Introduction
Sepsis is a systemic inflammatory response syndrome
(SIRS) caused by infection, which is an excessive inflammatory
response as a result of the uncontrolled release of inflammatory
mediators (1). In SIRS and secondary
tissue damage, cytokines are released into the circulation in a
dysregulated manner, causing hemodynamic instability, extensive
tissue damage and fatal multiple organ dysfunction (2). The number of organ failures in patients
with sepsis is significantly associated with mortality (3). The kidney is one of the most vulnerable
organs in the body to sepsis (4).
Acute kidney injury (AKI) is the most common and serious
complication of sepsis and it is often used as an independent risk
factor for predicting mortality (1).
According to research statistics, in the intensive care unit, ~42%
of patients with sepsis are afflicted with different degrees of AKI
(5). AKI is a clinical syndrome that
results in severe tubular damage with mortality rates ranging
between 24 and 62% (6). In the case
of AKI, the mortality rate of sepsis can reach 70% due to a lack of
effective therapy strategies (7,8).
The pathogenesis of septic AKI is closely associated
with renal hemodynamic abnormalities, inflammatory injury,
apoptosis and adaptive mechanisms. During adaptation, the host
reduces its sensitivity to inflammation-induced tissue damage which
mainly affects metabolism, resists injury, guides tissue repair and
promotes organ recovery by altering cell signaling pathways
(9). In particular, the main cause
of septic AKI is the lipopolysaccharide (LPS)-mediated apoptosis of
renal tubular epithelial cells (RTEC) (10). LPS is a component of the outer
membrane of Gram-negative bacteria that is involved in the
pathogenesis of sepsis-induced AKI (11,12). LPS
has the ability to stimulate severe inflammatory reactions,
frequently resulting in the release of a large number of
inflammatory cytokines, including tumor necrosis factor-α (TNF-α),
interleukin (IL)-1β and IL-6. The subsequent inflammatory reaction
can in turn lead to oxidative stress, mitochondrial damage and
energy depletion, and eventually the apoptosis of RTEC. It has been
previously reported that kidney injury in AKI animal models can be
markedly ameliorated by reducing the intensity of inflammatory
reactions; therefore, effective removal of the inflammatory
mediators, including TNF-α, IL-15 and IL-6, as an effective means
of treating septic AKI (13,14).
RTEC death in septic AKI is manifested as necrosis,
apoptosis and autophagy. Cell necrosis is a cell lysis process
typically due to pathology or trauma, apoptosis is a cell death
mechanism occurring in an orderly, controlled manner, while
autophagy is a metabolic process of cell self-defense that is
distinct from apoptosis, and is important for the turnover of
intracellular substances in eukaryotes (15). Autophagy is a process in which cells
form autophagic lysosomes to degrade their own damaged organelles
such as mitochondria, and other macromolecules. Autophagy can
satisfy cellular metabolic requirements and the renewal of
organelles, and is an important regulatory mechanism for cell
growth, differentiation and death (16). Regulation of the balance of pro- and
anti-inflammatory factors serves a pivotal role in the severity of
the inflammatory response. Studies have shown that autophagy can
antagonize apoptosis and protect RTECs from LPS-mediated damage,
and the inhibition of autophagy can aggravate LPS-mediated AKI
(17,18).
Ataxia-telangiectasia mutated (ATM) belongs to the
phosphatidyl inositol-3-kinase-like kinase family of proteins in
mammalian cells, which also includes ataxia telangiectasia and rad3
related, DNA-dependent protein kinase and mTOR. Mutated or
inactivated forms of ATM have been identified in ataxia
telangiectasia patients (19). ATM
is one of the key transducers of the DNA double-stranded break
response and serves critical roles in early signal transduction
through cell cycle checkpoints. Homologs of ATM are present in all
eukaryotic cell types examined to date, including budding and
fission yeasts (20). A previous
study suggested that persistent inflammation leads to DNA damage in
RTEC and further activation of ATM (21,22). In
addition, another study in ischemia reperfusion kidney injury
models reported that AMP-activated protein kinase (AMPK) activated
the ATM-AMPK-tuberous sclerosis complex 2 (TSC2)-mTOR pathway,
triggering autophagy (23). However,
it remains unclear whether the inflammatory reaction in septic AKI
leads to increased ATM expression or increased autophagy.
In the present study, changes in ATM expression and
levels of cell autophagy in an in vitro RTEC model of septic
AKI was assessed using lentiviral transfection to knock down ATM
expression in HK-2 cells. The results of immunofluorescence and
western blotting suggest that in septic AKI, ATM expression is
elevated, which increases autophagy in RTEC. In addition,
downregulation of ATM expression in HK-2 cells reduced the
expression levels of inflammatory factors and autophagy in
LPS-induced septic AKI cells. The aim of the current study was to
investigate the mechanism by which the inflammatory response of
septic AKI mediates RTEC damage, thus providing a new strategy for
the therapeutic intervention of septic AKI.
Materials and methods
Cell lines
The human RTEC line HK-2 was obtained from Cell
Culture Center of the Basic Institute of Medical Sciences, Peking
Union Medical College.
Cell culture and passage
The HK-2 cell line was cultured in DMEM (Gibco;
Thermo Fisher Scientific, Inc.) containing 10% fetal bovine serum
(FBS; Biochrom, Ltd.) and incubated at 37°C in a humidified
atmosphere with 5% CO2. The cells were sub-cultured at
80% confluence, which were removed from the incubator and the
original medium in the dish was discarded. Cells were rinsed using
3 ml PBS and digested by treatment with 1 ml trypsin for 1–2 min at
37°C. Digestion was terminated using 2 ml DMEM medium, and the cell
suspension was subsequently centrifuged at 450 × g for 5 min at
4°C. Supernatant was discarded and cells were resuspended in 2 ml
corresponding DMEM medium to obtain a single cell suspension. Cells
were seeded into different dishes/microplates at different
densities for subsequent experimentation, as described below.
Induction of HK-2 cell injury using
LPS
HK-2 cells were cultured under the above conditions.
At 100% confluency, cells were digested and centrifuged at 480 × g
for 8 min at room temperature. The supernatant was subsequently
discarded and the cells were resuspended in 10 ml PBS, counted and
seeded into six-well plates. At 80% confluency the cells were
washed three times in PBS and then cultured in DMEM/F12 (Gibco;
Thermo Fisher Scientific, Inc.) medium without FBS for 6 h. LPS
(Sigma-Aldrich; Merck KGaA) diluted in DMEM/F12 without antibiotics
was then added to the cells at final concentrations of 1, 10, 20
and 30 µg/ml followed by further incubation for 0, 6, 12, 24 h.
Finally, the optimal concentration (10 µg/ml) and the optimal
stimulation time (24 h) were selected for subsequent experiments.
In the control group PBS was added instead of LPS.
Cell proliferation assay
Cell proliferation was analyszd using the Cell
Counting Kit-8 (CCK-8; Dojindo Molecular Laboratories, Inc.). The
HK-2 cells in the logarithmic growth phase were seeded into a
96-well plate at a density of 2×104 cells/well.
Following incubation with different concentrations of LPS solution
(1, 10, 20 and 30 µg/ml) and whole medium (DMEM + 10% fetal bovine
serum), HK-2 cells were continuously stimulated. The control group
was treated with an equal volume of PBS, whereas the blank control
consisted of medium only with no cells. After the completion of LPS
treatment, CCK-8 solution (10 µl) was added into each well and the
mixture was incubated for 2 h. Absorbance values (OD value) at 450
nm was measured in each well using an enzyme-labeled instrument.
The results were obtained from three independent experiments in
triplicate. The OD value is considered to be directly proportional
to the number of viable cells contained in the culture system.
Inflammatory factor detection by
reverse transcription-quantitative PCR (RT-qPCR)
HK-2 cells in good condition were first collected
and cultured, and the levels of ATM, TNF-α, IL-1β and IL-6 mRNA
expression in HK-2 cells were detected using RT-qPCR. Following 24
h of LPS stimulation, total RNA was extracted using TRIzol reagent
according to the manufacturer's protocol (Invitrogen; Thermo Fisher
Scientific, Inc.). The concentration of the extracted RNA was
measured using an ultraviolet analyzer. Reverse transcription of
RNA into cDNA was performed using M-MLV reverse transcriptase
(Promega Corporation) in a reaction mix prepared according to
manufacturer's protocol (Table I),
and the resultant cDNA was stored at −80°C. The setup of a qPCR
reaction system (SYBR® Premix Ex Taq™; Takara
Biotechnology Co., Ltd.) is provided in Table II. The sequences of the primer pairs
used for RT-qPCR were as follows: ATM forward,
5′-ATAGATTGTGTAGGTTCCGATGG-3′ and reverse,
5′-CATCTTGTCTCAGGTCATCACG-3′; TNF-α forward,
5′-ACCTCTCTCTAATCAGCCCTCT-3′ and reverse,
5′-GGGTTTGCTACAACATGGGCTA-3′; IL-1β forward,
5′-GCAATGAGGATGACTTGTTCTTTG-3′ and reverse,
5′-CAGAGGTCCAGGTCCTGGAA-3′; IL-6 forward,
5′-AGCCACTCACCTCTTCAGAAC-3′ and reverse,
5′-ACATGTCTCCTTTCTCAGGGC-3′, β-actin (reference) forward,
5′-CCTGACTGACTACCTCATGAAG-3′ and reverse,
5′-GACGTAGCACAGCTTCTCCTTA-3′. The following thermocycling
conditions were used for qPCR: Initial denaturation at 95°C for 15
sec; 45 cycles of 95°C for 5 sec and 60°C for 30 sec. The
absorbance values of the fluorophores were read each time during
the extension phase. A melting curve was then prepared and an
initial denaturation of the template DNA at 95°C for 1 min after
the end of PCR. It was then cooled to 55°C to allow sufficient
binding to the DNA duplex. From 55°C to 95°C, each step was
increased by 0.5°C for 4 sec while the absorbance was being read.
The 2−ΔΔCq method (24)
was used to calculate the relative expression of ATM, TNF-α, IL-1β
and IL-6 mRNA against β-actin. All samples were tested in
triplicate.
 | Table I.RT reaction solution. |
Table I.
RT reaction solution.
| Reagent | Amount added per
reaction |
|---|
| 5X RT buffer | 4 µl |
| 10 mM dNTPs | 2 µl |
| RNasin | 0.5 µl |
| M-MLV-RT | 1 µl |
| DEPC
H2O | 3.5 µl |
| Table II.Quantitative PCR reaction system. |
Table II.
Quantitative PCR reaction system.
| Reagent | Amount added per
tube |
|---|
| SYBR premix ex
taq | 10 µl |
| ROX Reverse Dye
(50X) | 0.4 µl |
| Forard primer (2.5
µM) | 0.5 µl |
| Reverse primer (2.5
µM) | 0.5 µl |
| cDNA | 1.0 µl |
| H2O | 7.6 µl |
ELISA
Changes in expression levels of inflammatory
factors, including IL-1β, IL-6 and TNF-α, in response to LPS were
detected using ELISA kits (cat. nos. 558279, 555220 and 555212,
respectively; BD Pharmingen; BD Biosciences). Briefly, 50 µl of the
capture monoclonal antibody (mAb) was applied to the ELISA plate,
and the plate was incubated at 37°C for 1 h and washed three times
with PBS supplemented with Tween-20 (PBS-T). The plate was then
treated with 50 µl bovine serum albumin (BSA; 10 mg/ml; Proliant,
Inc.) and 50 µl FBS, followed by incubation at 37°C for 1 h. A
total of 50 µl biotin-conjugated detector mAb was added to each
well and incubated at 37°C for 1 h, followed by probing using an
avidin-horseradish peroxidase (HRP) solution (10 mg/ml). After
final rinsing with PBS-T, 50 µl tetramethylbenzidine substrate
solution (10 mg/ml) was added to start the color reaction of the
antigen-antibody complex, which was then stopped by adding 50 µl
H2SO4 (10 µmol/ml). Final absorbance at a
wavelength of 450 nm was measured using an automated ELISA reader
(Bio-Rad Laboratories, Inc.). The concentration of each cytokine
was determined by comparing the optical densities at 450 nm to a
standard curve. To further confirm the role of autophagy in HK-2
cell injury induced by LPS, HK-2 cells were pretreated with 10
mol/l 3-methyladenine (3-MA; Sigma-Aldrich; Merck KGaA) at room
temperature, followed by treatment with 10 µg/ml LPS for 30
min.
Construction of lentivirus-mediated
ATM-knockdown system
The mRNA sequence of ATM was obtained from the NCBI
database (https://www.ncbi.nlm.nih.gov/gene/472) and four shRNA
sequences were designed according to Table III, and the constructs were named
accordingly: PLVE2142, PLVE2143, PLVE2144 and PLVE2145. The
double-strand DNA oligonucleotide containing the interference
sequence was synthesized by Shanghai GeneChem Co., Ltd. and cloned
into the pLV-GFP lentiviral vector (Hanheng Biotechnology Shanghai
Co., Ltd.) using EcoRI-HF and AgeI-HF restriction
sites. The vectors were prepared using a Plasmid Minipreparation
kit (cat. no. KL060; Shanghai Kang Lang Biological Technology Co.,
Ltd.) and detected using 2% agarose gel electrophoresis with
ethidium bromide staining.
| Table III.Sequences of ATM shRNAs. |
Table III.
Sequences of ATM shRNAs.
| Primer | Sequence
(5′-3′) |
|---|
| SH-NC-F |
CCGGGAGGTCAAACCTAGAAAGCTCCTCGAGGAGCTTTCTAGGTTTGACCTCTTTTTTG |
| SH-ATM-R |
AATTCAAAAAAGAGGTCAAACCTAGAAAGCTCCTCGAGGAGCTTTCTAGGTTTGACCTC |
| SH-ATM-F1 |
CCGGGAGGTCAAACCTAGAAAGCTCCTCGAGGAGCTTTCTAGGTTTGACCTCTTTTTTG |
| SH-ATM-R1 |
AATTCAAAAAAGAGGTCAAACCTAGAAAGCTCCTCGAGGAGCTTTCTAGGTTTGACCTC |
| SH-ATM-F2 |
CCGGGCTGCAGAGTCAATCAATAGACTCGAGTCTATTGATTGACTCTGCAGCTTTTTTG |
| SH-ATM-R2 |
AATTCAAAAAAGCTGCAGAGTCAATCAATAGACTCGAGTCTATTGATTGACTCTGCAGC |
| SH-ATM-F3 |
CCGGGAGCTCTTCAGGTCTAAATCACTCGAGTGATTTAGACCTGAAGAGCTCTTTTTTG |
| SH-ATM-R3 |
AATTCAAAAAAGAGCTCTTCAGGTCTAAATCACTCGAGTGATTTAGACCTGAAGAGCTC |
| SH-ATM-F4 |
CCGGGTCATATAGGAAGTAGAGGAACTCGAGTTCCTCTACTTCCTATATGACTTTTTTG |
| SH-ATM-R4 |
AATTCAAAAAAGTCATATAGGAAGTAGAGGAACTCGAGTTCCTCTACTTCCTATATGAC |
The experiments were performed in five groups:
Negative control (NC), PLVE2142, PLVE2143, PLVE2144 and PLVE2145
groups. Using the Lipofectamine® 3000 reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to
manufacturer's protocol, DNA-liposome complexes were prepared at
4°C to a final volume of 1 µg/µl and added to HK-2 cells (1 µg/ml),
transfection was performed for 6 h at room temperature. Further
experiments were performed 48 h post-transfection.
Cellular infection with recombinant
lentiviruses expressing ATM shRNA
Cells were seeded into six-well plates at a density
of 1×105 cells/well. Once cells reached 30% confluence,
the recombinant lentiviruses PLVE2142, PLVE2143, PLVE2144 or
PLVE2145 were used to transfect HK-2 cells (8 µg/ml Polybrene;
Sigma-Aldrich; Merck KGaA). The cells were then transferred to DMEM
containing 2% horse serum (Gibco; Thermo Fisher Scientific, Inc.),
and the lentivirus:polybrene mixture was added to cells. The cells
were subsequently incubated in 37°C for 6 h before the media was
replaced with media free of lentivirus. Following 48 h of further
culture at 37°C, images of the cells were taken using a
fluorescence microscope. Western blot analysis and RT-qPCR were
used to determine the efficiency of ATM knockdown HK-2 cells, and
the appropriate multiplicity of infection of ATM lentiviral
interference vector was selected for subsequent experiments after
incubation of 24 h. After the ATM gene was silenced by shRNA (MOI,
10), HK-2 cells were stimulated with 10 µg/ml LPS for 24 h at 4°C,
following which the effects of ATM knockdown on the mRNA expression
of cellular inflammatory factors and autophagy was examined.
Immunofluorescence staining
HK-2 cells were transfected with pDSRed-LC3 the
lentivirus (MOI, 20) for 48 h and then fixed with 4%
paraformaldehyde (Sigma-Aldrich; Merck KGaA) for 15 min at room
temperature, washed three times with PBS for a total of 10 min each
and permeabilized with pre-chilled (−20°C) 70% ethanol for 20 min.
The cells were then blocked with 8% BSA diluted in PBS for 1 h and
washed with PBS. Following incubation for 2 h, transfected HK-2
cells were labeled with DAPI (5 mg/l) and incubated with pDSRed-LC3
(cat. no. 38171200626; purchased from REBIO; Shanghai Shengwu
Gongcheng Co., Ltd.) overnight at 4°C. Subsequently, the treated
cells were washed three times with PBS and incubated at room
temperature with AlexaFluor® 488-conjugated anti-rabbit
secondary antibody (dilution 1:1,000; cat. no. A-11008; Thermo
Fisher Scientific, Inc) for 1 h. The cells attached to glass slides
were removed from the 24-well plates in the dark room and placed
faced up on a blotting paper, where a drop of antifade mounting
medium (Invitrogen; Thermo Fisher Scientific, Inc.) was added.
After the cells were dried, they were imaged using a Leica TCS SP8
laser confocal microscope (Leica Microsystems, Inc.).
Western blot analysis of HK-2 cells
autophagy
Western blot analysis was performed to measure the
expression of beclin-1 and LC3-II/I, proteins associated with
autophagy (25). HK-2 cells were
lysed using RIPA lysis buffer (Teknova, Inc.) at 4°C for 10–15 min.
Protein concentration was determined using bicinchoninic acid assay
method. Equal amounts of 50 µg protein extract were separated by
10% SDS-PAGE and transferred onto polyvinylidene fluoride
membranes. The membranes were blocked with 5% nonfat milk diluted
in Tris-buffered saline containing 0.1% Tween-20 at room
temperature for 90 min. The membranes were then immunoblotted for
Beclin1 (1:1,000; cat. no. ab55878; Abcam), LC3 (1:1,000; cat. no.
ab48394; Abcam), and β-actin (1:1,000; cat. no. ab3280; Abcam) at
20–27°C for 2 h before incubation with secondary antibodies
conjugated to horseradish peroxidase-conjugated goat anti-rabbit
Immunoglobulin G (1:5,000, cat. no. ab7074; Abcam) at 37°C for 20
min and visualized with enhanced chemiluminescence reagent
(Vazyme). Immunoreactive bands were analyzed using Image Pro Plus
6.0 software (Media Cybernetics, Inc.).
Statistical analysis
Plots were constructed using GraphPad Prism 7
(GraphPad Software, Inc.), and the statistical analysis was
performed using SPSS 19.0 (IBM Corp.) software. Normally
distributed data are presented as the mean ± SD. The non-normally
distributed data are presented as the mean ± interquartile range or
as the median. One-way and two-way ANOVA were used for comparisons
between groups, and the Bonferroni test was used for multiple
comparisons. P<0.05 was considered to indicate a statistically
significant difference. All experiments were repeated three
times.
Results
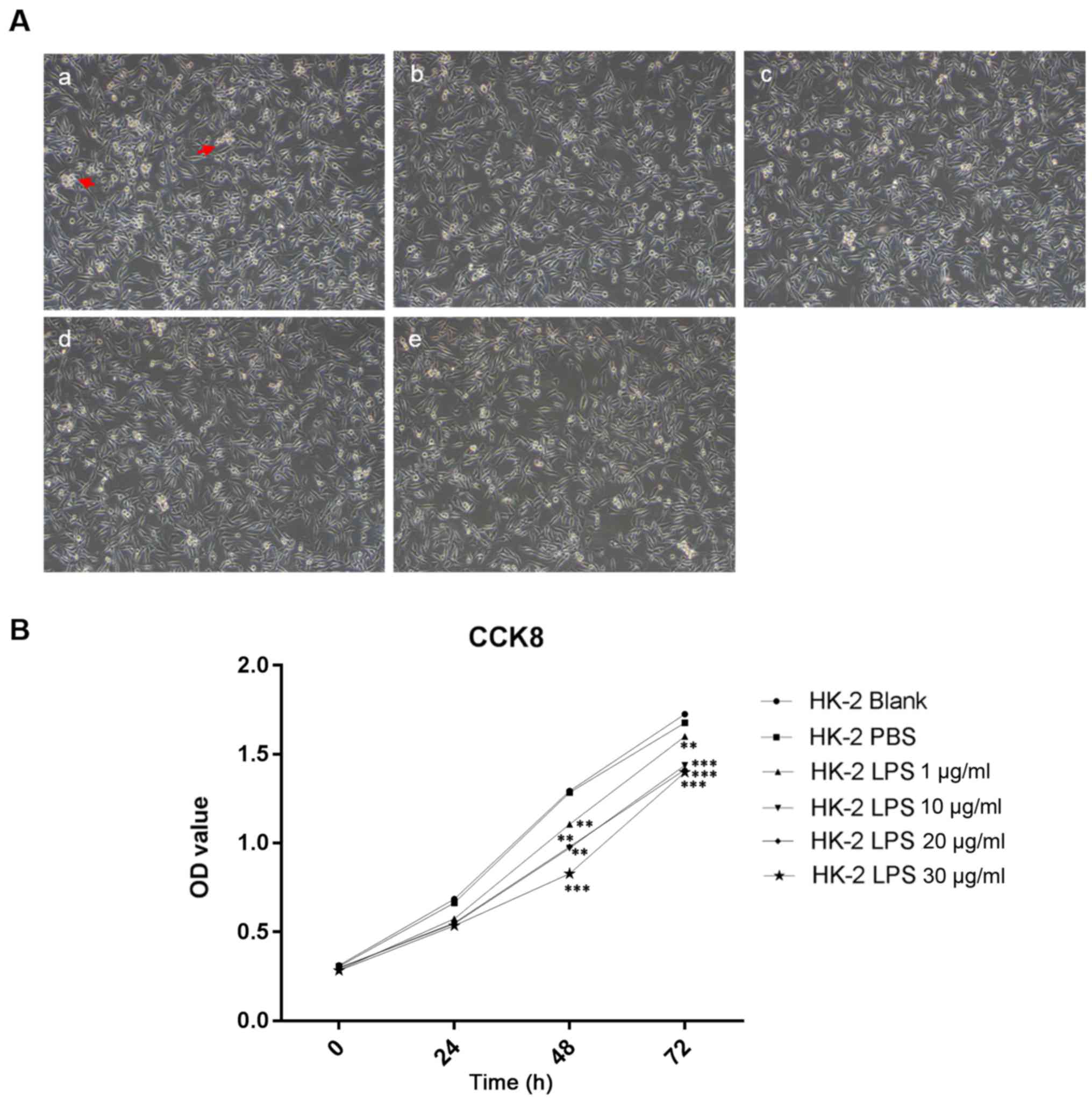
Effect of LPS on the morphology of
HK-2 cells
The morphology of HK-2 cells changed following LPS
treatment. Under the microscope, the HK-2 cells in the control
group exhibited good adherence, clear outlines, tight cell-cell
junctions and good cell growth (Fig.
1A). After the addition of LPS, in a time-dependent manner,
HK-2 cells were rounded into a slender or fusiform shape with
poorly defined outlines, weaker cell-cell junctions, reduced
adherence with reduced of HK-2 cell numbers (Fig. 1A). This finding suggested that LPS
inhibited the growth of HK-2 cells.
Effects of LPS on the viability of
HK-2 cells
After treatment of HK-2 cells with increasing
concentrations of LPS up to 30 µg/ml, CCK-8 assay results indicated
that HK-2 cell viability decreased in response to LPS in a
dose-dependent manner after 72 h (P<0.01, P<0.001; Fig. 1B).
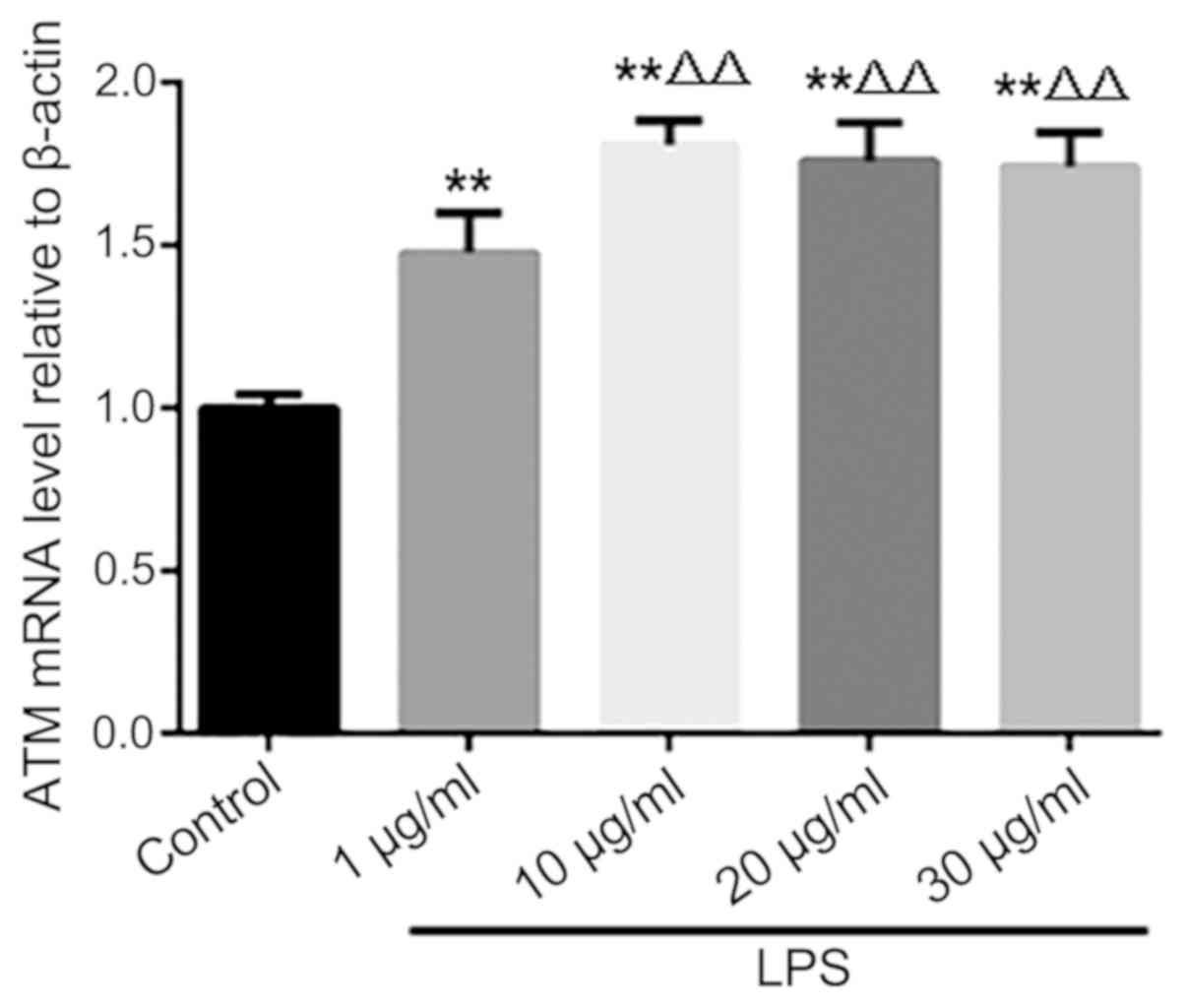
Effect of LPS on the expression of ATM
in HK-2 cells
Following LPS stimulation, ATM mRNA expression
increased in HK-2 cells, with the magnitude peaking when the dose
of 10 µg/ml LPS was used. Increasing the dose of LPS to 20 and 30
µg/ml produced no further increases in ATM expression (Fig. 2).
Effect of LPS on the expression of
inflammatory factors in HK-2 cells
Following 24 h of LPS treatment, the IL-6 and IL-1β
mRNA expression levels increased in a dose-dependent manner
compared with the control group (Fig. 3A
and B). The expression level of TNF-α mRNA was higher after 1
and 10 µg/ml LPS stimulation compared with the control group;
however, the expression level of TNF-α following treatment with 20
and 30 µg/ml LPS was lower compared with treatment with 1 µg/ml LPS
(Fig. 3C). The results of RT-qPCR
analysis were subsequently verified using ELISA. LPS treatment
increased IL-1β, IL-6 and TNF-α expression in a dose-dependent
manner. The levels of the three inflammatory factors aforementioned
peaked when a dose of 10 µg/ml LPS used, and were reduced with the
concomitant treatment with the autophagy inhibitor 3-MA (Fig. 3D-F). These results suggested that LPS
induced an inflammatory response in HK-2 cells. Based on the
findings of this experiment, LPS at a concentration of 10 µg/ml was
selected for subsequent experiments.
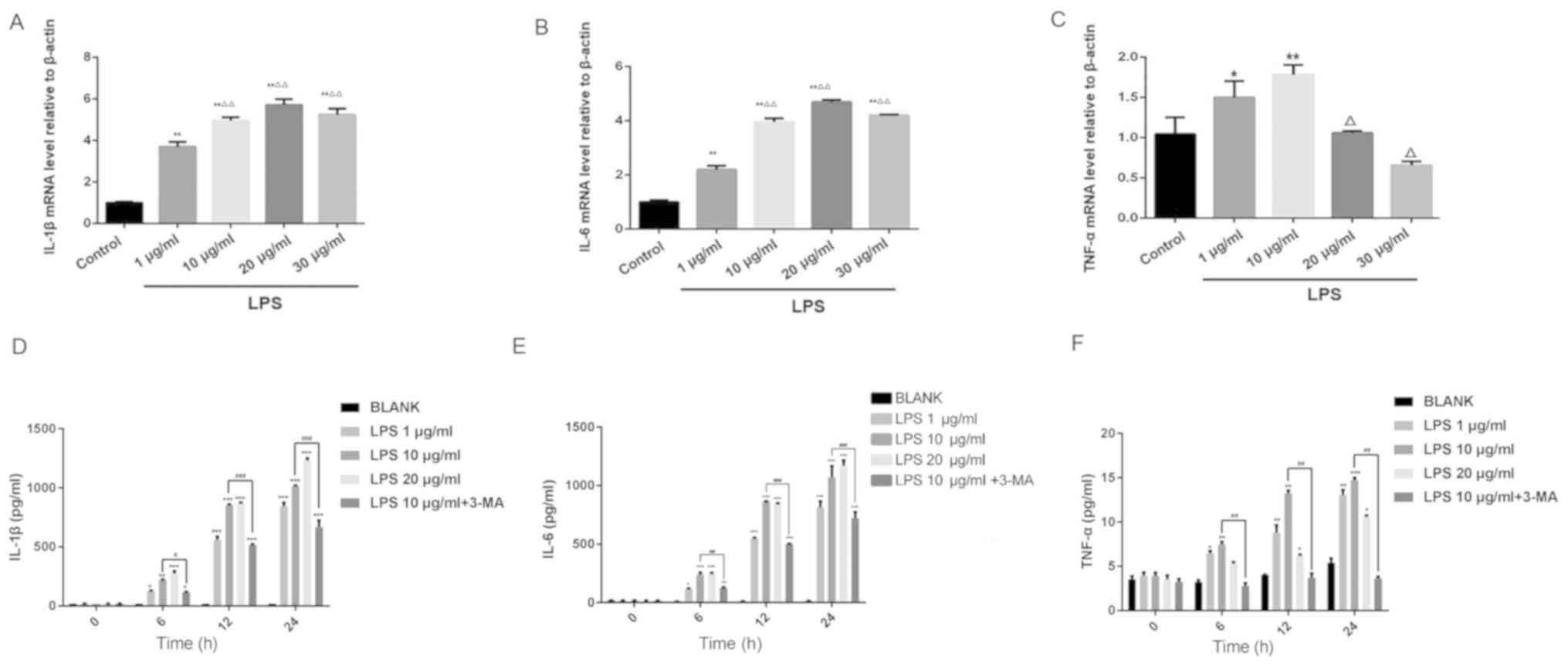 | Figure 3.Effects of different concentrations
of LPS on the expression of (A) IL-1β, (B) IL-6, (C) TNF-α mRNA in
HK-2 cells as detected using reverse transcription-quantitative
PCR. *P<0.05 and **P<0.01 vs. control and
ΔP<0.05 and ΔΔP<0.01 vs. 1 µg/ml LPS.
Effects of different concentrations of LPS and concomitant 3-MA
treatment on the concentrations of (D) IL-1β, (E) IL-6 and (F)
TNF-α in HK-2 cells as measured using ELISA. *P<0.05,
**P<0.01 and ***P<0.001, vs. BLANK, ##P<0.01
and ###P<0.001, vs. 10 µg/ml LPS. LPS,
lipopolysaccharide; IL, interleukin; TNF-α, tumor necrosis
factor-α. |
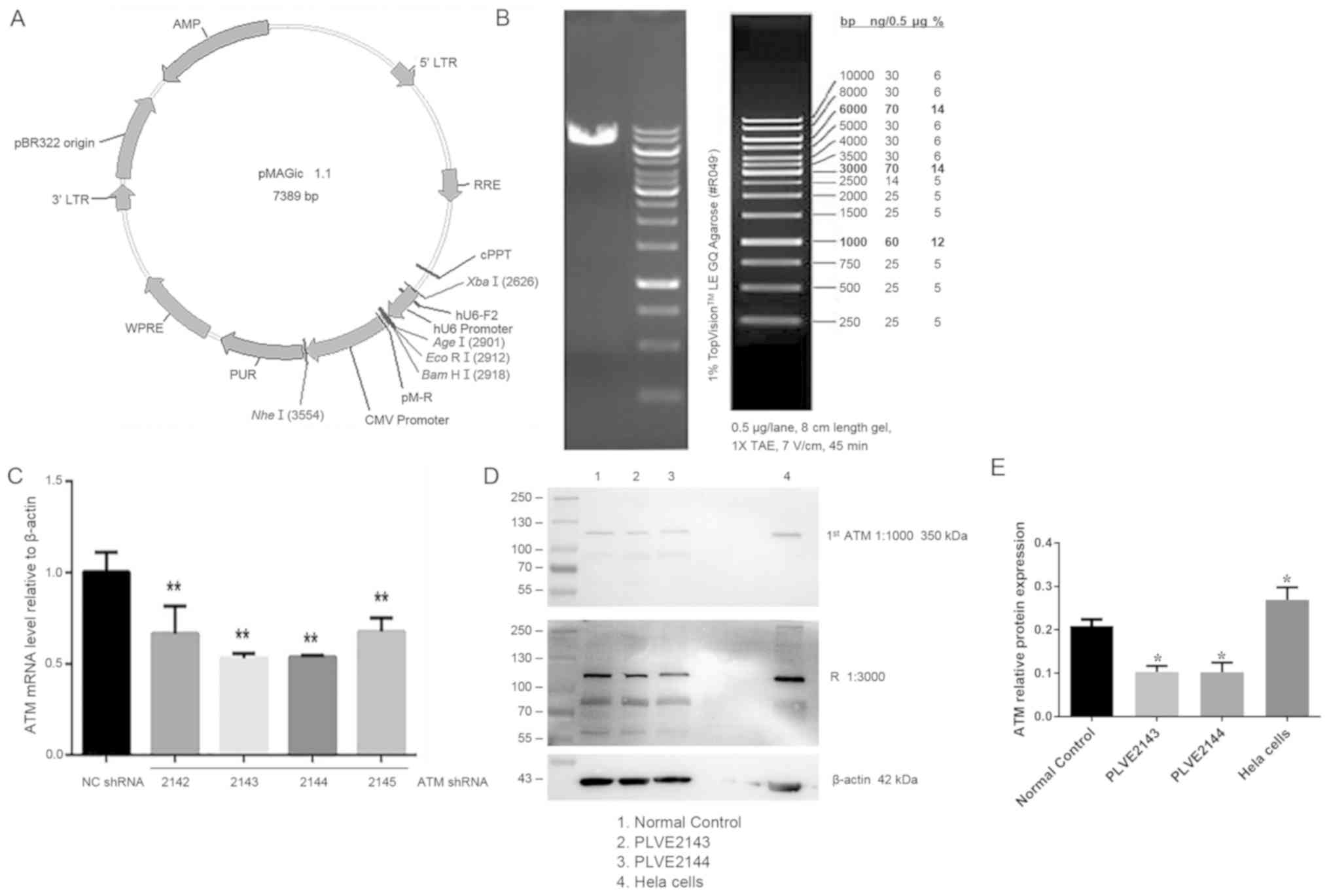
Effect of ATM lentiviral interference
vector on ATM-2 expression in HK-2 cells
Following the construction of lentivirus-mediated
ATM-knockdown system and using it to transfect HK-2 cells, the
linearization of the interference carrier and the carrier map are
shown in Fig. 4A, and the enzyme
digestion map is shown in Fig. 4B.
Lentiviral vectors encoding shRNA2143 and shRNA2144 were more
efficient in knocking down ATM expression and compared with
shRNA2142 and shRNA2145 (Fig. 4C).
To validate this observation, HK-2 cells were transfected with
lentiviruses encoding shRNA2143 and shRNA2144, followed by western
blot analysis. ATM protein expression was significantly reduced
following transfection with shRNA2143 and shRNA2144 (P<0.05;
Fig. 4D and E).
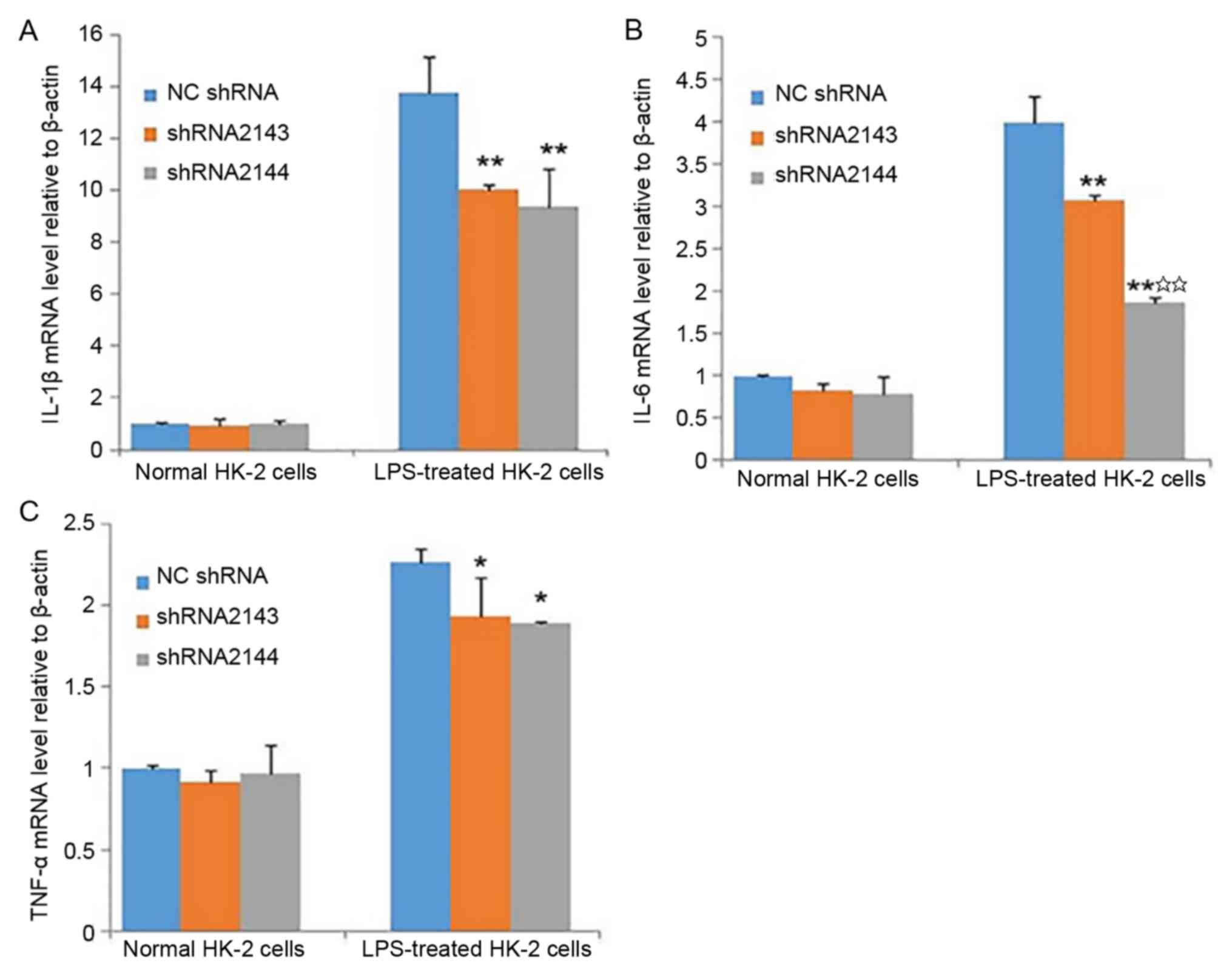
Effect of ATM knockdown on the levels
of inflammatory cytokines in HK-2 cells
After transfection of HK-2 cells with shRNA2143- and
shRNA2144-encoding lentiviral particles, no statistically
significant differences between the shRNA2143 and shRNA2144
interference groups were observed in the levels of IL-1β, IL-6 and
TNF-α in the absence of LPS. Following LPS stimulation and
transfection with shRNA2143 or shRNA2144, the levels of IL-1β, IL-6
and TNF-α were significantly lower compared with the NC shRNA group
(Fig. 5A-C). The result suggested
that downregulation of ATM expression can partially reduce the
expression of inflammatory cytokines caused by LPS.
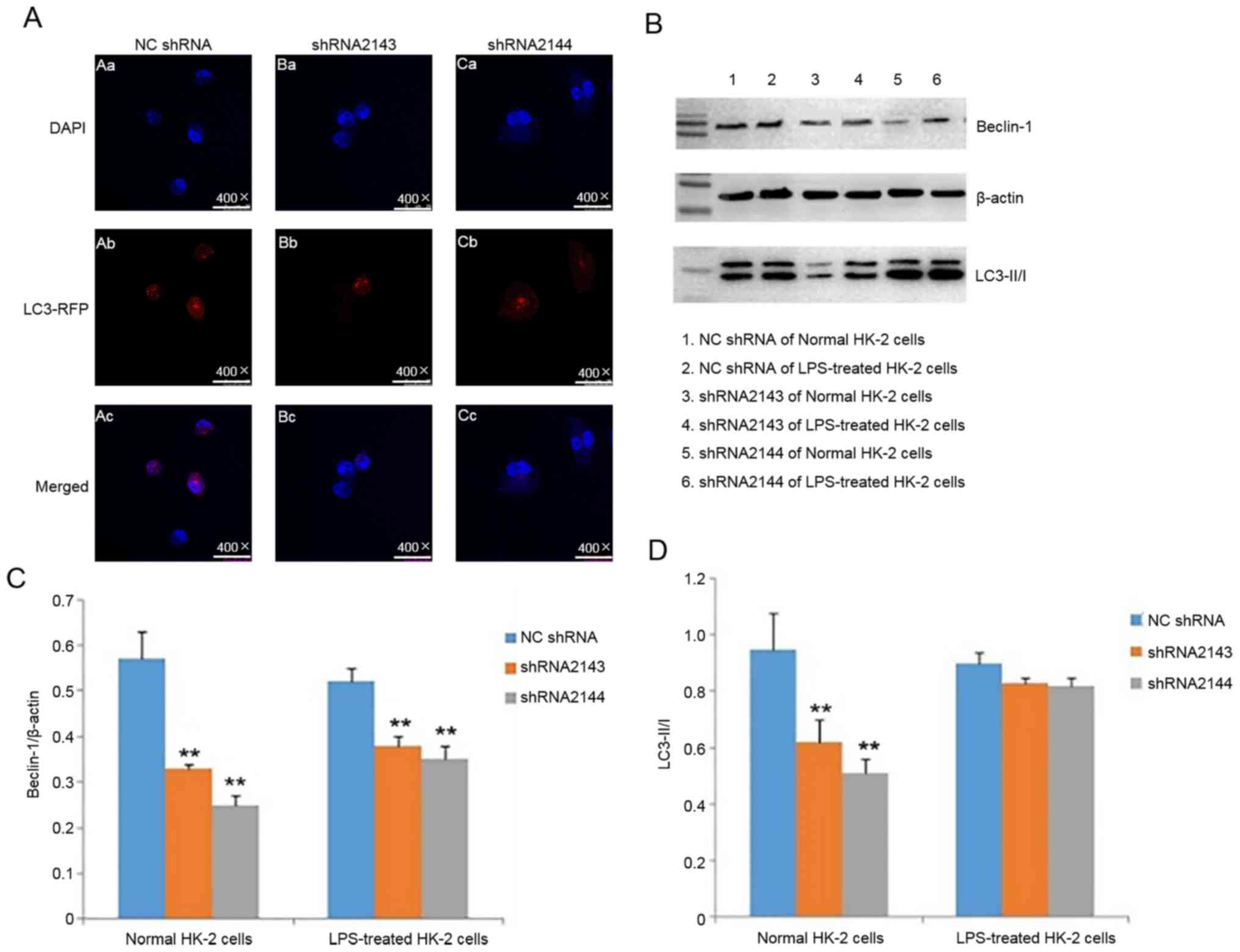
Effect of downregulating ATM
expression on in HK-2 cell autophagy
After HK-2 cells were transfected with shRNA2143 and
shRNA2144, of HK-2 cell autophagy was detected using
immunofluorescence and western blotting following LPS
stimulation.
After LPS stimulation of HK-2 cells for 24 h,
immunofluorescence assay showed that the levels of autophagy after
transfection with shRNA2143 and shRNA2144 were reduced compared
with the control group (Fig. 6A).
The result suggested that downregulation of ATM expression can
reduce autophagy caused by LPS.
The protein expression levels of beclin-1 in HK-2
cells transfected with shRNA2143 and shRNA2144 were significantly
lower compared with the NC shRNA group (Fig. 6B and C). Although the LC3I/II ratio
exhibited a certain degree of reduction in LPS-treated shRNA2143
and shRNA2144 groups compared with NC shRNA, no statistically
significant differences were observed (Fig. 6D). These observations suggested that
downregulation of ATM expression can significantly reduce the
beclin-1 expression. ATM may be involved in the autophagy of HK-2
cells induced by LPS.
Discussion
In the present study, a model of septic AKI was
established using LPS-stimulated HK-2 cells. Treatment with LPS
increased the expression of inflammatory factors in HK-2 cells. In
HK-2 cells with ATM expression knocked down, the levels of
autophagy and expression levels of inflammatory cytokines were
reduced. Therefore, it may be hypothesized that ATM can increase
the expression of inflammatory cytokines further by promoting
autophagy, resulting in HK-2 cell damage.
AKI is a severe clinical condition with high rates
of morbidity and mortality due to a lack of effective treatment
(26). The mortality rate of
patients with septic AKI is >70%, significantly higher compared
with patients with sepsis but without AKI (27). Conventionally the main
pathophysiological mechanism of septic AKI was considered to be
renal ischemia-induced hypoperfusion resulting from renal tubular
perivascular dysfunction and acute tubular necrosis (ATN). However,
renal pathology analysis in patients who succumbed to septic AKI
revealed that 70% of those patients exhibited no ATN; instead, RTEC
apoptosis was more common. In the case of constant or even
increased renal blood flow, RTEC still undergo apoptosis,
suggesting that renal hemodynamic changes are only part of the
cause of septic AKI (28).
Inflammation serves a key role in the pathophysiological mechanism
of septic AKI and septic AKI is directly associated with the
inflammatory response (29).
However, the mechanism of this cycle remains unclear due to the
complexity of the mechanism involved.
Bacterial endotoxin and inflammatory cytokines have
been reported to be direct and important causes of renal injury
(30). Endotoxins, of which LPS is
an example, can stimulate severe inflammatory reactions in the
body, resulting in the production of a large number of inflammatory
factors, including TNF-α, IL-1β and IL-6, in turn leading to
oxidative stress, mitochondrial damage and energy depletion and
finally RTEC apoptosis (31). LPS
induces AKI via the induction of tubular epithelial cell apoptosis
(32). It is crucial to explore
therapeutic strategies to inhibit LPS-induced RTEC apoptosis in
treating AKI. In the present study, LPS was used to stimulate HK-2
cells to establish a septic AKI model. After LPS stimulation, the
morphology of HK-2 cells changed. Untreated cells exhibited good
adherence, clear outlines and tight cell-cell junctions. By
contrast, cells treated with LPS changed from round to shuttle
shapes, the outline of the cells becoming blurred with the cell-
cell junctions appearing loose. LPS-treated cells also appear low
confluency with the number of cells reduced. The viability of HK-2
cells after LPS treatment was measured using CCK-8 assay, and it
was found that LPS reduced cell viability in a dose-dependent
manner. In addition, changes in the expression of inflammatory
factors TNF-α, IL-1β and IL-6 were examined in this model. It was
found that LPS increased the expression of TNF-α, IL-1β and IL-6
mRNA, with a dose of 10 µg/ml having the most potent effect,
consistent with previous reports (33–35).
These results suggest that the LPS-induced HK-2 cell injury model
was successful, providing basis for further research.
Autophagy is a general term for the process by which
intracellular materials are degraded by lysosomes under the
regulation of proteins associated with autophagy. It is the basic
catabolic mechanism that contributes to the routine recycling of
cell materials by the turnover of dysfunctional cellular components
(36). This process exists in
physiological and pathophysiological processes of a number of
different diseases (37). Indeed,
autophagy occurs in most tissues under both physiological and
pathological conditions (16,38,39),
which can be dramatically upregulated by unfavorable stimulus,
including hypoxia and nutrient depletion (40). A previous study has demonstrated that
excessive autophagy may lead to programmed cell death (41). However, autophagy is invariably
linked with disease regardless of whether it is excessively
inhibited or activated, since it serves a dual role in cell
survival and death (42). In
particular, similar studies have found that autophagy serves an
important role in the pathogenesis of sepsis (43,44).
The pathogenesis of LPS-induced AKI is closely
associated with excessive inflammation (45). Pro-inflammatory cytokines are the
major mediators of AKI induced by sepsis (46). Controlling the production of
pro-inflammatory factors and downstream pro-inflammatory mediators
may be an effective approach in AKI therapy (47). Kong et al (48) demonstrated that antithrombin III can
ameliorate serum amyloid P component-induced renal damage by
inhibiting inflammation, oxidative stress and apoptosis. In
addition, Lu et al (49) came
to the same conclusion in the medium contrast-induced AKI. In the
present study, the role of ATM in LPS-induced septic AKI and its
mechanism were explored. In a previous study, persistent
inflammatory response causes DNA damage in RTEC, further activating
the protein kinase ATM (21,22). In a model of ischemia-reperfusion
kidney injury, AMPK was found to activate the ATM-AMPK-TSC2-mTOR
pathway, causing autophagy (23). In
the present study, it was found that LPS stimulated HK-2 cells and
caused an increase in intracellular ATM mRNA levels. To explore the
role of ATM in LPS-induced HK-2 cell injury, an ATM-interfering
lentivirus was constructed, which was used to transfect HK-2 cells
to downregulate endogenous ATM expression. Following ATM knockdown,
LPS-induced increase in the expression of inflammatory cytokines
IL-6, IL-1β and TNF-α was partially reversed. Likewise, beclin-1
expression was significantly reduced compared with NC shRNA group,
but no significant changes were observed in the LC3 II/I ratio. The
role of autophagy in AKI has not been previously determined. At
present, some studies have supported the notion that autophagy can
protect RTEC cells and alleviate AKI deterioration (17,18,50),
whilst other studies suggest that autophagy will increase cell
death (37,51). It remains unclear whether autophagy
protects RTEC against AKI and any associated mechanism. A previous
study using a mouse model of sepsis induced by intraperitoneal LPS
injection reported that the expression levels of LC3, beclin-1 and
other genes associated with autophagy were elevated, and the
secretion of inflammatory factors IL-1β and IL-18 was also
significantly increased (52). It
has also been reported that reducing autophagy promotes
inflammatory responses and subsequent cell death (53). Autophagy has been shown to be
upregulated in numerous AKI models (54). In the present study, it was found
that autophagy aggravated the inflammatory response and cellular
damage in HK-2 cells treated with LPS for 24 h. When shRNA2143 and
shRNA2144 were used to knockdown ATM expression, it was found that
the levels of autophagy and inflammatory factors were also
significantly reduced following 24 h LPS treatment. This suggests
that the level of autophagy is reduced when ATM is downregulated.
Therefore, it can be hypothesized that LPS may induce autophagy in
HK-2 cells through the ATM pathway, resulting in the upregulation
of pro-inflammatory cytokines. To the best of our knowledge, this
was the first time that ATM was found to increase the expression of
inflammatory cytokines by promoting autophagy, resulting in HK-2
cell damage.
In conclusion, the present study revealed that LPS
can reduce HK-2 cell viability, and increase autophagy and the
expression of inflammatory cytokines IL-1β and IL-6, ATM. By
contrast, downregulation of ATM levels in HK-2 cells can reduce the
levels of inflammatory cytokines and autophagy in LPS-induced HK-2
cells. Autophagy can exacerbate inflammatory responses and cellular
damage, however, the mechanism by which ATM affects LPS-induced
inflammatory response and autophagy in AKI renal tubular epithelial
cells remains unclear. The present study provides a new direction
and lays a foundation for the future treatment of AKI.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the Wenzhou
Committee of Science and Technology of China (grant nos. Y20170055,
ZS2017008 and Y20180159) and the Zhejiang Province Natural Science
Foundation (grant nos. LQ19H050002 and LY15H050008).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CFZ and YZ wrote the first draft of manuscript. CFZ,
YYH and BCC contributed to the conception and design of the
research. MMW, YX, XC and MS contributed to the experiments and
analysis of the data. YZ, YL, CSC and JGP contributed to the
analysis and interpretation of the data. All authors critically
revised the manuscript, and agreed to be fully accountable for
ensuring the integrity and accuracy of the work, and read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bagshaw SM, Uchino S, Bellomo R, Morimatsu
H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, Gibney N, et al:
Septic acute kidney injury in critically ill patients: Clinical
characteristics and outcomes. Clin J Am Soc Nephrol. 2:431–439.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (Sepsis-3). JAMA.
315:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Holthoff JH, Wang Z, Patil NK, Gokden N
and Mayeux PR: Rolipram improves renal perfusion and function
during sepsis in the mouse. J Pharmacol Exp Ther. 347:357–364.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Howell GM, Gomez H, Collage RD, Loughran
P, Zhang X, Escobar DA, Billiar TR, Zuckerbraun BS and Rosengart
MR: Augmenting autophagy to treat acute kidney injury during
endotoxemia in mice. PLoS One. 8:e695202013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bagshaw SM, George C and Bellomo R; ANZICS
Database Management Committee, : Early acute kidney injury and
sepsis: A multicentre evaluation. Crit Care. 12:R472008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang F, Zhang G, Lu Z, Geurts AM, Usa K,
Jacob HJ, Cowley AW, Wang N and Liang M: Antithrombin III/SerpinC1
insufficiency exacerbates renal ischemia/reperfusion injury. Kidney
Int. 88:796–803. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen LW, Chen W, Hu ZQ, Bian JL, Ying L,
Hong GL, Qiu QM, Zhao GJ and Lu ZQ: Protective effects of growth
arrest-specific protein 6 (Gas6) on sepsis-induced acute kidney
injury. Inflammation. 39:575–582. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yohannes S and Chawla LS: Evolving
practices in the management of acute kidney injury in the ICU
(Intensive Care Unit). Clin Nephrol. 71:602–607. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gómez H, Kellum JA and Ronco C: Metabolic
reprogramming and tolerance during sepsis-induced AKI. Nat Rev
Nephrol. 13:143–151. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Luo CJ, Luo F, Zhang L, Xu Y, Cai GY, Fu
B, Feng Z, Sun XF and Chen XM: Knockout of interleukin-17A protects
against sepsis-associated acute kidney injury. Ann Intensive Care.
6:562016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chunzhi G, Zunfeng L, Chengwei Q, Xiangmei
B and Jingui Y: Hyperin protects against LPS-induced acute kidney
injury by inhibiting TLR4 and NLRP3 signaling pathways. Oncotarget.
7:82602–82608. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu C, Chang A, Hack BK, Eadon MT, Alper SL
and Cunningham PN: TNF-mediated damage to glomerular endothelium is
an important determinant of acute kidney injury in sepsis. Kidney
Int. 85:72–81. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ahn JM, You SM, Lee YM, Oh SW, Ahn SY, Kim
S, Chin HJ, Chae DW and Na KY: Hypoxia-inducible factor activation
protects the kidney from gentamicin-induced acute injury. PLoS One.
7:e489522012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sutton TA, Hato T, Mai E, Yoshimoto M,
Kuehl S, Anderson M, Mang H, Plotkin Z, Chan RJ and Dagher PC: p53
Is renoprotective after ischemic kidney injury by reducing
inflammation. J Am Soc Nephrol. 24:113–124. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Leventhal JS, Ni J, Osmond M, Lee K,
Gusella GL, Salem F and Ross MJ: Autophagy limits endotoxemic acute
kidney injury and alters renal tubular epithelial cell cytokine
expression. PLoS One. 11:e01500012016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mei S, Livingston M, Hao J, Li L, Mei C
and Dong Z: Autophagy is activated to protect against endotoxic
acute kidney injury. Sci Rep. 6:221712016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu JH, Cho SO, Lim JW, Kim N and Kim H:
Ataxia telangiectasia mutated inhibits oxidative stress-induced
apoptosis by regulating heme oxygenase-1 expression. Int J Biochem
Cell Biol. 60:147–156. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Moser BA, Subramanian L, Khair L, Chang YT
and Nakamura TM: Fission yeast Tel1 (ATM) and Rad3 (ATR) promote
telomere protection and telomerase recruitment. PLoS Genet.
5:e10006222009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Abraham RT: Cell cycle checkpoint
signaling through the ATM and ATR kinases. Genes Dev. 15:2177–2196.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bencokova Z, Kaufmann MR, Pires IM, Lecane
PS, Giaccia AJ and Hammond EM: ATM activation and signaling under
hypoxic conditions. Mol Cell Biol. 29:526–537. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang LT, Chen BL, Wu CT, Huang KH, Chiang
CK and Hwa Liu S: Protective role of AMP-activated protein
kinase-evoked autophagy on an in vitro model of
ischemia/reperfusion-induced renal tubular cell injury. PLoS One.
8:e798142013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2 (-Delta Delta C (T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jung G, Roh J, Lee H, Gil M, Yoon DH, Suh
C, Jang S, Park CJ, Huh J and Park CS: Autophagic markers BECLIN 1
and LC3 are associated with prognosis of multiple myeloma. Acta
Haematol. 134:17–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dellepiane S, Marengo M and Cantaluppi V:
Detrimental cross-talk between sepsis and acute kidney injury: New
pathogenic mechanisms, early biomarkers and targeted therapies.
Crit Care. 20:612016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jackson WL Jr: Acute renal failure and
sepsis. N Engl J Med. 351:2347–2349. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Langenberg C, Wan L, Egi M, May CN and
Bellomo R: Renal blood flow in experimental septic acute renal
failure. Kidney Int. 69:1996–2002. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fani F, Regolisti G, Delsante M,
Cantaluppi V, Castellano G, Gesualdo L, Villa G and Fiaccadori E:
Recent advances in the pathogenetic mechanisms of sepsis-associated
acute kidney injury. J Nephrol. 31:351–359. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen L, Yang S, Zumbrun EE, Guan H,
Nagarkatti PS and Nagarkatti M: Resveratrol attenuates
lipopolysaccharide-induced acute kidney injury by suppressing
inflammation driven by macrophages. Mol Nutr Food Res. 59:853–864.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Deng SY, Zhang LM, Ai YH, Pan PH, Zhao SP,
Su XL, Wu DD, Tan HY, Zhang LN and Tsung A: Role of interferon
regulatory factor-1 in lipopolysaccharide-induced mitochondrial
damage and oxidative stress responses in macrophages. Int J Mol
Med. 40:1261–1269. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li T, Zhao J, Miao S, Xu Y, Xiao X and Liu
Y: Dynamic expression and roles of sequestome-1/p62 in LPS-induced
acute kidney injury in mice. Mol Med Rep. 17:7618–7626.
2018.PubMed/NCBI
|
|
33
|
Jiao XY, Shen YQ and Li KS: The
correlation between cytokine production by cerebral cortical glial
cells and brain lateralization in mice. Neuromodulation. 11:23–32.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Johnson RL, Murray ST, Camacho DK and
Wilson CG: Vagal nerve stimulation attenuates IL-6 and TNFα
expression in respiratory regions of the developing rat brainstem.
Respir Physiol Neurobiol. 229:1–4. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee WS, Shin JS, Jang DS and Lee KT:
Cnidilide, an alkylphthalide isolated from the roots of Cnidium
officinale, suppresses LPS-induced NO, PGE2, IL-1β, IL-6 and TNF-α
production by AP-1 and NF-κB inactivation in RAW 264.7 macrophages.
Int Immunopharmacol. 40:146–155. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim KH and Lee MS: Autophagy as a
crosstalk mediator of metabolic organs in regulation of energy
metabolism. Rev Endoc Metab Disord. 15:11–20. 2014. View Article : Google Scholar
|
|
37
|
Shen HM and Codogno P: Autophagic cell
death: Loch Ness monster or endangered species? Autophagy.
7:457–465. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: A double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Levine B and Yuan J: Autophagy in cell
death: An innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Casado P, Bilanges B, Rajeeve V,
Vanhaesebroeck B and Cutillas PR: Environmental stress affects the
activity of metabolic and growth factor signaling networks and
induces autophagy markers in MCF7 breast cancer cells. Mol Cell
Proteomics. 13:836–848. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shi R, Weng J, Zhao L, Li XM, Gao TM and
Kong J: Excessive autophagy contributes to neuron death in cerebral
ischemia. CNS Neurosci Ther. 18:250–260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Livingston MJ and Dong Z: Autophagy in
acute kidney injury. Semin Nephrol. 34:17–26. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chien WS, Chen YH, Chiang PC, Hsiao HW,
Chuang SM, Lue SI and Hsu C: Suppression of autophagy in rat liver
at late stage of polymicrobial sepsis. Shock. 35:506–511. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hsieh CH, Pai PY, Hsueh HW, Yuan SS and
Hsieh YC: Complete induction of autophagy is essential for
cardioprotection in sepsis. Ann Surg. 253:1190–1200. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Şen V, Uluca Ü, Ece A, Güneş A, Zeytun H,
Arslan S, Kaplan I, Türkçü G and Tekin R: Role of Ankaferd on
bacterial translocation and inflammatory response in an
experimental rat model of intestinal obstruction. Int J Clin Exp
Med. 7:2677–2686. 2014.PubMed/NCBI
|
|
46
|
Sang HS, Lee KE, Kim IJ, Kim O, Kim CS,
Choi JS, Choi HI, Bae EH, Ma SK, Lee JU and Kim SW: Alpha-lipoic
acid attenuates lipopolysaccharide-induced kidney injury. Clin Exp
Nephrol. 19:82–91. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Xiang H, Hu B, Li Z and Li J:
Dexmedetomidine controls systemic cytokine levels through the
cholinergic anti-inflammatory pathway. Inflammation. 37:1763–1770.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kong Y, Yin J, Cheng D, Lu Z, Wang N, Wang
F and Liang M: Antithrombin III attenuates AKI following acute
severe pancreatitis. Shock. 49:572–579. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lu Z, Cheng D, Yin J, Wu R, Zhang G, Zhao
Q, Wang N, Wang F and Liang M: Antithrombin III protects against
contrast-induced nephropathy. EBioMedicine. 17:101–107. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen K, Dai H, Yuan J, Chen J, Lin L,
Zhang W, Wang L, Zhang J, Li K and He Y: Optineurin-mediated
mitophagy protects renal tubular epithelial cells against
accelerated senescence in diabetic nephropathy. Cell Death Dis.
9:1052018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lu B, Capan E and Li C: Autophagy
induction and autophagic cell death in effector T cells. Autophagy.
3:158–159. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nakahira K, Haspel JA, Rathinam VA, Lee
SJ, Lam HC, Rabinovitch M, et al: Autophagy proteins regulate
innate immune response by inhibiting NALP3 inflammasome-mediated
mitochondrial DAN release. American Thoracic Society 2011
International Conference, May 13–18, 2011 • Denver Colorado.
2011.A1077
|
|
53
|
Carchman EH, Rao J, Loughran PA, Rosengart
MR and Zuckerbraun BS: Heme oxygenase-1-mediated autophagy protects
against hepatocyte cell death and hepatic injury from
infection/sepsis in mice. Hepatology. 53:2053–2062. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kaushal GP: Autophagy protects proximal
tubular cells from injury and apoptosis. Kidney Int. 82:1250–1253.
2012. View Article : Google Scholar : PubMed/NCBI
|