Introduction
Atherosclerosis (AS) and the resulting coronary
heart disease (CHD) are threatening human health both in developed
and developing countries. In China, with the improvement of
people's living standards and changes in dietary habits, the
incidence rates of AS and its related diseases also show an
increasing trend year by year, which not only seriously endangers
people's health, but also lay as huge economic burden on the
society. Therefore, it is extremely urgent to fully understand the
pathogenesis of AS and CHD. It is well known that the occurrence of
AS is not simply due to dyslipidemia, but also closely related to
chronic inflammatory diseases. The inflammatory responses occur
throughout AS, and is one of the main causes of AS, exerting
effects on the progression of AS mainly through affecting lipid
metabolism and biological activity of the vascular wall (1).
The complex interaction among arterial vascular
cells, lipoproteins and inflammatory cells is involved in AS. The
key factor in the initiation of AS is the abundance of lipoprotein
in apolipoprotein B under the endothelium, followed by recruitment
and differentiation of inflammatory monocytes into macrophages
(2). Macrophages take in
lipoproteins and turn into lipid-loaded ‘foam cells’, which
gradually exacerbate the inflammatory state along with other immune
cells and intimal smooth muscle cells. The dead and dying
macrophages, combined with insufficient scavenging ability of dead
cells, lead to development of the so-called lipid-rich necrotic
core, which is a complex process closely related to the rupture of
atherosclerotic plaques, thereby causing acute myocardial
infarction and sudden death (3). The
lipid-rich necrotic core is associated with plaque instability,
probably because it is the aggregation place of matrix
metalloproteinase, inflammatory mediators and pro-thrombosis
molecules. Therefore, apoptosis of macrophages may be a key factor
for facilitating the formation of lipid-rich necrotic core and
promoting the transformation of benign pathological changes into
unstable lesion phenotype. In advanced AS, an important cause of
macrophage apoptosis is the cell death caused by chronic
endoplasmic reticulum stress (ERS) pathway (4). The effect of ERS on cells in AS is
poorly understood, but currently there is evidence that ERS can
regulate the survival of smooth muscle cells and endothelial cells
(5,6).
Recent studies have confirmed that adenosine
monophosphate-activated protein kinase (AMPK) is a potential target
for resisting AS. AMPK is a kind of important protein kinase,
which, in the case of cascade activation, will enhance the
protective effect of cells under stress state. The main target
cells in cardiovascular diseases are cardiovascular endothelial
cells and smooth muscle cells, and they are regulated by AMPK
directly and indirectly. Most of the evidence comes from the
research results on the close association between dysregulation of
AMPK pathway and vascular disease. Therefore, AMPK is a key target
in the prevention and treatment of AS (7). Studies have demonstrated that the
activity of AMPK significantly increases when many cardiovascular
organs have no adequate blood flow in the early stage under such
acute and chronic stimuli as load increase, but the activation of
AMPK will be inhibited by excessive injury, especially under the
action of reactive oxygen species (ROS) (8). Therefore, activating the AMPK pathway
has become a potential target for drug therapy of AS.
Atorvastatin is a clinically common selective
3-hydroxy-3- methylglutaryl coenzyme A (HMG-CoA) reductase
inhibitor. In addition to its lipid-regulating effect in resisting
AS, atorvastatin also slows down the occurrence and development of
AS through non-lipid lowering effects, such as anti-inflammation,
anti-oxidation and immunoregulation (9,10). It is
known that atorvastatin can promote the phosphorylation of AMPK
(10), while the AMPK pathway can
reduce the adhesion of inflammatory cells to the vascular
endothelium and the proliferation of inflammatory cells caused by
lipid oxidation, thereby stimulating the expression of related
genes in antioxidant defense system in cells and the production of
endothelial nitric oxide synthase (11), and exerting the corresponding anti-AS
effect.
To better determine the anti-AS mechanism of
atorvastatin, it is hypothesized that atorvastatin can reduce the
ERS in endothelial cells in apolipoprotein E
(ApoE)−/− mice, and such a protective effect
of atorvastatin is realized through activating AMPK.
Materials and methods
Animals and reagents
A total of 20, 8-week-old male Specific Pathogen
Free (SPF) ApoE−/− mice, weighing 18–22 g,
were from Beijing Vital River Laboratory Animal Technology Co.,
Ltd. High-fat diet (containing 0.25% cholesterol and 21% lard) was
from Beijing Keao Xieli Feed Co., Ltd. Rabbit anti-phospho-PERK
antibody, and spliced XBP-1 antibody, phosphorylated eIF2a antibody
were from Cell Signaling Technology, oxidized low-density
lipoprotein (ox-LDL) was from Yiyuan Biotechnologies, and H&E
staining kit was from Beyotime Biotechnology.
Modeling and grouping
This study was approved by the Animal Ethics
Committee of Fudan University Animal Center (Qingpu, China). A
total of 20 ApoE−/− mice were fed with
high-fat diet added with 0.15% cholesterol and 21% lard for 2 weeks
to establish the mouse model of hyperlipidemia. After successful
modeling, the mice were randomly divided into control group and
atorvastatin group. The mice in atorvastatin group were
intraperitoneally injected with atorvastatin solution (5 mg/kg)
every day, while those in control group were intraperitoneally
injected with the same dose of phosphate-buffered saline (PBS).
They were fed under the temperature of 21±2°C, relative humidity of
50±15% and 12/12 h light/dark cycle. After 6 weeks, the mice were
sacrificed for further data analysis.
Hematoxylin and eosin (H&E)
staining
After the mice were sacrificed, the blood vessels
were first perfused with PBS and then 4% paraformaldehyde fixative.
The blood vessels in the brachial artery and aortic root were
separated, fixed in 4% paraformaldehyde fixative for 12 h and
embedded in OCT. Then the specimens were serially sliced into 20
sections at an interval of 50 µm, washed with distilled water for 5
min, stained with hematoxylin for 5 min, and washed with water for
3 min, followed by differentiation with 0.5% hydrochloric acid
alcohol for 20 sec, washing with tap water for 2 min, eosin
staining for 3 min, and washing with tap water for 1 min again to
remove the excess dye. After dehydration with gradient alcohol, 80%
ethanol for 5 sec, 95% ethanol I for 2 min and 95% ethanol II for 2
min, absolute ethanol I for 3 min and absolute ethanol II for 3
min, and transparent reagent I for 5 min and transparent reagent II
for 5 min, the sections were sealed with neutral balsam, and the
transparent reagent around the tissues was absorbed using absorbent
paper. Then a small drop of balsam was added dropwise at the center
of tissues, and carefully covered with a clean cover glass using
tweezers. Finally, the morphological structure of aortic vessels
and plaques was observed under a microscope.
Immunohistochemistry
The sections were taken from the refrigerator
(−80°C), placed at room temperature for 20 min, soaked in distilled
water for 20 min, washed with PBS, immersed in 3%
H2O2 for 10 min, washed with PBS for 5 min, 3
times, sealed with 5% goat serum and incubated at room temperature
for 20 min. After the serum was removed, the sections were added
dropwise with primary antibodies (1:50) for incubation at 4°C
overnight, washed with PBS for 5 min, 3 times, incubated with
horseradish peroxidase-labeled secondary antibodies at 37°C for 40
min, and washed again with PBS. After color development using
diaminobenzidine (DAB) in the dark, the sections were washed with
running water, stained with Harris hematoxylin dye for 2 min,
washed again with running water for 30 sec, and differentiated with
1% hydrochloric acid alcohol for 5–10 sec, followed by dehydration
with ethanol at different concentrations, and transparentization.
Then the sections were added dropwise with neutral balsam and
covered with the cover glass. The antigen-positive cells were
stained dark brown.
Western blotting
The total proteins required for the assay were
extracted from the tissues as follows: Approximately 30 mg of
tissues were taken, ground with liquid nitrogen using a clean
mortar, and lysed with cell lysis buffer (150 µl/30 mg) for 30 min,
followed by centrifugation at 12,000 × g for 5 min at 4°C. The
supernatant was taken and stored at −80°C. The protein
concentration in the sample was detected using bicinchoninic acid
(BCA) colorimetry (Pierce; Thermo Fisher Scientific, Inc.). Protein
diluent (25 µl) and 25 µl of standards were added into each well,
200 µl of developing solution (A:B=50:1) was also added, and the
mixture was incubated in an incubator at 37°C for 30 min. The
optical density value was measured using a microplate reader, and
the concentration and loading volume of the protein sample were
calculated. The samples were taken out from the refrigerator
(−80°C) and placed on ice. Electrophoresis solution (500 ml)
already prepared was poured into the electrophoresis tank, and the
electrophoresis gel was also placed into the electrophoresis tank.
The protein was loaded and subjected to electrophoresis under
constant pressure of 100 V for 30 min. After the sample ran through
the spacer gel, electrophoresis was performed again under constant
pressure of 120 V for ~1.5 h, after which the gel plate was taken
out. The polyvinylidene fluoride (PVDF) membranes (Millipore) was
first immersed in methanol for 10 sec and in distilled water for 2
min, and then placed into the transfer buffer. The transfer holder
was opened, and the fiber pad, filter paper, PVDF membranes,
electrophoresis gel, filter paper and fiber pad were placed in
order from bottom to top. Then the transfer holder was placed in
the transfer tank containing transfer buffer for membrane transfer.
After the electrode was connected, the protein was transferred onto
the membrane under constant pressure of 80 V for 2 h, sealed with
5% skim milk for 2 h, and incubated with primary antibodies
overnight. After the membrane was washed, the protein was incubated
again with secondary antibodies for 1 h, and the color was
developed.
Statistical analysis
GraphPad Prism 6.0 software (GraphPad Software,
Inc.) was used for plotting, and Statistical Product and Service
Solutions (SPSS) 22.0 software (IBM, Corp.) for statistical
analysis. Measurement data were expressed as mean ± standard
deviation (mean ± SD). Differences between two groups were analyzed
by using the Student's t-test. Comparison between multiple groups
was done using One-way ANOVA test followed by Post Hoc Test (Least
Significant Difference). P<0.05 indicates a statistically
significant difference.
Results
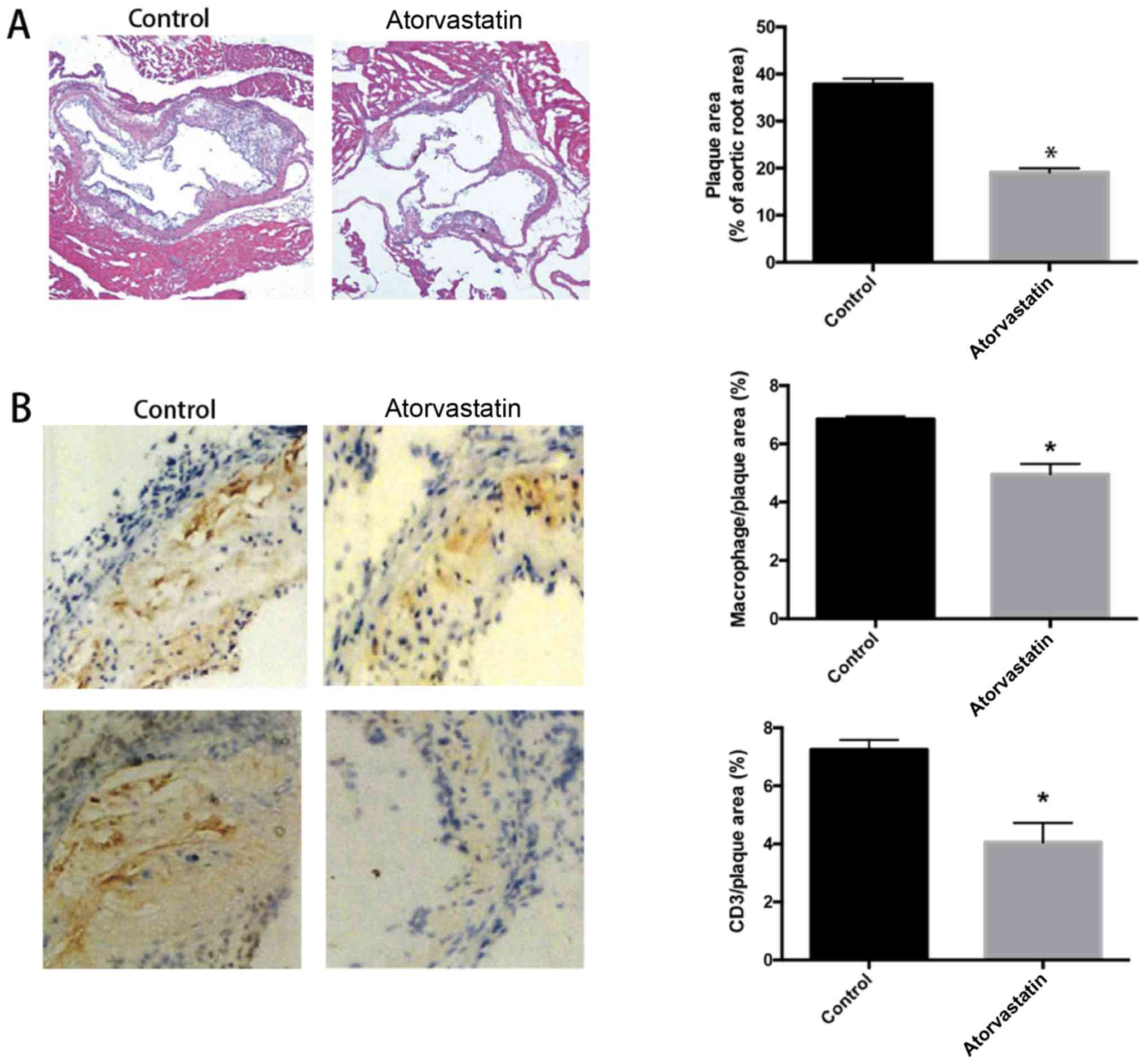
Atorvastatin treatment reduces
atherosclerotic plaque formation and increases plaque stability in
ApoE−/− mice
To detect the effect of atorvastatin on
atherosclerotic plaque formation, the aortic root sections were
H&E stained. The results showed that the area of
atherosclerotic plaques in the thoracic aorta were significantly
reduced in atorvastatin group (P<0.05) (Fig. 1A), indicating that atorvastatin
applied in vivo can reduce the atherosclerotic plaque
formation. To explore the effect of atorvastatin on plaque
stability, the aortic root sections of T cells and macrophages were
stained. The results revealed that the content of macrophages
declined, and the CD3+ T cell infiltration was also
significantly reduced in atorvastatin group (P<0.05) (Fig. 1B), suggesting that the plaque
stability in the atorvastatin group was significantly higher than
that in the control group.
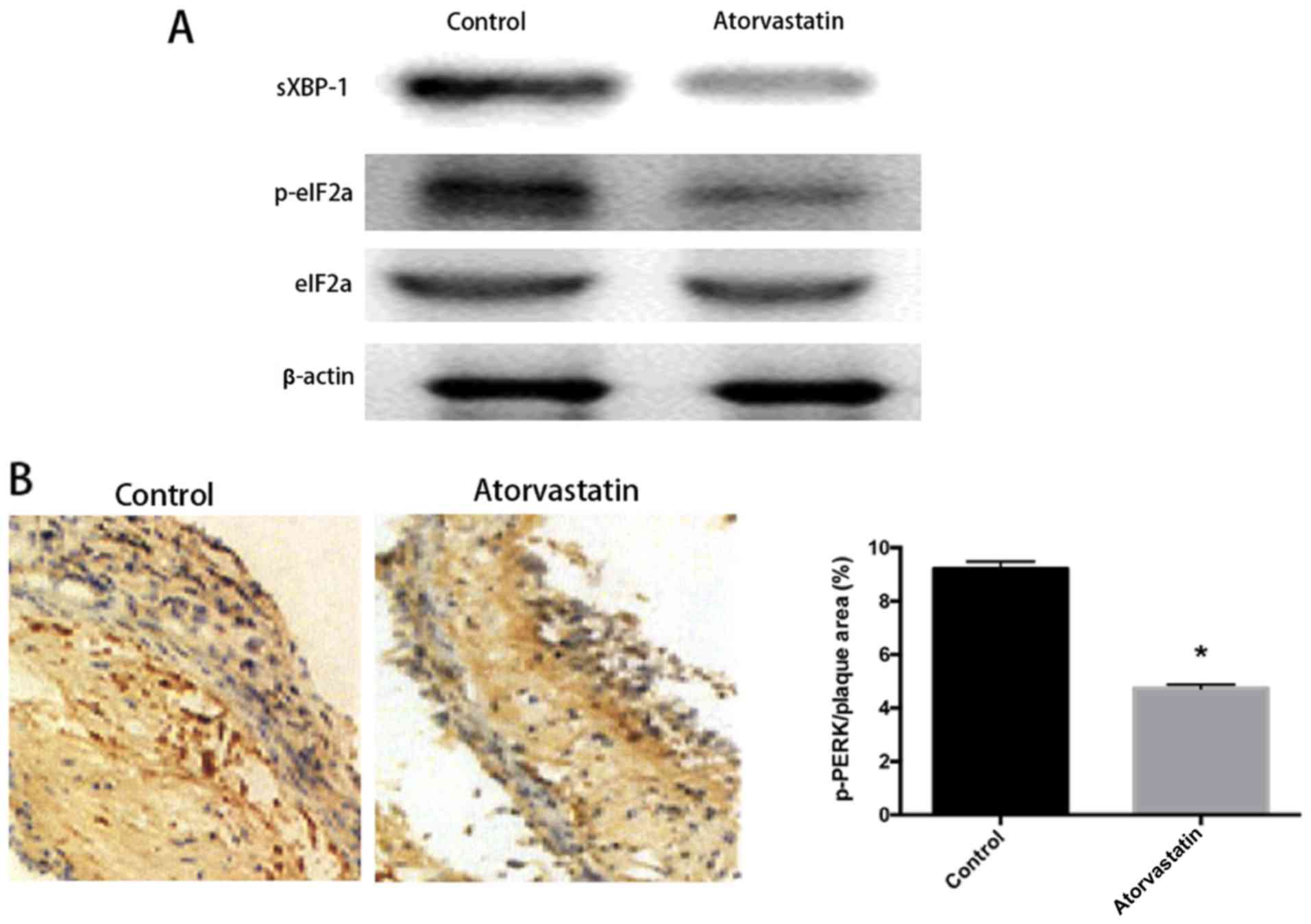
Atorvastatin alleviates ERS in local
vascular walls and plaques in ApoE−/−
mice
The level of ERS in local vascular walls and plaques
was first detected after intraperitoneal injection of atorvastatin.
The thoracic aorta was ground and smashed to extract the proteins,
and the ERS-related proteins spliced X box-binding protein 1
(sXBP-1) and phospho-eukaryotic initiation factor-2α (p-eIF2α) were
selected as detection markers for ERS and subjected to western
blotting. It was found that the levels of ERS-related proteins
sXBP-1 and p-eIF2α were obviously reduced in the thoracic aorta in
atorvastatin group, while the level of total eIF2α had no changes
(Fig. 2A), demonstrating that
atorvastatin can effectively inhibit activation of the above
proteins, alleviating the occurrence of ERS. To clarify the local
conditions of plaques, immunohistochemistry was used to detect the
level of another ERS-related protein phospho-protein kinase-like ER
kinase (p-PERK) in local plaques. The results showed that the level
of p-PERK in local plaques obviously declined (P<0.05) (Fig. 2B). The above findings indicate that
atorvastatin applied in vivo can relieve ERS in local
vascular walls and plaques.
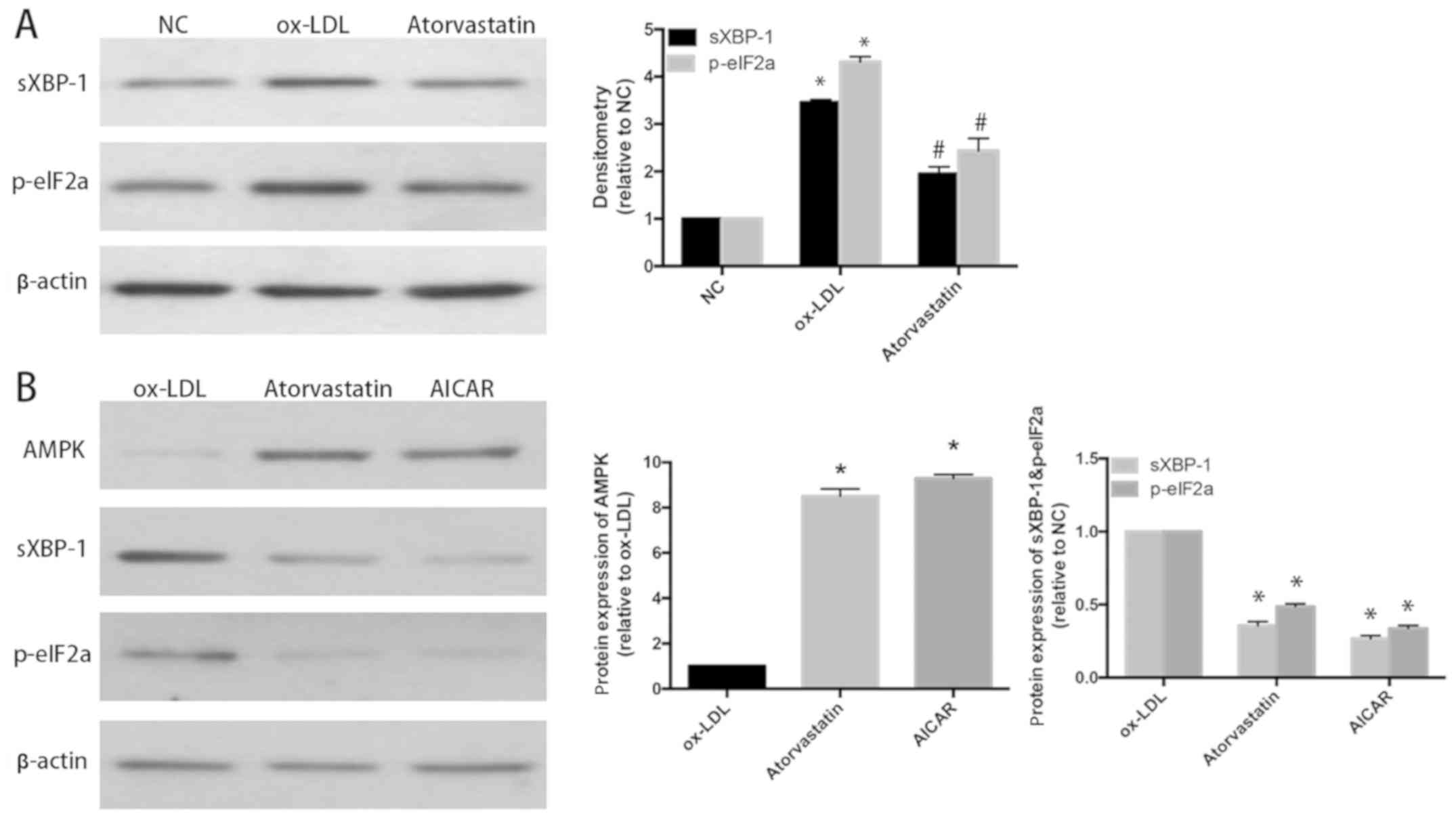
Atorvastatin attenuates ox-LDL-induced
ERS in human umbilical vein endothelial cells (HUVECs) through
activating AMPK
HUVECs were divided into control (NC) group, ox-LDL
group (treated with ox-LDL at a concentration of 100 µg/ml for 24
h) and atorvastatin group (treated with ox-LDL at a concentration
of 100 µg/ml and atorvastatin at a concentration of 10 µmol/l for
24 h), and the levels of ERS-related molecules p-elF2a and sXBP-1
were determined using western blotting. It was found that the
levels of p-elF2a and sXBP-1 were increased in ox-LDL group, while
the elevated protein levels declined in atorvastatin group
(P<0.05) (Fig. 3A). Furthermore,
HUVECs were divided into ox-LDL group, atorvastatin group and AMPK
agonist 5-aminoimidazole-4-carboxamide-I-β-D-ribofuranoside (AICAR)
group (treated with ox-LDL at a concentration of 100 µg/ml and 1 mM
of AICAR for 24 h). The results manifested that after application
of atorvastatin and AMPK agonist AICAR, the level of AMPK was
increased, and the protein expression levels of ERS-related
molecules were also inhibited in AICAR group, indicating that AMPK
agonist and atorvastatin are able to exert the same inhibitory
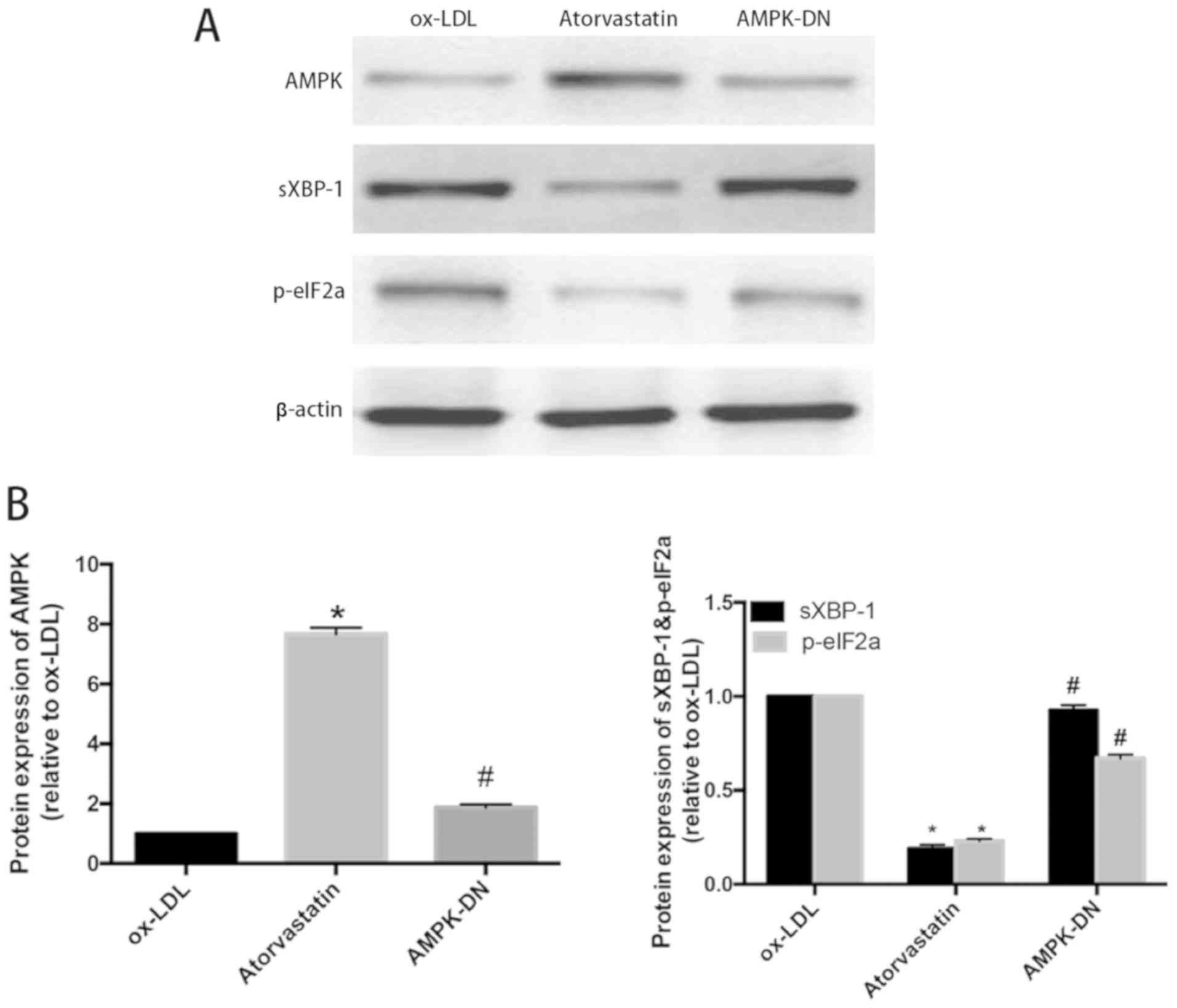
effect on ox-LDL-induced ERS in HUVECs (P<0.05) (Fig. 3B). To explore the role of AMPK in the
inhibition of atorvastatin on ERS, HUVECs were transfected with
AMPK-DN (treated with ox-LDL at a concentration of 100 µg/ml and
AICAR plasmid for 24 h). The results showed that AMPK-DN could
effectively suppress the atorvastatin-induced increase of AMPK
level, and in AMPK-DN group, the inhibitory effect of atorvastatin
on ox-LDL-induced ERS in HUVECs was evidently weakened, and the
decreased protein levels of ERS-related molecules p-elF2a and
sXBP-1 increased again (P<0.05) (Fig.
4A and B). It is concluded that atorvastatin inhibits the
ox-LDL-induced ERS in HUVECs through AMPK.
Discussion
ER is an important organelle and calcium ion
reservoir for protein synthesis, folding, modification and
transport in eukaryotic cells, and is also closely related to the
lipid synthesis and the maintenance of redox balance. ER is very
sensitive to a variety of stimuli, such as oxidative stress,
imbalance of calcium homeostasis, cholesterol overload,
glycosylation changes and other changes in physicochemical
environment, which can lead to dysfunction of ER, and cause ERS
mainly characterized by accumulation of unfolded and/or misfolded
proteins and imbalance of calcium homeostasis. The massive
accumulation of unfolded and/or misfolded proteins in the ER lumen
will result in the activation of a series of intracellular signal
transduction pathways, known as unfolded protein response (UPR).
UPR to a certain degree is beneficial for maintaining the ER
function and cell survival, but excessive or over long-term stress
will induce apoptosis through activating ERS-related signaling
pathways (12).
Cardio-cerebrovascular diseases with AS as the pathological basis
seriously harm human health. In recent years, a large number of
basic and clinical studies have confirmed that ERS plays an
important role in the occurrence and development of AS, and it is
expected to become a new therapeutic target for AS (13). Currently, UPR is the most
well-studied ERS signaling pathway, which is sensed and mediated by
3 kinds of ER transmembrane proteins, namely double-stranded
RNA-dependent PERK, inositol-requiting enzyme 1 (IRE1) and
activating transcription factor 6 (ATF6).
PERK is type I ER transmembrane protein with
serine/threonine kinase activity. Under ERS, activated PERK
facilitates the phosphorylation of eukaryotic translation
initiation factor 2α (eIF2α), thereby reducing the overall level of
protein translation and lowering the ER unfolded protein load
(14). IRE1 is type I ER
transmembrane protein with dual activity of serine/threonine kinase
and endonuclease. After dissociation from GRP78, IRE1 forms dimers
and leads to activation of its protein kinase activity and
autophosphorylation, thereby activating its endonuclease activity.
Then a 26 bp intron in XBP1 precursor mRNA is sliced, producing the
active sXBP-1 (XBP1S). XBP1S enters the nucleus to bind to the
promoter of ERS response element (ERSE), inducing the expression of
molecular chaperones and foldase genes, and up-regulate the
ER-associated degradation (ERAD) pathway-related proteins, thus
promoting the correct folding and maturation of protein and the
degradation of misfolded proteins (15).
Recent studies have shown that the ERS response
exists throughout the occurrence and development of AS,
participates in the activity regulation and apoptosis of vascular
endothelial cells, vascular smooth muscle cells and macrophages,
and plays an important role in AS caused by such risk factors such
as hyperlipidemia, homocysteine and hyperglycemia (16–19). The
activation of AMPK is associated with phosphorylation of eIF2α, and
studies have revealed that atorvastatin can activate AMPK and
reduce homocysteine-induced ERS response, thereby alleviating
vascular wall damage and progression of AS (20).
In conclusion, this study confirmed that
atorvastatin can relieve ERS and reduce atherosclerotic plaque
formation in ApoE−/− mice fed with high-fat
diets. Atorvastatin exerts its inhibitory effect on ERS through
AMPK, but its further signaling mechanism still needs in-depth
study.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
WX, MF and ZZ designed the study and performed the
experiments, WX and CW established the animal models, MF and WW
collected the data, RL and LS analyzed the data, WX, MF and ZZ
prepared the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was approved by the Animal Ethics
Committee of Fudan University Animal Center (Qingpu, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Zhang ZY, Hu CF, Wang MX, Lin J, Li JM and
Wang RZ: Research on mechanism of PCS in damaging vascular
endothelial cells and promoting formation of atherosclerosis via
TLR4/TREM-1. Eur Rev Med Pharmacol Sci. 22:7533–7542.
2018.PubMed/NCBI
|
|
2
|
Williams KJ and Tabas I: The
response-to-retention hypothesis of atherogenesis reinforced. Curr
Opin Lipidol. 9:471–474. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tabas I: Macrophage death and defective
inflammation resolution in atherosclerosis. Nat Rev Immunol.
10:36–46. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moore KJ and Tabas I: Macrophages in the
pathogenesis of atherosclerosis. Cell. 145:341–355. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kedi X, Ming Y, Yongping W, Yi Y and
Xiaoxiang Z: Free cholesterol overloading induced smooth muscle
cells death and activated both ER- and mitochondrial-dependent
death pathway. Atherosclerosis. 207:123–130. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nakano T, Watanabe H, Ozeki M, Asai M,
Katoh H, Satoh H and Hayashi H: Endoplasmic reticulum
Ca2+ depletion induces endothelial cell apoptosis
independently of caspase-12. Cardiovasc Res. 69:908–915. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Levine YC, Li GK and Michel T:
Agonist-modulated regulation of AMP-activated protein kinase (AMPK)
in endothelial cells. Evidence for an AMPK -> Rac1 -> Akt
-> endothelial nitric-oxide synthase pathway. J Biol Chem.
282:20351–20364. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Y, Huang Y, Lam KS, Li Y, Wong WT, Ye
H, Lau CW, Vanhoutte PM and Xu A: Berberine prevents hyperglycemia-
induced endothelial injury and enhances vasodilatation via
adenosine monophosphate-activated protein kinase and endothelial
nitric oxide synthase. Cardiovasc Res. 82:484–492. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bao XM, Wu CF and Lu GP: Atorvastatin
inhibits homocysteine- induced oxidative stress and apoptosis in
endothelial progenitor cells involving Nox4 and p38MAPK.
Atherosclerosis. 210:114–121. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Heeba G, Hassan MK, Khalifa M and Malinski
T: Adverse balance of nitric oxide/peroxynitrite in the
dysfunctional endothelium can be reversed by statins. J Cardiovasc
Pharmacol. 50:391–398. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Arad M, Seidman CE and Seidman JG:
AMP-activated protein kinase in the heart: Role during health and
disease. Circ Res. 100:474–488. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hetz C: The unfolded protein response:
Controlling cell fate decisions under ER stress and beyond. Nat Rev
Mol Cell Biol. 13:89–102. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Minamino T, Komuro I and Kitakaze M:
Endoplasmic reticulum stress as a therapeutic target in
cardiovascular disease. Circ Res. 107:1071–1082. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma Y and Hendershot LM: Delineation of a
negative feedback regulatory loop that controls protein translation
during endoplasmic reticulum stress. J Biol Chem. 278:34864–34873.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lerner AG, Upton JP, Praveen PV, Ghosh R,
Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, et
al: IRE1α induces thioredoxin-interacting protein to activate the
NLRP3 inflammasome and promote programmed cell death under
irremediable ER stress. Cell Metab. 16:250–264. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ishiyama J, Taguchi R, Akasaka Y, Shibata
S, Ito M, Nagasawa M and Murakami K: Unsaturated FAs prevent
palmitate-induced LOX-1 induction via inhibition of ER stress in
macrophages. J Lipid Res. 52:299–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thorp E, Li G, Seimon TA, Kuriakose G, Ron
D and Tabas I: Reduced apoptosis and plaque necrosis in advanced
atherosclerotic lesions of Apoe−/− and
Ldlr−/− mice lacking CHOP. Cell Metab. 9:474–481. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Luo Y, Li SJ, Yang J, Qiu YZ and Chen FP:
HMGB1 induces an inflammatory response in endothelial cells via the
RAGE-dependent endoplasmic reticulum stress pathway. Biochem
Biophys Res Commun. 438:732–738. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Larroque-Cardoso P, Swiader A, Ingueneau
C, Nègre-Salvayre A, Elbaz M, Reyland ME, Salvayre R and Vindis C:
Role of protein kinase Cδ in ER stress and apoptosis induced by
oxidized LDL in human vascular smooth muscle cells. Cell Death Dis.
4:e5202013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jia F, Wu C, Chen Z and Lu G: Atorvastatin
inhibits homocysteine-induced endoplasmic reticulum stress through
activation of AMP-activated protein kinase. Cardiovasc Ther.
30:317–325. 2012. View Article : Google Scholar : PubMed/NCBI
|