown
to improve cognitive function in experimental animal models of AD
(16,17). To evaluate the therapeutic effects of
GBE, tacrine was used as a positive reference drug. Furthermore,
the effects of GBE on the cholinergic nervous system, mRNA
expression of memory-related genes, and antioxidant activities were
further examined to understand its role in memory impairment.
Materials and methods
Quantification of ginsenoside Re
GBE were prepared and supplied from Aribio Co.,
Ltd., as previously reported (12).
Ginsenoside Re was purchased from ChemFaces (Wuhan, China).
Ginsenoside Re concentration in GBE was quantified using
high-performance liquid chromatography (HPLC) (Agilent 1100,
Agilent Technologies) equipped with reversed phase column (Capcell
Pak C18 UG120, 4.6×250 mm, 5 µm; Shiseido) and diode array detector
system (Agilent Technologies). GBE was dissolved in 60% of
methanol, and ginsenoside Re in 100% of methanol. GBE and
ginsenoside Re were eluted using 20–30% acetonitrile gradient
solution containing 0.01% of phosphoric acid. Ginsenoside Re was
detected at the wavelength of 203 nm. Ginsenoside Re in GBE was
quantified according to peak area and retention time.
Cell viability assay
HT-22 cells, a murine normal hippocampal neuronal
cell line, were obtained from Millipore and maintained at 37°C with
5% CO2. After HT-22 cells (1×104 cells/well)
were grown for 24 h, the cells were treated with GBE (0.01–100
µg/ml) for 72 h to examine the effect of GBE on the viability of
HT-22 cells. In another experiments, GBE-pretreated HT-22 cells
(0.01–100 µg/ml, 0.5 h) were subsequently exposed to 5 mM of
glutamate or 500 µM of hydrogen peroxide
(H2O2) for 12 h. Cell viability was measured
at wavelength of 450 nm using EZ-Cytox cell viability assay kit
(Daeil Labservice) and automated microplate reader (VersaMax™;
Molecular Devices). The cell viability was calculated as relative
to the untreated control cells.
Animal husbandry and treatment
C57BL/6NCrljOri mice (n=120; male, 18–21 g, 6 weeks
old) were supplied from Orient Bio, Inc., maintained with a supply
of filtered pathogen-free air, and provided with standard rodent
chow (Purinafeed) and water ad libitum at standard condition
(temperature, 20–25°C; light/dark cycle, 12/12 h; relative
humidity, 40–45%). After 1 week of acclimatization, mice were
divided into 6 groups (n=20/group); vehicle, SCP, SCP + tacrine 10
mg/kg, SCP + GBE 400 mg/kg, SCP + GBE 200 mg/kg, and SCP + GBE 100
mg/kg. GBE or tacrine dissolving in distilled water was
administered orally once a day for 28 days. To induce memory
impairment, 1 mg/kg of SCP dissolving in sterilized saline was
intraperitoneally injected three times 1 h after GBE (or tacrine)
administration on days 6, 13 and 27. Instead of GBE and SCP,
vehicle group was given an equal volume of distilled water and
saline. And SCP group administered distilled water to induce same
stresses.
Measurement of body weight
To reduce the differences from feeding, all mice
were fasted for 12 h (water was not) at initiation of first GBE (or
tacrine) administration. Body weight was measured on days −1, 0, 1,
13, 20, 27 and 28 using an automatic electronic balance (XB320M;
Precisa Instrument).
Passive Avoidance Test
The step-through passive avoidance test (n=10/group)
was conducted, as previously described (16,18).
After the last SCP injection, each mouse was placed in the
light/noise compartment 30 min for training mice. Light and noise
was applied until the mouse escaped into the neighboring grid floor
compartment. An electric shock (3.0 mA) for 3 sec was applied to
the grid floor. A retention test was performed 24 h after training
test using the same condition, and the moving time from the
light/noise compartment to the grid floor was recorded as a
step-through latency time.
Morris water maze test
The Morris water maze test (n=10/group) was carried
out using separated animals from the passive avoidance test, as
previously reported (19). For
training, each mouse was placed in circular pool (diameter, 100 cm;
depth, 27 cm; temperature, 22°C) 30 min after the last SCP
injection, and began to find the submerged escape platform
(diameter, 10 cm) in one of the pool quadrants. When the mouse was
successfully placed on the platform, it was allowed to remain on
the platform for 10 sec. However, if the mouse was failed to find
the platform within 150 sec, the mouse was placed on the platform
for 10 sec. A retention test was performed 24 h after training
test, and the escape latency time from the water to the escape
platform was recorded using a video tracking system (Smart
junior).
Isolation of total RNA and RT-PCR
After the passive avoidance test, the mouse was
anesthetized under inhalation anesthesia with 2–3% of isoflurane
(Hana Pharm. Co.), euthanized by cervical dislocation, and the
hippocampus (n=10/group) was collected. Total RNA was isolated
using Trizol reagent (Invitrogen), and then reacted with
recombinant DNase I (Ambion) to remove contaminated DNA. cDNA was
synthesized using oligo-dT16 primer and High-Capacity
cDNA Reverse Transcription Kit (Applied Biosystems). RT-PCR was
carried out using an ABI Step One Plus Sequence Detection System
(Applied Biosystems). Gene specific primer pairs were used as
followed: Choline acetyltransferase (ChAT) forward
5′-CTTGGATGGTCCAGGCAC-3′, backward 5′-GTCATACCAACGATTCGCTCC-3′;
brain-derived neurotrophic factor (BDNF) forward
5′-GACAAGGCAACTTGGCCTAC-3′, backward 5′-CTGTCACACACGCTCAGCTC-3′;
phosphoinositide 3-kinase (PI3K) forward
5′-TCCAAATACCAGCAGGATCA-3′, backward 5′-ATGCTTCGATAGCCGTTCTT-3′;
Akt forward 5′-TACTCATTCCAGACCCACGA-3′, backward
5′-GAGGTTCTCCAGCTTCAGGT-3′; extracellular signal-regulated kinase 1
(ERK1) forward 5′-TGGCTTTCTGACGGAGTATG-3′, backward
5′-GGTCCAGGTAGTGCTTGC-3′; ERK2 forward 5′-CCTCAAGCCTTCCAACCTC-3′,
backward 5′-GCCCACAGACCAAATATCAATG-3′; cAMP response element
binding protein (CREB) forward 5′-TACCCAGGGAGGAGCAATAC-3′, backward
5′-GAGGCAGCTTGAACAACAAC-3′; Ca2+/calmodulin-dependent
protein kinase (CaMK) IV forward 5′-AAATCAGCCTGGTCCTTGAG-3′,
backward 5′-GAAGCATTTGCGGTGCACGATG-3′; β-actin forward
5′-GCTGAGAGGGAAATCGTGCGT-3′, backward 5′-TCTGGTTTGAGGTCACGATG-3′.
Expression level of β-actin mRNA was used as endogenous control,
and the relative expression level of specific gene was calculated
by 2−ΔΔCq (20).
Quantification of acetylcholine (ACh)
and measurement of AChE activity
The hippocampus (n=10/group) was homogenized in
ice-cold 0.01 M Tris-HCl (pH 7.4) using bead beater (Taco™ Prep;
GeneResearch Biotechnology Corp.) and ultrasonic cell disruptor
(KS-750; Madell Technology Corp.), and then centrifuged at 12,000 ×
g for 15 min. ACh concentration and AChE activity in the
hippocampal homogenates were measured using an Amplex Red ACh/AChE
assay kit (Invitrogen), according to manufacturer's instruction.
Fluorescence intensities at 560 nm (excitation wavelength) and 590
nm (emission wavelength) were detected using an automated
microplate reader (Infinite 200 Pro; Tecan).
Measurement of antioxidant activities
in the cerebral cortex
After mouse that had performed the Morris water maze
test was euthanized by cervical dislocation under inhalation
anesthesia (2–3% of isoflurane), the cerebral cortex (n=10/group)
was collected, homogenized with a buffer consisting of 10 mM
sucrose, 10 mM Tris-HCl (pH 7.4), and 0.1 M
ethylenediaminetetraacetic acid, and then clarified by
centrifugation. Protein concentration of tissue homogenates was
determined using bovine serum albumin as a standard protein. Lipid
peroxidation was measured at 525 nm of wavelength by quantifying
concentration of malondialdehyde using a thiobarbituric acid, and
was expressed as malondialdehyde (ng) per tissue (g). After
homogenates were precipitated by 25% of trichloroacetic acid,
reduced glutathione (GSH) contents in resulting supernatants were
determined at 412 nm of wavelength using a 2-nitrobenzoic acid, and
were expressed as nM/mg protein. Decomposition of
H2O2 by tissue homogenates was measured at
240 nm. One unit of catalase (CAT) activity was defined as the
amount of enzyme required to degrade 1 nM of
H2O2 for 1 min. After homogenates were
reacted with nitroblue tetrazolium to form formazan, absorbance at
560 nm was measured for determining superoxide dismutase (SOD)
activity. One unit of SOD activity was defined as the amount of
enzyme that can reduce initial absorbance of nitroblue tetrazolium
by 50% for 1 min. Specific activity was expressed as U/mg
protein.
Statistical analysis
All numerical values are expressed as the mean ±
standard deviation (SD) (n=6 for in vitro assay; n=10 or 20
for in vivo assay). One-way ANOVA was used to assess the
significance among experimental groups, followed by Tukey's honest
significance difference or Dunnett's T3 as post hoc
analysis. P values less than 0.05 were considered as statistical
differences of significance.
Results
GBE prevents oxidative stress-mediated
cytotoxicity in HT-22 cells
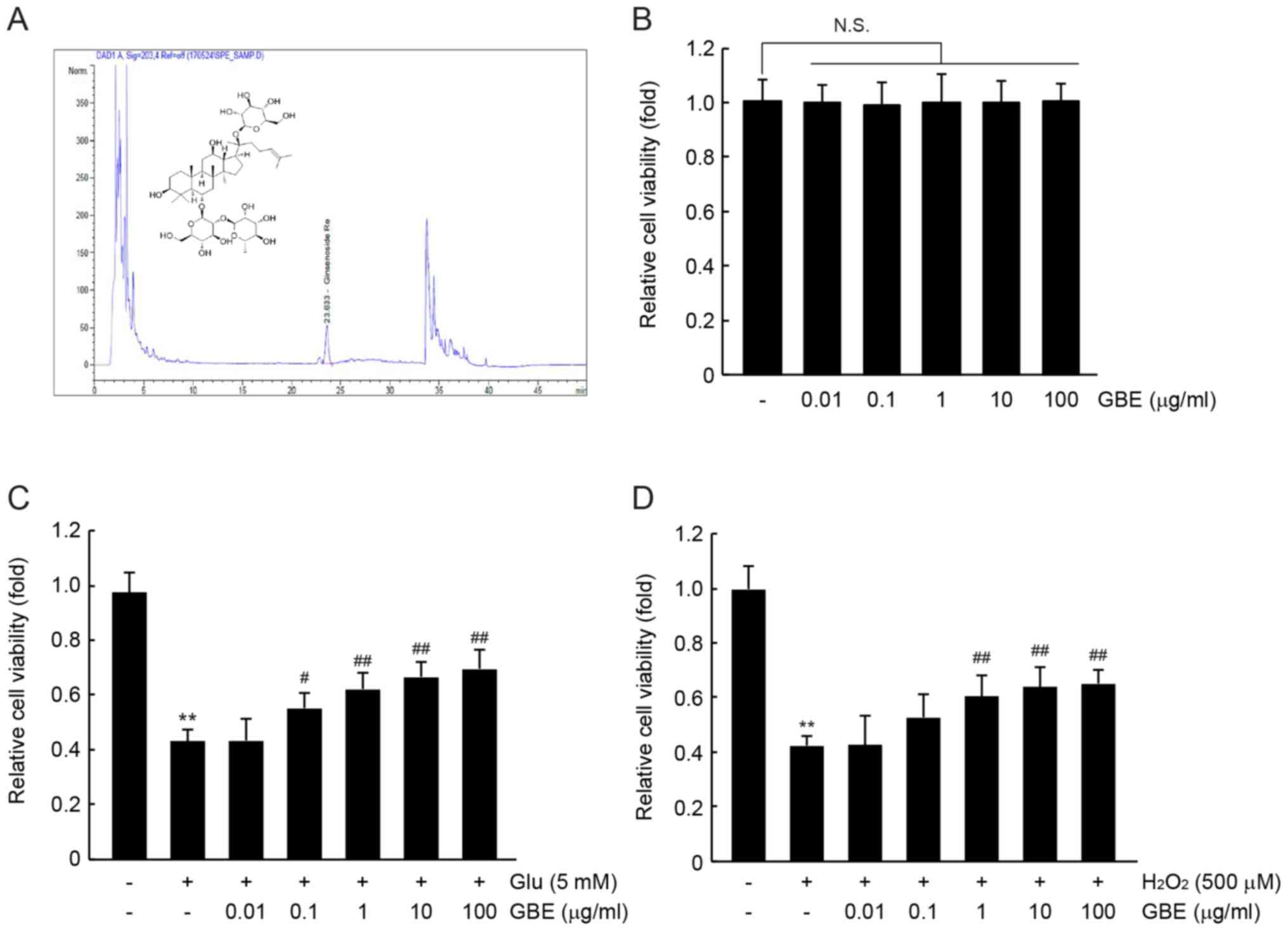
Ginsenoside Re is one of the most abundant
ginsenosides found in the ginseng berry (12,21).
Before investigating the beneficial effects of GBE, we quantified
the concentration of ginsenoside Re in GBE using HPLC under
optimized conditions. HPLC analysis indicated that the GBE used in
this study contained 29.13±0.15 mg/g of ginsenoside Re (Fig. 1A). Next, we examined whether GBE
exerted cytotoxic effects against HT-22 cells. Using a cell
viability assay, no statistical differences were found when HT-22
cells were exposed to up to 100 µg/ml of GBE for 72 h (Fig. 1B). To explore the cytoprotective
effects of GBE, GBE-pretreated HT-22 cells were continuously
exposed to 5 mM of glutamate or 500 µM of
H2O2, representative oxidative stressors of
neuronal cells, for 12 h. As expected, treatment with glutamate or
H2O2 alone significantly decreased the cell
viability of HT-22 cells (P<0.01; Fig. 1C and D). However, pretreatment with
GBE tended to increase cell viability in a concentration-dependent
manner. Compared with glutamate-treated cells, a significant
difference in cell viability was observed in cells pretreated with
0.1–100 µg/ml of GBE (P<0.05, 0.01 µg/ml GBE; P<0.01, 1–100
µg/ml GBE; Fig. 1C). Relative cell
viability after treatment with glutamate, glutamate + 0.01 µg/ml
GBE, glutamate + 0.1 µg/ml GBE, glutamate + 1 µg/ml GBE, glutamate
+ 10 µg/ml GBE, and glutamate + 100 µg/ml GBE was 43.0±4.4,
43.2±8.0, 55.0±5.5, 62.0±6.3, 66.5±5.7 and 69.5±7.2% of control
cells, respectively. In addition, 1–100 µg/ml of GBE significantly
reduced H2O2-mediated cytotoxicity in HT-22
cells (P<0.01; Fig. 1D). Relative
cell viability after treatment with H2O2,
H2O2 + 0.01 µg/ml GBE,
H2O2 + 0.1 µg/ml GBE,
H2O2 + 1 µg/ml GBE,
H2O2 + 10 µg/ml GBE, and
H2O2 + 100 µg/ml GBE was 42.3±3.4, 42.5±1.1,
52.7±8.4, 60.3±8.0, 63.7±7.6 and 65.0±4.9% of control cells,
respectively. These results suggest that GBE can protect neuronal
cells from oxidative stress in vitro.
GBE mitigates SCP-induced memory
impairment in mice
The hippocampus is involved in cognitive function
including memory, learning, and emotion. To expand the findings
that GBE can protect hippocampal neuronal cells from oxidative
stress in vitro, we tested its effects in vivo using
a SCP-induced memory impairment murine model. C57BL/6NCrljOri mice
(6 weeks old, male) were orally administered one of three dosages
of GBE (100–400 mg/kg) or tacrine (10 mg/kg; positive control) once
per day for 28 days (i.e., initial GBE administration on day 0). To
induce memory impairment, SCP was intraperitoneally injected three
times, 1 h after GBE administration on days 6, 13, and 27 (Fig. 2A). All mice experienced a decrease in
body weight on day 0 due to an overnight fasting period of 12 h,
and body weight gradually increased during the 28-days of the
experimental period. There were no significant differences in body
weight among the experimental groups (Fig. 2B). Two behavior tests were conducted
to investigate the neuroprotective effects associated with the
hippocampus. In the retention trial, the passive avoidance test
indicated that SCP decreased the step-through latency time compared
with the vehicle-treated control group (P<0.01). However,
administration of all three dosages of GBE or tacrine significantly
prevented the decrease in step-through latency time by SCP
(P<0.01). Specifically, the increase in step-through latency
time in mice treated with 400 mg/kg of GBE was significantly higher
than in mice treated with tacrine (P<0.05; Fig. 2C). Similarly, SCP injection resulted
in increased latency time in the Morris water maze (P<0.01), and
treatment with the three dosages of GBE significantly decreased the
escape time (P<0.01). There were no statistical differences of
latency time between mice administered GBE vs. tacrine
(Fig. 2D). Collectively, these
results suggest that GBE can improve cognitive function in mice
with SCP-induced memory impairment.
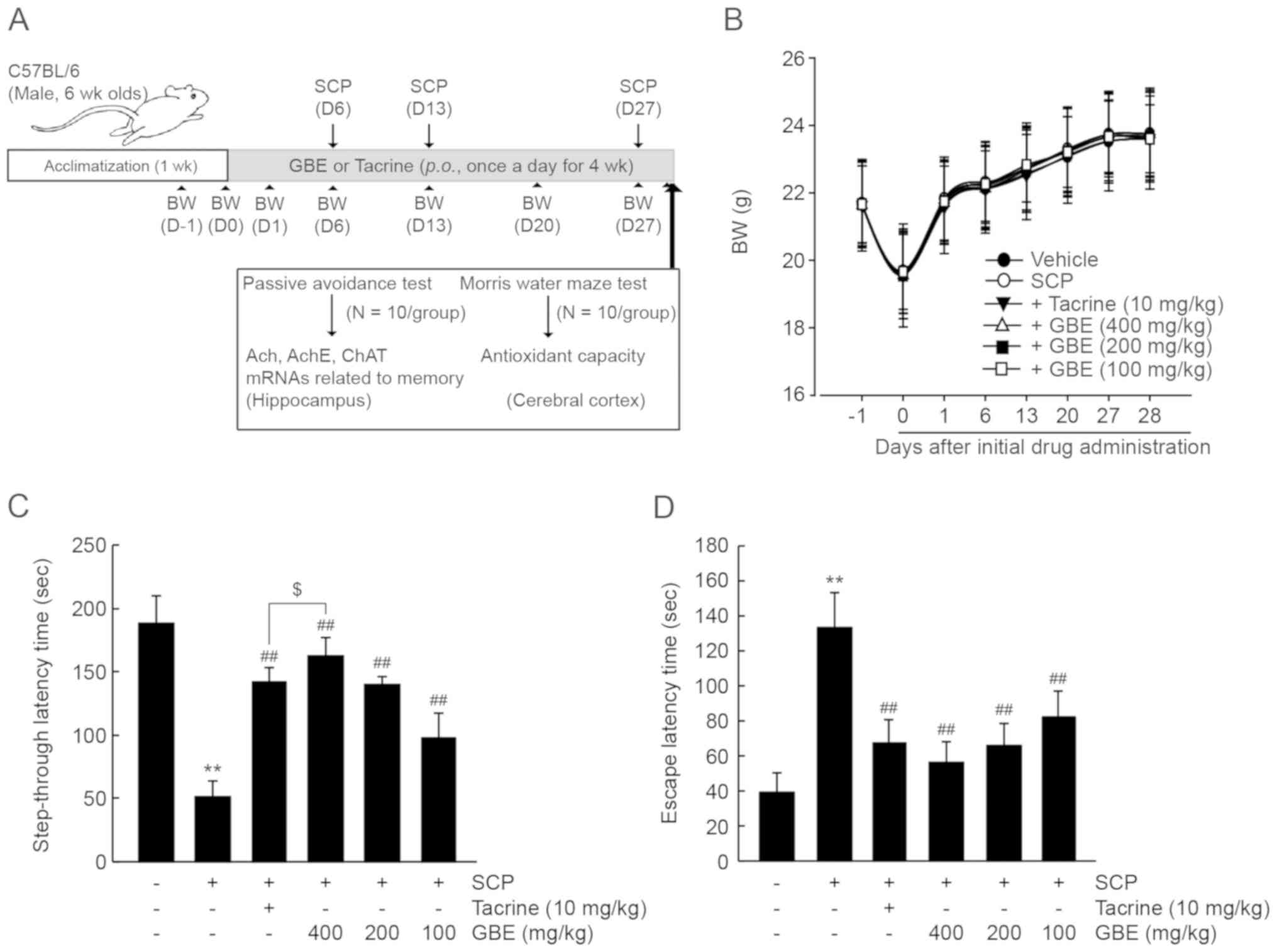 | Figure 2.GBE prevents SCP-induced memory
impairment in mice. (A) In vivo experimental designs for
drug treatment, behavior testing, and biochemical analysis; 100–400
mg/kg of GBE or 10 mg/kg of tacrine was orally administered once
per day for 4 weeks, and SCP was injected three times 1 h after
GBE/tacrine administration on days 6, 13 and 27. (B) Mouse BW. All
mice were fasted for 12 h before the first GBE/tacrine
administration. BW (n=20/group) was measured on days −1, 0, 1, 13,
20, 27 and 28 after the initial drug administration. (C) Passive
avoidance test. The time from the light/noise compartment to the
electric grid floor for each individual mouse was considered a
measure of step-through latency time (n=10/group). (D) Morris water
maze test. The time from the water to the submerged platform for
each individual mouse considered a measure of escape latency time
(n=10/group). All values are expressed as the mean ± SD.
**P<0.01 vs. vehicle-treated group; ##P<0.01 vs.
SCP-injected group; $P<0.05 vs. tacrine-treated
group). BW, body weight; GBE, ginseng berry aqueous extract; SCP,
scopolamine; SD, standard deviation. |
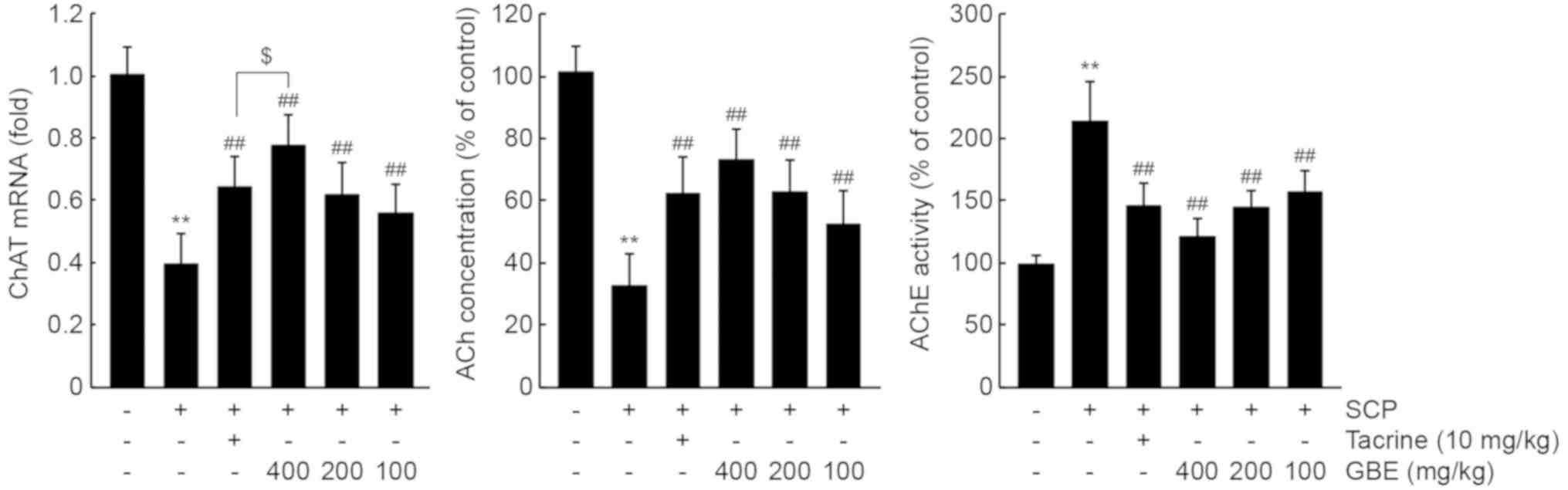
GBE enhances cholinergic nervous
system functioning in the hippocampus
To investigate whether GBE prevents memory
impairment by enhancing cholinergic signaling in the hippocampus,
we measured mRNA levels of ChAT, which produces ACh from acetyl-CoA
and choline. As expected, SCP injection significantly decreased
ChAT mRNA levels (P<0.01). However, all three dosages of GBE or
tacrine administration prevented this reduction in hippocampal ChAT
mRNA (P<0.01). Specifically, ChAT mRNA levels in mice treated
with 400 mg/kg of GBE were higher than those in mice treated with
10 mg/kg of tacrine (P<0.05; Fig.
3, left). In addition, GBE administration significantly
alleviated the reduction of Ach by SCP in the hippocampus
(P<0.01; Fig. 3, middle).
Moreover, the increase in hippocampal AChE activity after SCP
injection was significantly decreased following GBE administration
with all three dosages (P<0.01; Fig.
3, right). ACh concentration and AChE activity after treatment
with the three dosages of GBE were comparable to those after
treatment with tacrine.
GBE increases mRNA levels of
memory-related genes in the hippocampus
BDNF is a representative neurotrophic factor
associated with formation and storage of memory that activates CREB
through transducing cellular signaling involving PI3K, Akt, ERK1,
ERK2, and CaMK VI (22–24). To examine the effects of GBE on mRNA
levels of memory-related genes, RT-PCR analysis was conducted using
hippocampal RNAs. SCP significantly decreased mRNA levels of BDNF,
PI3K, Akt, ERK1, ERK2, CaMK VI, and CREB (P<0.01; Table I). Administration of 200 and 400
mg/kg of GBE prevented the reduction of mRNA levels of all genes
related to memory in the hippocampus (PI3K in 200 mg/kg GBE:
P<0.05; P<0.01 for the others), while treatment with 100
mg/kg of GBE only changed BDNF, Akt, and ERK1 mRNA levels compared
with the SCP-injected group (Akt: P<0.05; BDNF and ERK1:
P<0.01). Moreover, the mRNA levels of PI3K, Akt, and ERK2 in the
400 mg/kg GBE group were significantly higher than those in the
tacrine group (PI3K: P<0.05; Akt and ERK2: P<0.01; Table I).
 | Table I.The effect of GBE administration on
mRNA levels related to memory in SCP-induced memory-impaired
mice. |
Table I.
The effect of GBE administration on
mRNA levels related to memory in SCP-induced memory-impaired
mice.
| Group | BDNF | PI3K | Akt | ERK1 | ERK2 | CaMK IV | CREB |
|---|
| Vehicle | 1.01±0.13 | 1.01±0.10 | 1.00±0.11 | 0.99±0.07 | 1.02±0.09 | 1.01±0.14 | 0.99±0.12 |
| SCP |
0.37±0.10a |
0.30±0.11a |
0.38±0.08a |
0.30±0.09a |
0.28±0.09a |
0.31±0.12a |
0.33±0.09a |
| + Tacrine 10
mg/kg |
0.63±0.13b |
0.46±0.10c |
0.60±0.10b |
0.53±0.06b |
0.53±0.10b |
0.51±0.08b |
0.55±0.13b |
| + GBE 400
mg/kg |
0.75±0.11b |
0.67±0.14b,e |
0.80±0.12b,d |
0.71±0.15b |
0.72±0.12b,d |
0.62±0.13b |
0.69±0.14b |
| + GBE 200
mg/kg |
0.65±0.11b |
0.47±0.13c |
0.60±0.13b |
0.54±0.07b |
0.52±0.10b |
0.50±0.07b |
0.54±0.09b |
| + GBE 100
mg/kg |
0.56±0.08b | 0.42±0.08 |
0.55±0.11c |
0.48±0.07b | 0.39±0.08 | 0.44±0.05 | 0.46±0.10 |
GBE increases antioxidant activity in
the cerebral cortex
To investigate whether GBE increases antioxidant
capacity in vivo, we first measured the level of
malondialdehyde (a marker of lipid peroxidation) in the cerebral
cortex. SCP injection significantly increased the level of
malondialdehyde (P<0.01), showing that SCP promoted lipid
peroxidation in the brain. By contrast, administration of all three
doses of GBE or tacrine significantly reduced lipid peroxidation
(P<0.01). The decrease in lipid peroxidation after treatment
with the three doses of GBE was comparable to that by tacrine
(Fig. 4A). In addition, SCP
decreased GSH levels and CAT and SOD activities in the cerebral
cortex (P<0.01), and administration of GBE or tacrine
significantly prevented the decreases in GSH levels and CAT and SOD
activity (CAT activity in tacrine: P<0.05; P<0.01 for the
others). In contrast, the decrease in CAT activity was not
prevented in the group following administration with 100 mg/kg of
GBE. GSH and SOD activity levels in mice treated with 400 mg/kg of
GBE were higher than those in mice treated with tacrine (P<0.05;
Fig. 4B-D).
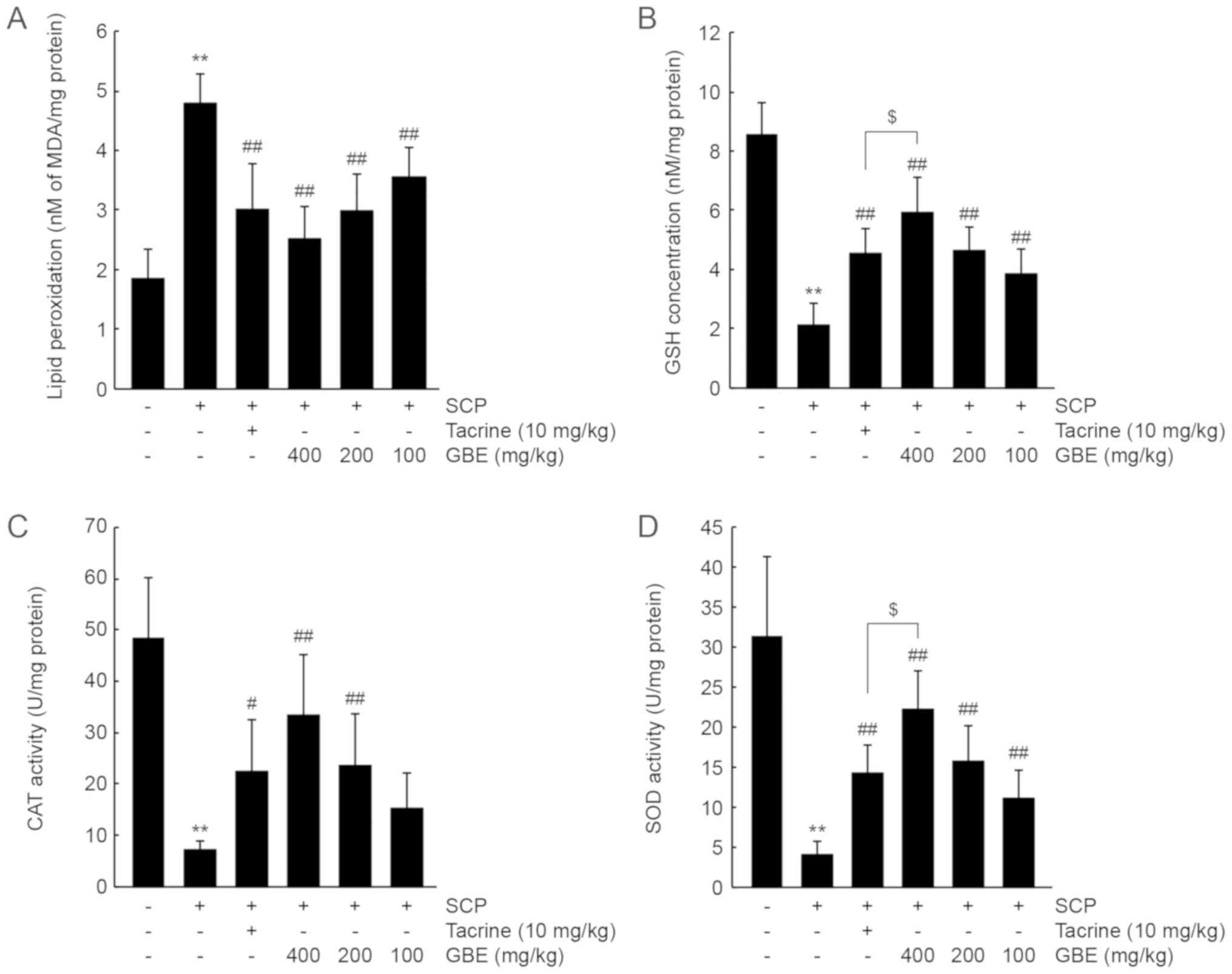 | Figure 4.GBE increases antioxidant activity in
the SCP-injected mice. (A) Lipid peroxidation, (B) GSH
concentration, and (C) CAT and (D) SOD activities in the cerebral
cortex were determined from mice that had performed the Morris
water maze test. All values are expressed as the mean ± SD of 10
mice. **P<0.01 vs. vehicle-treated group;
##P<0.01, #P<0.05 vs. SCP-injected
group; $P<0.05 vs. tacrine-treated group. CAT,
catalase; GBE, ginseng berry aqueous extract; GSH, glutathione;
MDA, malondialdehyde; SCP, scopolamine; SD, standard deviation;
SOD, superoxide dismutase. |
Discussion
It has been reported that ginseng root, its enriched
fractions, and isolated bioactive compounds can attenuate
SCP-induced memory impairment in experimental animals (16,18,25–30).
Protection of the cholinergic nervous system via antioxidant and
anti-inflammatory actions is associated with the pharmacological
effect of ginseng root in animals with SCP-induced amnesia
(16,25,26,28).
However, the effects of ginseng berries on memory impairment remain
poorly understood. Although similar beneficial effects to those of
ginseng root have been reported in other parts of the P.
ginseng plant (6,8,9), certain
bioactive compounds are selectively distributed throughout P.
ginseng (21). Before
investigating the anti-amnesic effect of GBE, we performed an HPLC
analysis to assess the quality of GBE and found that GBE used in
the present study contained 29.13±0.15 mg/g of ginsenoside Re.
Lee et al (2017) reported that 29 of the 58
ginsenosides previously tested are found in ginseng berry
methanolic extract (21).
Ginsenosides F1 and Rg4 are found only in the berry at low
concentrations, while ginsenoside Re, Rg2, F4, Rg5, malonylated Rd,
malonylated Rb2, malonylated Rc, and malonylated Rb1 are
concentrated in berry compared to other parts of the ginseng plant
(21). Moreover, previous results
from different groups have shown that ginsenoside Re, Rf, Rb1, Rc,
Rb2, and Rd are contained in ginseng berry aqueous extract
(12), which is the identical to the
extract used in this study. Ginsenoside Re is found to be the most
abundant ginsenoside in ginseng berry (12,21).
Although there is no direct evidence that ginsenoside Re
ameliorates SCP-induced amnesia, an accumulation of evidence
suggests that ginsenoside Re enhances cognitive function through
the reduction of amyloid β (31,32).
Additionally, other ginsenosides found in GBE have been reported to
exhibit neuroprotective activities (33–35).
Therefore, ginsenoside Re and other unidentified compounds in GBE
are thought to contribute cooperatively to mitigating cognitive
deficits caused by SCP. Further studies are needed to identify the
major compounds involved in GBE-mediated neuroprotection.
In the present study, SCP (1 mg/kg) was
intraperitoneally injected three times in mice to induce memory
loss. SCP is a nonselective antagonist of the muscarinic ACh
receptor, which reduces cholinergic transmission in the central
nervous system and causes cognitive dysfunction, including
long-term memory loss (36).
Therefore, SCP is one of the most extensively used neurotoxins to
elucidate possible therapeutic agents for modulating memory
impairment (16,18,26–30). To
study the neuroprotective effects of GBE related to memory, we
conducted two behavioral tests after SCP injection. The passive
avoidance test has been performed to investigate non-spatial
long-term memory after an aversive experience (37). In parallel with previous reports
(16,18,26,29), SCP
significantly decreased the step-through latency time. However, GBE
administration significantly prolonged the step-through latency
time in a dose-dependent manner. The Morris water maze test is
another behavioral test used to assess long-term and spatial memory
in the hippocampus (19,38). In the present study, SCP injection
significantly increased the latency time to the escape platform in
the water maze, whereas GBE mitigated the SCP-induced spatial
memory impairment as shown by a decreased escape time.
ACh, which is synthesized by ChAT, is a critical
neurotransmitter for regulating cognitive function. In addition,
memory loss by SCP is closely correlated with an increase in AChE
(an ACh-hydrolyzing enzyme in the cholinergic synaptic cleft)
activity and a subsequent reduction in ACh in the hippocampus
(26,27). To confirm whether GBE restores
long-term memory via the cholinergic nervous system, we further
measured hippocampal levels of ACh and its metabolizing enzymes.
SCP increased AChE activity and decreased ACh and ChAT mRNA levels
in the hippocampus, which was consistent with previous reports
(26,27). However, GBE administration
significantly reduced these changes. Therefore, these results
suggest that GBE attenuates SCP-induced memory deficit by enhancing
cholinergic signaling in the hippocampus.
BDNF is a neurotrophic factor that contributes to
neuronal plasticity and synaptic transmission (23). BDNF enhances memory function by
promoting the formation of long-term potentiation and by
up-regulating the sensitivity of N-methyl-D-aspartate receptors in
the hippocampus (22,39,40).
Binding of BDNF to tropomyosin receptor kinase B (TrkB) facilitates
dimerization and phosphorylation of TrkB tyrosine residues, and
recruits diverse Src homology 2 adaptor molecules, including
phospholipase C, insulin receptor substrate, and Shc (23,41).
Phospholipase C activates Ca2+-dependent calmodulin
kinase by increasing intracellular Ca2+ levels. In
addition, insulin receptor substrate and Shc activate the
phosphoinositide 3-kinase-dependent Akt and Ras/Raf/MEK-dependent
ERK signaling pathways (23).
Specifically, the BDNF signaling cascades regulate CREB, which is
an essential transcription factor necessary for late-stage of
long-term potentiation and long-term memory formation in the
hippocampus (42,43). The present results demonstrated that
SCP significantly decreased BDNF mRNA levels and its related
signaling molecules, and the administration of GBE prevented these
reductions in the hippocampus. Although GBE may restore long-term
memory by upregulating the expression of BDNF-related signaling
molecules in the hippocampus, further studies on the covalent
modifications (e.g., phosphorylation) of signaling molecules are
needed to establish the role of the GBE in signaling pathway
related to memory.
We also showed that GBE protected HT-22 cells from
glutamate- and H2O2-induced cytotoxicity.
Because of its low endogenous antioxidant capacity, an abundance of
polyunsaturated fatty acids, and high oxygen consumption rate, the
brain is one of the most susceptible organs to oxidative stress
(44,45). Therefore, oxidative stress is
important in the progression of neurodegenerative diseases,
including AD (44). It has been
reported that ginseng protects the brain from oxidative stress via
activation of nuclear factor-E2 related factor 2 (Nrf2) (16,25,46).
Moreover, genetic deletion of Nrf2 abolishes the protective effect
of compound K (a major aglycosylated metabolite of
protopanaxadiol-type ginsenosides) against SCP-induced amnesia
(16). In the resting state, Nrf2
resides in the cytoplasm by binding to Kelch-like ECH-associated
protein 1 and is autonomously degraded by the ubiquitin-dependent
proteasome system. Antioxidants as well as oxidative stress disturb
protein-protein interaction between Nrf2 and Kelch-like
ECH-associated protein 1, and translocate Nrf2 into the nucleus.
Nuclear Nrf2 heterodimerizes with Jun or small Maf and subsequently
binds to the antioxidant response elements of many antioxidant
genes, including γ-glutamate cysteine ligase (a rate-limiting
enzyme of GSH synthesis), CAT, or SOD (47). In this study, SCP injection increased
lipid peroxidation, decreased GSH levels, and reduced CAT and SOD
activity, which is in agreement with previous observations that SCP
provokes oxidative stress in the brain (26,48). GBE
administration inhibited lipid peroxidation significantly and
elevated the levels of GSH while also elevating CAT and SOD
activities in a concentration-dependent manner. Therefore, these
results suggest that GBE may attenuate SCP-induced memory
impairment, potentially by protecting neurons from oxidative
stress. However, further studies investigating the detailed
cellular mechanisms involved, including Nrf2 activation, are
warranted.
In this study, we compared the pharmacological
effect of GBE with tacrine. Although some statistical results
(e.g., step-through latency time, GSH level, SOD activity, and
ChAT, PI-3K, Akt, and ERK2 mRNA levels) showed that GBE (400 mg/kg)
was more potent than tacrine, most of the anti-amnesic effects of
GBE (200–400 mg/kg) were comparable to those of tacrine (10 mg/kg).
In conclusion, the results of this study suggest that GBE
alleviates SCP-induced memory impairment by restoring the
cholinergic nervous system, mRNA expression of BDNF-related
signaling molecules, and antioxidant capacity. Therefore, ginseng
berry may be a promising complementary medicine for managing
various neurodegenerative disorders with memory impairment.
Acknowledgments
Not applicable.
Funding
The current study was supported by the National
Research Foundation of Korea (NRF) funded by Korean government
(MSIP) (grant no. 2018R1A5A2025272).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SKK designed the study. JRH, YSC, JKK and SKK
conducted the research. JRH, IJC and SKK analyzed the data. JRH,
IJC and SKK co-wrote the manuscript. IJC and SKK had primary
responsibility for the final content. All authors approved the
final draft of the manuscript.
Ethics approval and consent to
participate
Animal experiments were conducted according to the
national regulations regarding the use and welfare of laboratory
animals and were approved by the Institutional Animal Care and Use
Committee in Daegu Haany University (approval no. DHU2017-081).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing of
interests.
Glossary
Abbreviations
Abbreviations:
|
ACh
|
acetylcholine
|
|
AChE
|
acetylcholine esterase
|
|
AD
|
Alzheimer's disease
|
|
BDNF
|
brain derived neurotropic factor
|
|
CaMK
|
Ca2+/calmodulin-dependent
protein kinase
|
|
CAT
|
catalase
|
|
ChAT
|
choline acetyltransferase
|
|
CREB
|
cAMP response element binding
protein
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
GBE
|
ginseng berry extract
|
|
GSH
|
glutathione
|
|
H2O2
|
hydrogen peroxide
|
|
HPLC
|
high-performance liquid
chromatography
|
|
Nrf2
|
nuclear factor-E2 related factor 2
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
SCP
|
scopolamine
|
|
SD
|
standard deviation
|
|
SOD
|
superoxide dismutase
|
|
TrkB
|
tropomyosin receptor kinase B
|
References
|
1
|
Burns A and Iliffe S: Alzheimer's disease.
BMJ. 338:b1582009. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Golde TE: Disease modifying therapy for
AD? J Neurochem. 99:689–707. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Francis PT, Palmer AM, Snape M and Wilcock
GK: The cholinergic hypothesis of Alzheimer's disease: A review of
progress. J Neurol Neurosurg Psychiatry. 66:137–147. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ibach B and Haen E: Acetylcholinesterase
inhibition in Alzheimer's Disease. Curr Pharm Des. 10:231–251.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rountree SD, Chan W, Pavlik VN, Darby EJ,
Siddiqui S and Doody RS: Persistent treatment with cholinesterase
inhibitors and/or memantine slows clinical progression of Alzheimer
disease. Alzheimers Res Ther. 1:72009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim CK, Cho DH, Lee KS, Lee DK, Park CW,
Kim WG, Lee SJ, Ha KS, Goo Taeg O, Kwon YG and Kim YM: Ginseng
berry extract prevents atherogenesis via anti-inflammatory action
by upregulating phase II gene expression. Evid Based Complement
Alternat Med. 2012:4903012012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim YK, Yoo DS, Xu H, Park NI, Kim HH,
Choi JE and Park SU: Ginsenoside content of berries and roots of
three typical Korean ginseng (Panax ginseng) cultivars. Nat
Prod Commun. 4:903–906. 2009.PubMed/NCBI
|
|
8
|
In-Ho C, Byung-Woo K, Yun-Jae P, Han-Joo
L, Sok P and Namju L: Ginseng berry extract increases nitric oxide
level in vascular endothelial cells and improves cGMP expression
and blood circulation in muscle cells. J Exerc Nutrition Biochem.
22:6–13. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dey L, Xie JT, Wang A, Wu J, Maleckar SA
and Yuan CS: Anti-hyperglycemic effects of ginseng: Comparison
between root and berry. Phytomedicine. 10:600–605. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Seo E, Kim S, Lee SJ, Oh BC and Jun HS:
Ginseng berry extract supplementation improves age-related decline
of insulin signaling in mice. Nutrients. 7:3038–3053. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang SO, Park HR, Sohn ES, Lee SW, Kim HD,
Kim YC, Kim KH, Na SW, Choi HK, Arasu MV and Kim YO: Classification
of ginseng berry (Panax ginseng C.A. MEYER) extract using 1H
NMR spectroscopy and its inhibition of lipid accumulation in 3T3-L1
cells. BMC Complement Altern Med. 14:4552014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim MH, Lee J, Jung S, Kim JW, Shin JH and
Lee HJ: The involvement of ginseng berry extract in blood flow via
regulation of blood coagulation in rats fed a high-fat diet. J
Ginseng Res. 41:120–126. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ma ZN, Liu Z, Wang Z, Ren S, Tang S, Wang
YP, Xiao SY, Chen C and Li W: Supplementation of American ginseng
berry extract mitigated cisplatin-evoked nephrotoxicity by
suppressing ROS-mediated activation of MAPK and NF-κB signaling
pathways. Food Chem Toxicol. 110:62–73. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu XY, Wang Z, Ren S, Leng J, Hu JN, Liu
Z, Chen C and Li W: Improved protective effects of American ginseng
berry against acetaminophen-induced liver toxicity through
TNF-α-mediated caspase-3/-8/-9 signaling pathways. Phytomedicine.
51:128–138. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang W, Xu L, Cho SY, Min KJ, Oda T,
Zhang L, Yu Q and Jin JO: Ginseng berry extract attenuates dextran
sodium sulfate-induced acute and chronic colitis. Nutrients.
8:1992016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Seo JY, Ju SH, Oh J, Lee SK and Kim JS:
Neuroprotective and cognition-enhancing effects of compound K
isolated from red ginseng. J Agric Food Chem. 64:2855–2864. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maurice T, Lockhart BP and Privat A:
Amnesia induced in mice by centrally administered beta-amyloid
peptides involves cholinergic dysfunction. Brain Res. 706:181–193.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim J, Kim SH, Lee DS, Lee DJ, Kim SH,
Chung S and Yang HO: Effects of fermented ginseng on memory
impairment and β-amyloid reduction in Alzheimer's disease
experimental models. J Ginseng Res. 37:100–107. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Morris R: Development of a water maze
procedure for studying spatial learning in the rat. J Neurosci
Methods. 11:47–60. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee JW, Choi BR, Kim YC, Choi DJ, Lee YS,
Kim GS, Baek NI, Kim SY and Lee DY: Comprehensive profiling and
quantification of ginsenosides in the root, stem, leaf, and berry
of Panax ginseng by UPLC-QTOF/MS. Molecules. 22:E21472017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nakajo Y, Miyamoto S, Nakano Y, Xue JH,
Hori T and Yanamoto H: Genetic increase in brain-derived
neurotrophic factor levels enhances learning and memory. Brain Res.
1241:103–109. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cunha C, Brambilla R and Thomas KL: A
simple role for BDNF in learning and memory? Front Mol Neurosci.
3:12010.PubMed/NCBI
|
|
24
|
Martin SJ, Grimwood PD and Morris RG:
Synaptic plasticity and memory: An evaluation of the hypothesis.
Annu Rev Neurosci. 23:649–711. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang Q, Lin J, Zhang H, Liu Y, Kan M, Xiu
Z, Chen X, Lan X, Li X, Shi X, et al: Ginsenoside compound K
regulates amyloid β via the Nrf2/Keap1 signaling pathway in mice
with scopolamine hydrobromide-induced memory impairments. J Mol
Neurosci. 67:62–71. 2019.PubMed/NCBI
|
|
26
|
Lu C, Lv J, Dong L, Jiang N, Wang Y, Wang
Q, Li Y, Chen S, Fan B, Wang F and Liu X: Neuroprotective effects
of 20(S)-protopanaxatriol (PPT) on scopolamine-induced cognitive
deficits in mice. Phytother Res. 32:1056–1063. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim J, Shim J, Lee S, Cho WH, Hong E, Lee
JH, Han JS, Lee HJ and Lee KW: Rg3-enriched ginseng extract
ameliorates scopolamine-induced learning deficits in mice. BMC
Complement Altern Med. 16:662016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xu T, Shen X, Yu H, Sun L, Lin W and Zhang
C: Water-soluble ginseng oligosaccharides protect against
scopolamine-induced cognitive impairment by functioning as an
antineuroinflammatory agent. J Ginseng Res. 40:211–219. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Peña ID, Yoon SY, Kim HJ, Park S, Hong EY,
Ryu JH, Park IH and Cheong JH: Effects of ginseol k-g3, an
Rg3-enriched fraction, on scopolamine-induced memory impairment and
learning deficit in mice. J Ginseng Res. 38:1–7. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jin SH, Park JK, Nam KY, Park SN and Jung
NP: Korean red ginseng saponins with low ratios of protopanaxadiol
and protopanaxatriol saponin improve scopolamine-induced learning
disability and spatial working memory in mice. J Ethnopharmacol.
66:123–129. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li J, Liu Y, Li W, Wang Z, Guo P, Li L and
Li N: Metabolic profiling of the effects of ginsenoside Re in an
Alzheimer's disease mouse model. Behav Brain Res. 337:160–172.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen F, Eckman EA and Eckman CB:
Reductions in levels of the Alzheimer's amyloid beta peptide after
oral administration of ginsenosides. FASEB J. 20:1269–1271. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Du Y, Fu M, Wang YT and Dong Z:
Neuroprotective effects of ginsenoside Rf on amyloid-β-induced
neurotoxicity in vitro and in vivo. J Alzheimers Dis. 64:309–322.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu JF, Yan XD, Qi LS, Li L, Hu GY, Li P
and Zhao G: Ginsenoside Rd attenuates Aβ25-35-induced oxidative
stress and apoptosis in primary cultured hippocampal neurons. Chem
Biol Interact. 239:12–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang Q, Sun LH, Jia W, Liu XM, Dang HX,
Mai WL, Wang N, Steinmetz A, Wang YQ and Xu CJ: Comparison of
ginsenosides Rg1 and Rb1 for their effects on improving
scopolamine-induced learning and memory impairment in mice.
Phytother Res. 24:1748–1754. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Klinkenberg I and Blokland A: The validity
of scopolamine as a pharmacological model for cognitive impairment:
A review of animal behavioral studies. Neurosci Biobehav Rev.
34:1307–1350. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lorenzini CA, Baldi E, Bucherelli C,
Sacchetti B and Tassoni G: Role of dorsal hippocampus in
acquisition, consolidation and retrieval of rat's passive avoidance
response: A tetrodotoxin functional inactivation study. Brain Res.
730:32–39. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Barnes CA, Danysz W and Parsons CG:
Effects of the uncompetitive NMDA receptor antagonist memantine on
hippocampal long-term potentiation, short-term exploratory
modulation and spatial memory in awake, freely moving rats. Eur J
Neurosci. 8:565–571. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Madara JC and Levine ES: Presynaptic and
postsynaptic NMDA receptors mediate distinct effects of
brain-derived neurotrophic factor on synaptic transmission. J
Neurophysiol. 100:3175–3184. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Korte M, Carroll P, Wolf E, Brem G,
Thoenen H and Bonhoeffer T: Hippocampal long-term potentiation is
impaired in mice lacking brain-derived neurotrophic factor. Proc
Natl Acad Sci USA. 92:8856–8860. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Segal RA: Selectivity in neurotrophin
signaling: Theme and variations. Annu Rev Neurosci. 26:299–310.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bozon B, Kelly A, Josselyn SA, Silva AJ,
Davis S and Laroche S: MAPK, CREB and zif268 are all required for
the consolidation of recognition memory. Philos Trans R Soc Lond B
Biol Sci. 358:805–814. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Korte M, Kang H, Bonhoeffer T and Schuman
E: A role of BDNF in the late-phase of hippocampal long-term
potentiation. Neuropharmacology. 37:553–559. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Singh A, Kukreti R, Saso L and Kukreti S:
Oxidative stress: A key modulator in neurodegenerative diseases.
Molecules. 24:E15832019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Olanow CW: An introduction to the free
radical hypothesis in Parkinson's disease. Ann Neurol. 32
(Suppl):S2–S9. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu L, Vollmer MK, Ahmad AS, Fernandez VM,
Kim H and Doré S: Pretreatment with Korean red ginseng or dimethyl
fumarate attenuates reactive gliosis and confers sustained
neuroprotection against cerebral hypoxic-ischemic damage by an
Nrf2-dependent mechanism. Free Radic Biol Med. 131:98–114. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sivandzade F, Prasad S, Bhalerao A and
Cucullo L: NRF2 and NF-κB interplay in cerebrovascular and
neurodegenerative disorders: Molecular mechanisms and possible
therapeutic approaches. Redox Biol. 21:1010592019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lee MR, Yun BS, Park SY, Ly SY, Kim SN,
Han BH and Sung CK: Anti-amnesic effect of Chong-Myung-Tang on
scopolamine-induced memory impairments in mice. J Ethnopharmacol.
132:70–74. 2010. View Article : Google Scholar : PubMed/NCBI
|