1. Introduction
Glaucoma is defined as a group of diseases with
progressive loss of the neuroretinal margin of the optic disc that
causes characteristic degenerative optic neuropathy (1). There is a sufficient number of studies
in the literature focusing on the topic of neuroprotection in
glaucoma (2-5).
Therefore, we will not deal with the issue of antioxidants,
adenosine receptor antagonists, nicotinic acetylcholine agonists,
neurotrophic factors, metabolic products in ganglion cell necrosis
and apoptosis, etc.
2. Electroretinogram and visual evoked
potentials
One of the first stimuli that led us to the study of
glaucoma was the simultaneous measurement of the pattern
electroretinogram (PERG) and pattern visual evoked potential (PVEP)
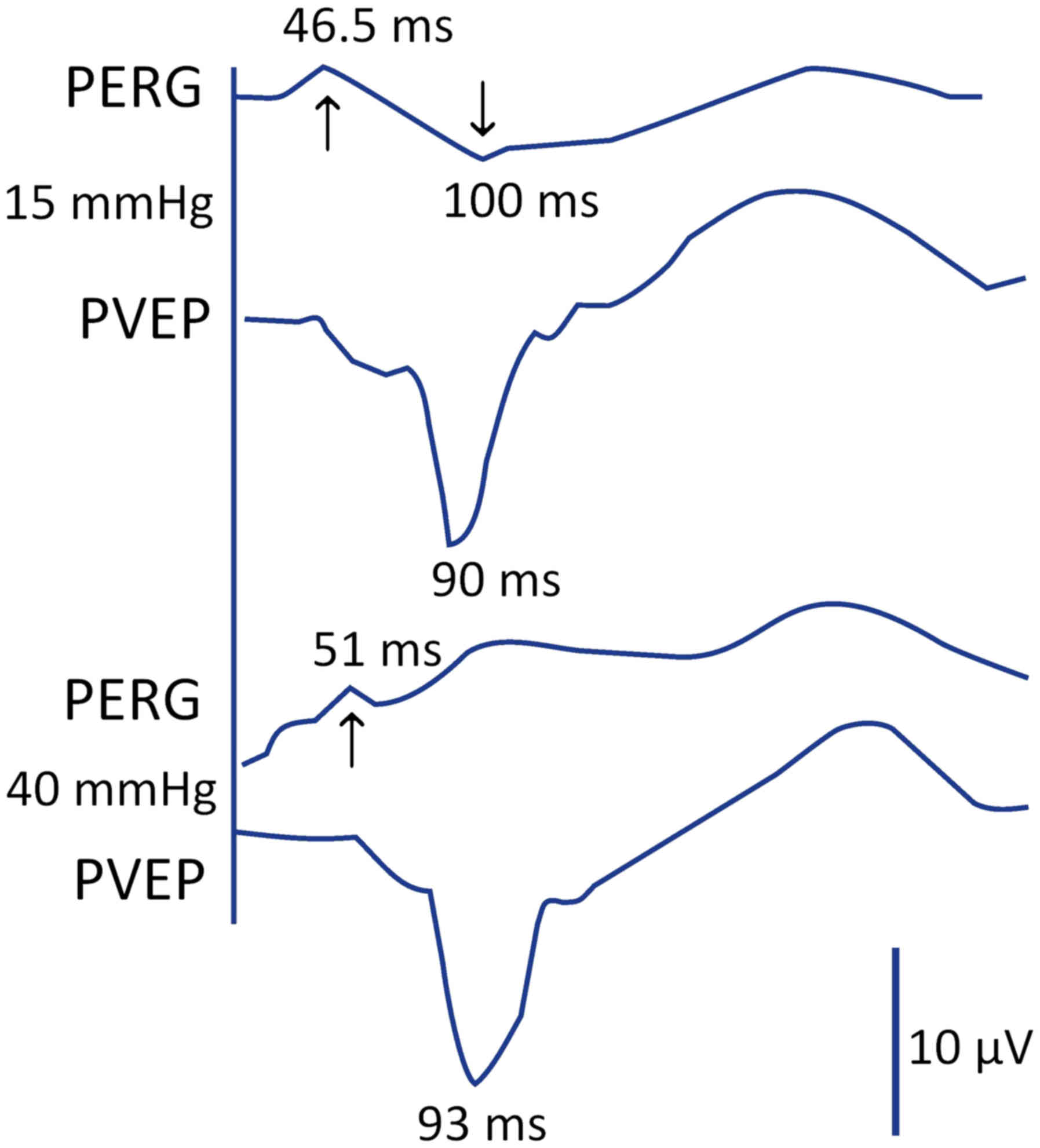
in a 20-year-old healthy individual. At first, the intraocular
pressure (IOP) was 15mmHg and it subsequently increased to 40 mmHg.
Surprisingly, neurotransmission was blocked at the level of the
retinal ganglion cell level, while PVEP changed slightly (Fig. 1). This fact did not correspond to the
existing definitions of glaucoma regarding impairment of the
retinal ganglion cell axons with excavation on the optic disc and
changes in the visual field. With the blockade of transport at the
level of the ganglion cells, we expected the absence of, or at
least abnormal PVEP response. Measurements were taken in
1987(6).
Therefore, we searched for an answer to this
response of the visual analyser. Several questions remained
unanswered. Why did the retinal ganglion cells not respond and what
happened to the central visual pathway, when we get an almost
normal response following the blockade at the level of the retinal
ganglion cells in the brain? What is the reason for us not noticing
the first changes at the level of the axons of the retinal ganglion
cell, when all the previously available glaucoma definitions
indicated this? There is one explanation for an
electrophysiologist. Following the stabilisation of the binocular
functions, the visual cortex is set up to receive a certain amount
of action potentials. When it is decreased at any level from the
photoreceptors to the cortical cells, it starts to use the feedback
processes to determine at which level this lesion occurred
(7-10).
Before explaining the above mechanisms, I need to
briefly explain the process of transmission of the electrical
changes in the visual pathway, from the photoreceptors to the
visual centres of the brain.
3. Transmission of the electrical changes in
visual pathway
Following the impact of light on the retina, a
chemical change occurs in the outer photoreceptor segments
(cis-retinal is changed to trans-form). This causes their
hyperpolarisation (11).
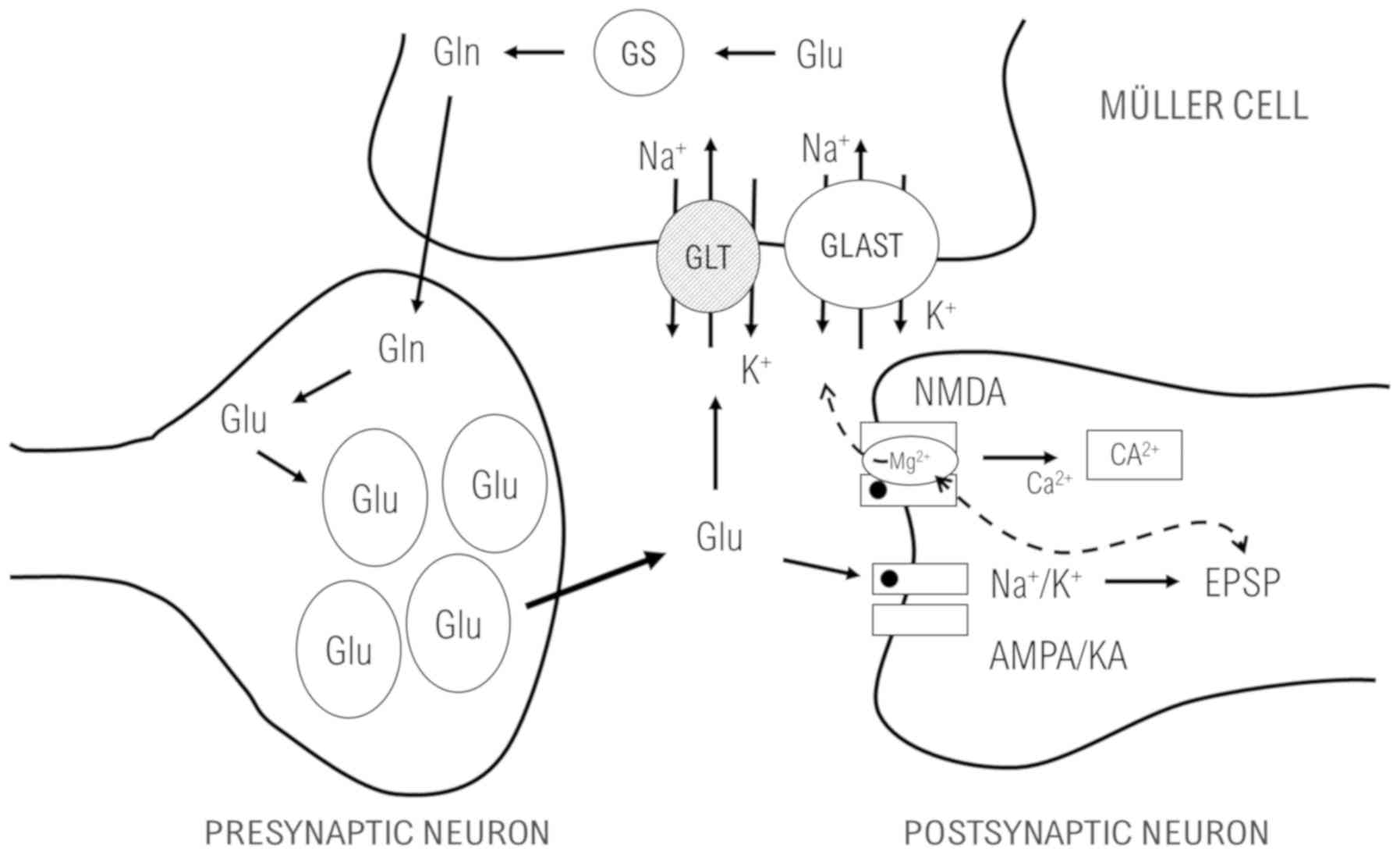
Hyperpolarisation of the photoreceptors during the
synaptic transfer causes a release of glutamate from the
presynaptic neuron into the synaptic cleft and subsequent binding
to the receptors located on the membrane of the postsynaptic neuron
(12).
Glutamate is bound to the receptors which were named
based on their selective agonists. N-methyl-D-aspartate is a
typical agonist for the NMDA receptors. A typical agonist for the
AMPA receptors is α-amino-3-hydroxy-5-methyl-4-8 isoxazolpropionate
(AMPA), and for the third type, kainate receptors, kainate. The
AMPA and kainate receptors are also called non-NMDA (13).
The NMDA receptors represent ion channels permeable
for calcium (Ca) ions. Calcium flow through the NMDA receptors is
blocked by the magnesium (Mg) ions at a normal membrane potential.
This block can be eliminated by strong depolarisation (14).
Excessive calcium influx into the cells through the
NMDA voltage-gated channel can be caused by hypoxia, hypoglycaemia,
etc. Under these conditions, the level of glutamate in the synaptic
cleft remains elevated for a long time, with sustained activation
of the NMDA receptors, resulting in such intracellular calcium
concentrations that are cytotoxic. Inhibition of the NMDA receptors
can delay this dying, using their antagonists (15).
Concentration of free glutamate in the synaptic
cleft achieves approximately 1.1 mM during the synaptic transfer.
However, its concentration quickly drops and it breaks down in the
NMDA receptors during 1.2 ms. However, glutamate is dissociated
much faster from the AMPA receptors. Thus, the time course of free
glutamate predicts that dissociation contributes to the breakdown
of the postsynaptic flow mediated by the AMPA receptors. Otherwise,
the voltage-gated channels would open (12).
Glutamate in the mammalian central nervous system is
eliminated from the synapsis mainly by the glutamate transporters
of the excitatory amino acid transporter type (EEAT) and glutamate
aspartate transporter (GLAST) as glutamate transporter to the
Muller cells (MC) and glutamine synthetase (GS) as glutamate to
glutamine in MC (16,17) (Fig.
2).
Subsequently, in glial cells, glutamate is converted
to glutamine, which no longer acts as a neurotransmitter and can
thus be released back into the synapsis, from where it is
subsequently taken up by the presynaptic neuron, which converts it
back to glutamate (18).
To date, there is no evidence of the presence of an
enzyme that would convert glutamate directly in the synapsis
(19).
All of the above is to explain the processes
involved in the transmission of the electrical voltage changes in
the visual pathway.
4. Restoring of action potentials
We have two possibilities to recover the amount of
action potentials coming to the brain to the baseline values. The
first is the release of a greater amount of neurotransmitter at the
level of the ‘damaged’ cell, and the second is to keep this
neurotransmitter in the synaptic cleft for a longer period. Both
possibilities were experimentally proven in glaucoma.
In the vitreous humour of the glaucoma eyes of
experimental animals, the glutamate (27 µM) value was up to 3-fold
higher compared to the control group. These values are toxic both
for the ganglion cell layer and for the internal plexiform layer
(20).
The GLAST and GS values were increased after just 3
weeks, following the increase of the IOP in rats. The number of
ganglion cells was decreased to 6 and 44% after 4-60 weeks from the
increase of IOP, respectively (21).
Glutamate receptors are expressed not only in the retinal ganglion
cells, but also in the photoreceptors, as well as in the horizontal
and bipolar cells (22).
The long-term effect of glutamate on the non-NMDA
receptors increases the postsynaptic potential and opens the
voltage-gated receptors that are normally closed by magnesium and
the entry of calcium into the cell. This process takes places in
all cells which have glutamate receptors. Therefore, not only the
retinal ganglion cells are impaired, but also the cells in the
internal core layer and the layer of photoreceptors (23).
The question remains why the signal transduction
failure occurred at the level of the retinal ganglion cells. We
found the explanation in the study by Shou et al (24). They qualitatively studied alpha and
beta retinal ganglion cells following acute increase of IOP. The
analysis found that cell density, size of the body, maximum
diameter of the dendritic field, total dendritic length and the
number of branches of dendritic bifurcations were significantly
decreased in the glaucoma eyes, compared to the healthy group. Loss
of cells and shrinking of dendrites in the type alpha retinal
ganglion cells were more pronounced compared to the beta cells. The
density of all types of retinal cells and corpus geniculatum
laterale declined over time if the IOP was increased, and the loss
of cells was more significant in large cells (alpha) compared to
small cells (beta). Ischaemia has a major influence on the decrease
of the dendritic diameter and cells alone. Larger ganglion cells
are more sensitive to the environmental changes (ischaemia) because
of their energy performance (24).
The nerve cells do not die immediately following the
influx of calcium to the cells. As stated above, their size is
first reduced. If they have a sufficient energetic reserve, they
will cope with this state. As soon as the energy is depleted, the
apoptotic or necrotic process is initiated and the cell dies.
5. Damage of the visual brain centres
If the visual pathway, including the visual cortex,
is involved in the process of hypertensive glaucoma, then we should
also find changes in the brain. The standard structural examination
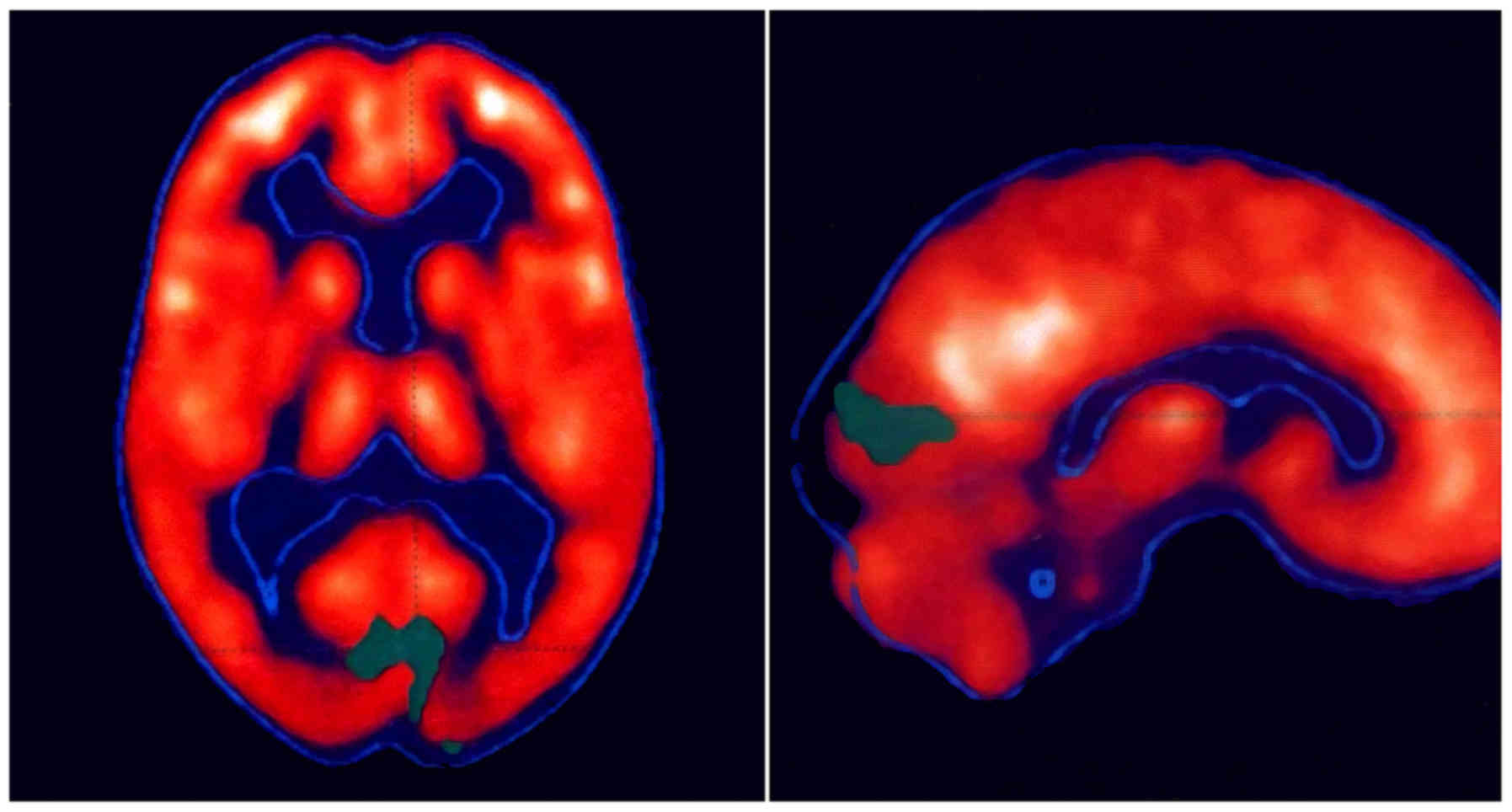
techniques do not make this diagnosis possible. For this reason, we
used positron emission tomography. Radioactive glucose (18
fluorodeoxyglucose), which is taken up in healthy cells, is used to
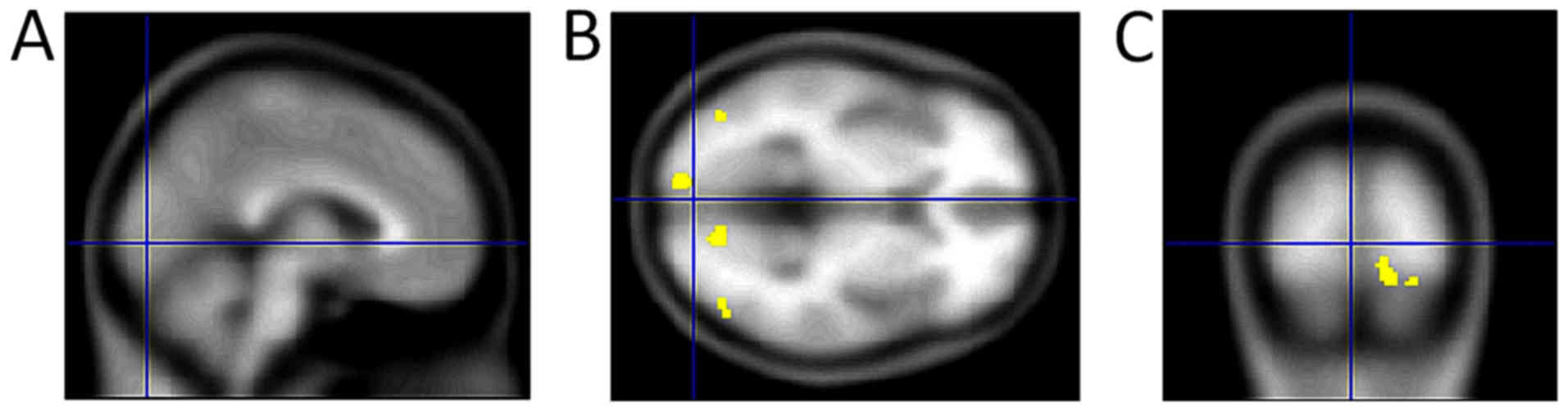
examine brain activity. Fig. 3 shows
the absence of glucose radioactivity in the area of the occipital
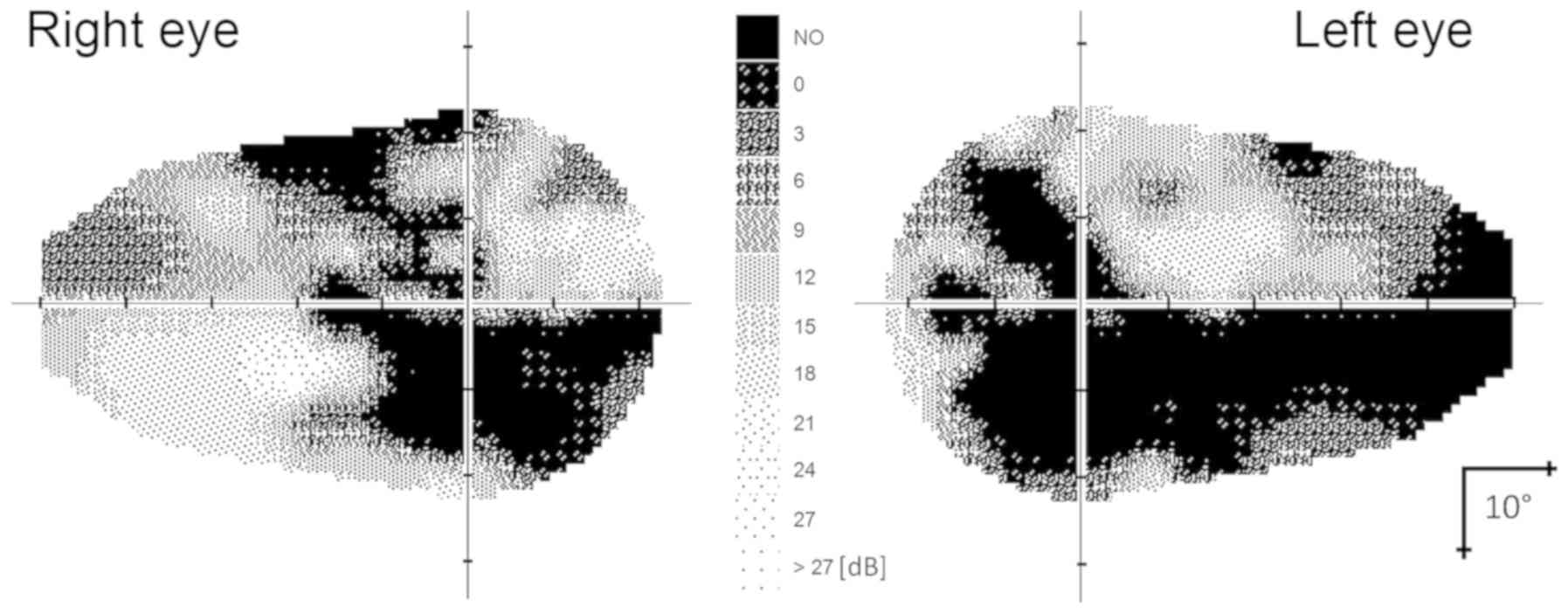
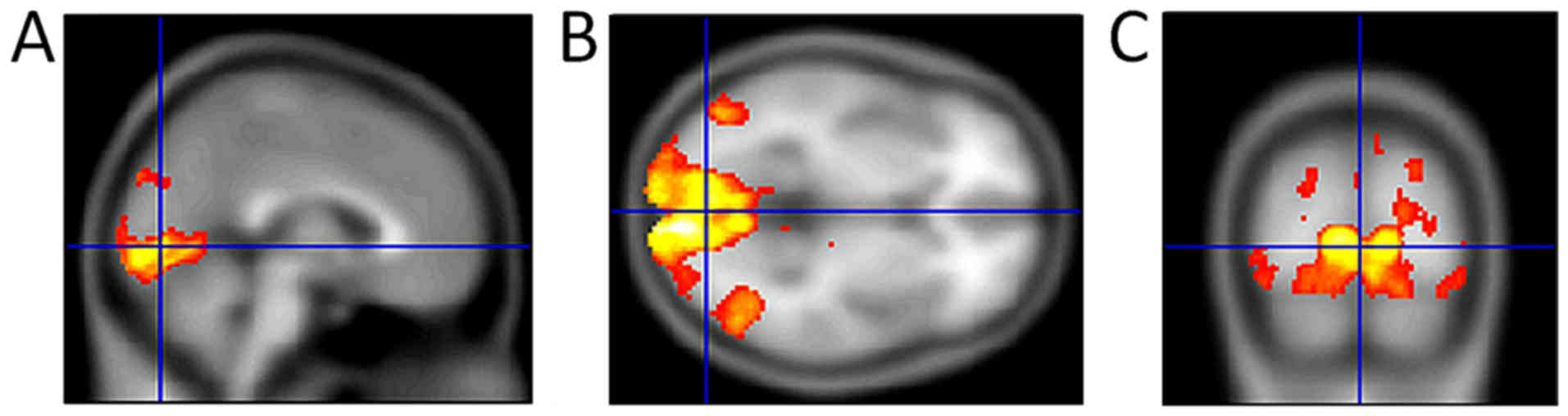
lobe. The examination was performed in 2001(25). Visual field and image of functional
magnetic resonance imaging (fMRI) are shown in Figs. 4 and 5. For comparison, we also present the
normal fMRI findings in a female patient with normotensive glaucoma
(Fig. 6). Using positron emission
tomography and fMRI, we found that damage of the visual brain
centres occurs in hypertensive glaucoma as well.
6. Determining the level and depth of
damage
During the experimental glaucoma, the
electroretinographic changes (decrease of the amplitudes by up to
50%) preceded the changes in the retinal neuronal fibre layer
(26). These facts, as well as the
conclusions of other authors (9,24,26),
forced us to use electrophysiological methods (PERG and PVEP) to
determine the level and depth of damage in various types of
hypertensive glaucoma (27-29).
Based on these examinations regarding the changes in PERG and PVEP,
we concluded that glaucoma causes damage not only to retinal
ganglion cells and subsequently their axons, but also to the visual
centres in the brain (30).
At the level of neuronal membrane, the mutual
relationship of both neurotransmitter systems is documented by
direct inhibition of the NMDA receptor by dopamine and the
inhibitory effect of glutamate on the release of dopamine. This
means that a higher level of dopamine blocks the NMDA receptor and,
conversely, glutamate blocks the release of dopamine (31,32).
We used the examination of the oscillation
potentials of the electroretinogram for verification of this
biochemical information. Amacrine cells are divided into
dopaminergic and GABAergic, based on the neurotransmitter.
Dopaminergic cells generate oscillation potentials in the
electroretinogram and GABAergic cells take part in the development
of the threshold scotopic potential (33).
We performed the examinations in 2001, using the
Primus (Lacce Elettronica) device according to the ISCEV
methodology (1989). Following a 30-min adaptation to dark, we
examined the oscillation potentials. Stimulation of the retina
during artificial mydriasis (0, 5% tropicamid) was performed, using
the flashlight of 5 ms in length and luminous flux of 2.5
cd/m2/s. Ten responses (stimuli in 15-sec intervals)
were averaged, using filters from 80 to 500 Hz. We evaluated the
latency and amplitude of the P2 oscillation.
The first group consisted of 23 eyes of healthy
people. In the second group, there were 36 glaucoma eyes with
imminent changes in the visual field with compensated IOP. The mean
age of the persons included in the groups was 40.3 years (range,
35-56). The results showed a significant prolongation of latency of
the P2 oscillation (P=0.049) and a decrease of the oscillation
amplitude (P=0.001) in glaucoma eyes, compared to the healthy
group.
We demonstrated indirectly increased values of
glutamate in the glaucoma eyes. We were also interested in how the
retinal ganglion cells (PERG) would behave during modification of
the anti-glaucoma therapy and subsequently the complete visual
analyser (PVEP).
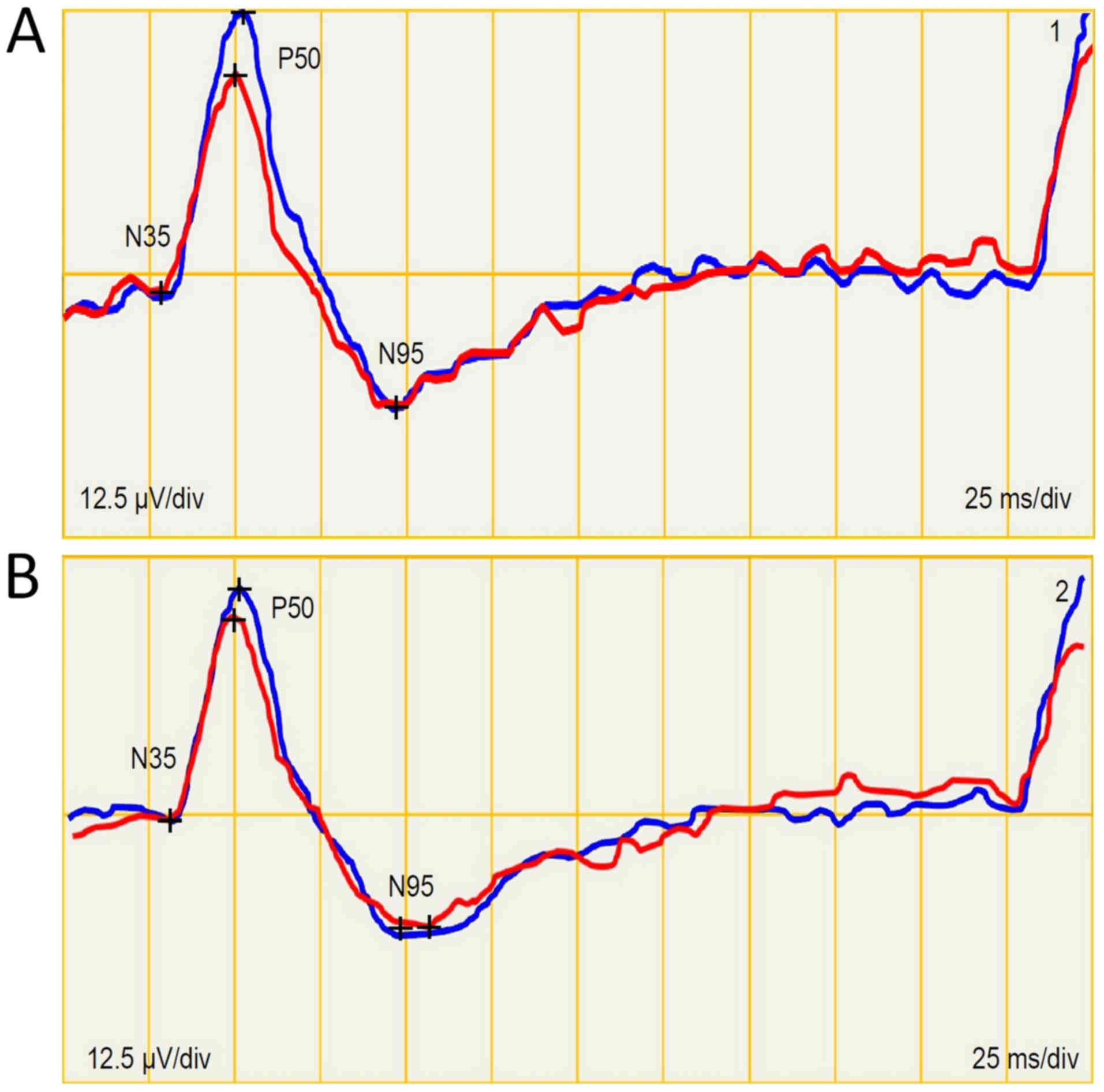
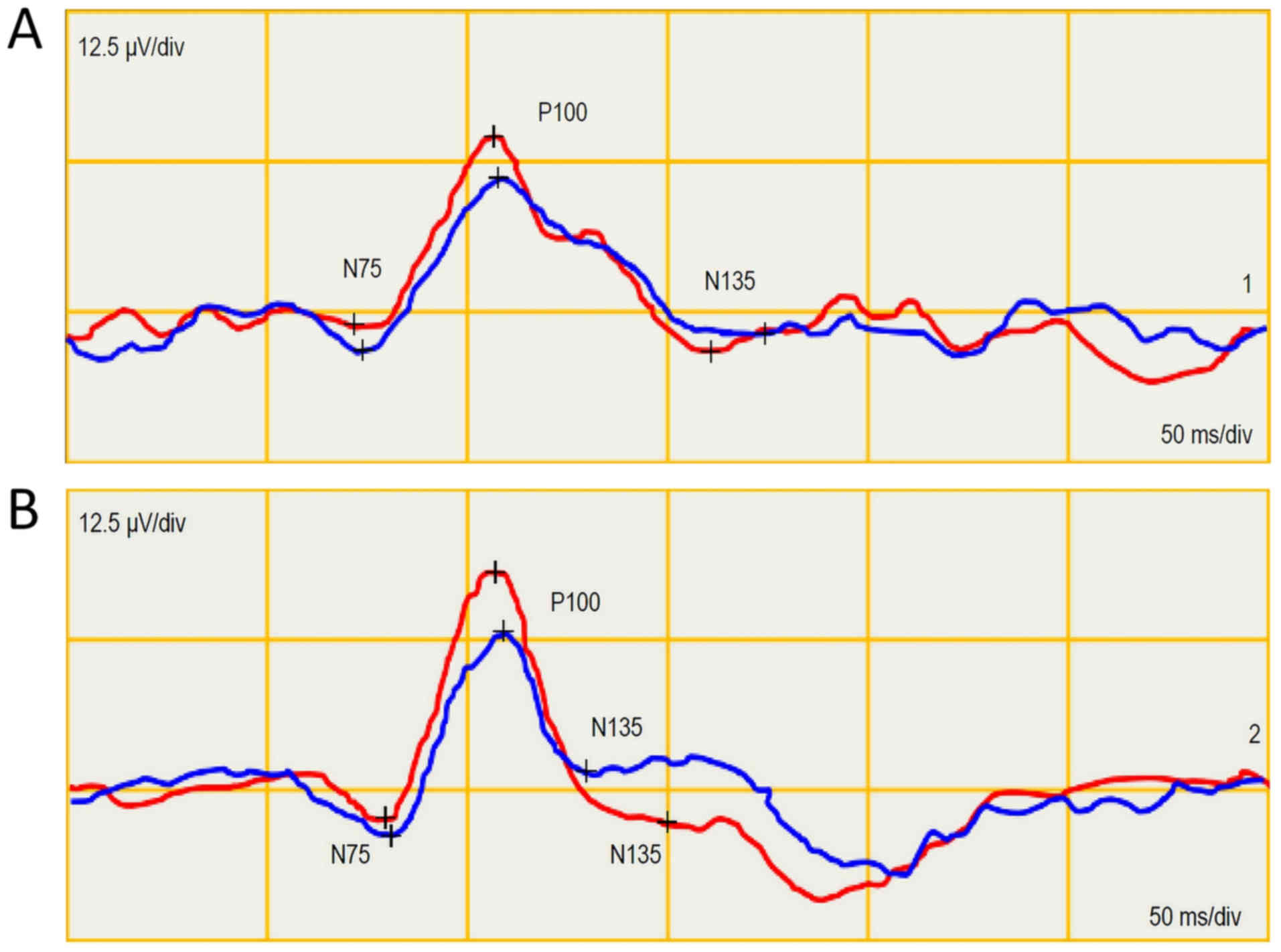
We performed the PERG and PVEP examination (using
the ISCEV methodology-2012 on the Roland Consult SRN device) in a
female patient (64 years) with glaucoma, compensated with
dorzolamid, timolol meleas and brominidin to the IOP of 18/18 mmHg.
The visual field was within the normal ranges, c/d=0.4 and nerve
fiber layer index 11/20 (normal values). With regard to these
values, we repeated the examination in one month, following
discontinuation of both anti-glaucoma medications. IOP was
increased to 23/29 mmHg. The PERG amplitude P50 and N95 was reduced
by 3.2/1.1 µV following discontinuation of the medication (Fig. 7) and, on the contrary, it was
increased in PVEP by 1.4/4.7 µV (Fig.
8).
This finding also shows alteration of the retinal
ganglion cells and, on the contrary, potentiation of the visual
pathway with glutamate.
Because, even with IOP controlled, the number of
action potentials is reduced due to the loss of cells involved in
processing of the electrical changes in the visual pathway, these
cells are ‘bombarded’ to a higher response by the feedback
mechanisms. Excessive release and decreased absorption of glutamate
from the synaptic cleft result in increase of the postsynaptic
potential. Subsequently, the voltage-gated channels are then
unblocked for influx of calcium into the cells and the whole
process progresses. With disease progression, the response to
releasing more neurotransmitter is also higher.
We also confirmed this in the study in which we
observed progression of changes in the visual fields in the
compensated glaucoma eyes in a five-year period. We found that the
bigger the initial perimetric changes were, the bigger was their
progression in five years (34).
Electrophysiological methods are not commonly used
to diagnose glaucoma. They are used in our clinic in questionable
cases, but mainly to verify normotensive glaucoma. Based on this
knowledge, neuroprotective therapy may be offered. We put a
decrease of the IOP first. This is followed by a decrease of
glutamate in the synaptic cleft and a blockade of its binding to
the NMDA receptors. Supply of the energy substrates to altered
nerve cells is also indispensable. Local ophthalmologic treatment
will not affect subcortical and cortical visual centers.
Neuroprotective treatment should be systemic because of impairment
of the complete visual pathway. However, attention should be drawn
to the side effect of NMDA receptor antagonists, which induce
symptoms like schizophrenia (35).
7. Conclusion
Impairment of the whole visual pathway occurs in
hypertensive glaucoma. Therefore, early diagnosis of the disease is
important. Treatment should be based not only on a reduction of the
IOP, but also on a decrease in glutamate levels in the synaptic
cleft and their binding to glutamate receptors. Delivery of the
energy substrate to the nerve cells, with the possibility of
dealing with the intracellular processes is an important part of
therapy. This therapy should be systemic.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All datasets generated and/or analysed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
JL is the author of the main idea, and designed and
created the main theoretical parts of this review. MF contributed
to the design and implementation of research, examination image
results analysis and to writing of the manuscript. JL explained the
ophthalmological and electrophysiological context.
Ethics approval and consent to
participate
All patient results and images included in this
review were retrospectively used with prior patient consent. The
consent was in accordance with the principles stated in the
Helsinki Declaration and as approved by the Internal Ethics
Committee of the Eye Clinic JL Faculty of Biomedical Engineering
CTU in Prague.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tham YC, Li X, Wong TY, Quigley HA, Aung T
and Cheng CY: Global prevalence of glaucoma and projections of
glaucoma burden through 2040: A systematic review and
meta-analysis. Ophthalmology. 121:2081–2090. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Osborne NN, Chidlow G, Wood J and Casson
R: Some current ideas on the pathogenesis and the role of
neuroprotection in glaucomatous optic neuropathy. Eur J Ophthalmol
13. (Suppl 3):S19–S26. 2003.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Gauthier AC and Liu J: Neurodegeneration
and neuroprotection in glaucoma. Yale J Biol Med. 89:73–79.
2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Almasieh M and Levin LA: Neuroprotection
in glaucoma: animal models and clinical trials. Ann Rev Vis Sci.
3:91–120. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Pardue MT and Allen RS: Neuroprotective
strategies for retinal disease. Prog Retin Eye Res. 65:50–76.
2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lešták J, Tintěra J, Kynčl M, Svatá Z and
Rozsíval P: High tension glaucoma and normal tension glaucoma in
brain MRI. J Clin Exp Ophthalmol. 4(291)2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Sherman SM and Guillery RW: Exploring the
thalamus and its role in cortical function. 2nd edition MIT Press,
Boston. 2006.
|
|
8
|
Shou TD: The functional roles of feedback
projections in the visual system. Neurosci Bull. 26:401–410.
2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Briggs F and Usrey WM: Corticogeniculate
feedback and visual processing in the primate. J Physiol.
589:33–40. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Thompson AD, Picard N, Min L, Fagiolini M
and Chen C: Cortical feedback regulates feedforward
retinogeniculate refinement. Neuron. 91:1021–1033. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kiser PD, Golczak M, Maeda A and
Palczewski K: Key enzymes of the retinoid (visual) cycle in
vertebrate retina. Biochim Biophys Acta. 182:137–151.
2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Clements JD, Lester RA, Tong G, Jahr CE
and Westbrook GL: The time course of glutamate in the synaptic
cleft. Science. 258:1498–1501. 1992.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Kew JN and Kemp JA: Ionotropic and
metabotropic glutamate receptor structure and pharmacology.
Psychopharmacology (Berl). 179:4–29. 2005.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Johnson JW and Ascher P: Voltage-dependent
block by intracellular Mg2+ of N-methyl-D-aspartate-activated
channels. Biophys J. 57:1085–1090. 1990.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Choi DW, Koh JY and Peters S: Pharmacology
of glutamate neurotoxicity in cortical cell culture: attenuation by
NMDA antagonists. J Neurosci. 8:185–196. 1988.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Rothstein JD, Martin L, Levey AI,
Dykes-Hoberg M, Jin L, Wu D, Nash N and Kuncl RW: Localization of
neuronal and glial glutamate transporters. Neuron. 13:713–725.
1994.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Amara SG and Fontana AC: Excitatory amino
acid transporters: Keeping up with glutamate. Neurochem Int.
41:313–318. 2002.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Danbolt NC: Glutamate uptake. Prog
Neurobiol. 65:1–105. 2001.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Huang YH and Bergles DE: Glutamate
transporters bring competition to the synapse. Curr Opin Neurobiol.
14:346–352. 2004.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Vorwerk CK, Gorla MS and Dreyer EB: An
experimental basis for implicating excitotoxicity in glaucomatous
optic neuropathy. Surv Ophthalmol 43. (Suppl 1):S142–S150.
1999.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Woldemussie E, Wijono M and Ruiz G: Muller
cell response to laser-induced increase in intraocular pressure in
rats. Glia. 47:109–119. 2004.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Shen Y, Liu XL and Yang XL:
N-methyl-D-aspartate receptors in the retina. Mol Neurobiol.
34:163–179. 2006.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Pavlidis M, Stupp T, Naskar R, Cengiz C
and Thanos S: Retinal ganglion cells resistant to advanced
glaucoma: A postmortem study of human retinas with the carbocyanine
dye DiI. Invest Ophthalmol Vis Sci. 44:5196–5205. 2003.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Shou T, Liu J, Wang W, Zhou Y and Zhao K:
Differential dendritic shrinkage of alpha and beta retinal ganglion
cells in cats with chronic glaucoma. Invest Ophthalmol Vis Sci.
44:3005–3010. 2003.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Lestak J, Tintera J, Svata Z, Ettler L and
Rozsival P: Glaucoma and CNS. Comparison of fMRI results in high
tension and normal tension glaucoma. Biomed Pap Med Fac Univ
Palacky Olomouc Czech Repub. 158:144–153. 2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Fortune B, Bui BV, Morrison JC, Johnson
EC, Dong J, Cepurna WO, Jia L, Barber S and Cioffi GA: Selective
ganglion cell functional loss in rats with experimental glaucoma.
Invest Ophthalmol Vis Sci. 45:1854–1862. 2004.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Holder GE: Pattern electroretinography
(PERG) and an integrated approach to visual pathway diagnosis. Prog
Retin Eye Res. 20:531–561. 2001.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Parisi V, Miglior S, Manni G, Centofanti M
and Bucci MG: Clinical ability of pattern electroretinograms and
visual evoked potentials in detecting visual dysfunction in ocular
hypertension and glaucoma. Ophthalmology. 113:216–228.
2006.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Nebbioso M, Gregorio FD, Prencipe L and
Pecorella I: Psychophysiological and electrophysiological testing
in ocular hypertension. Optom Vis Sci. 88:E928–E939.
2011.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lestak J, Nutterova E, Pitrova S, Krejcova
H, Bartosova L and Forgacova V: High tension versus normal tension
glaucoma. A comparison of structural and functional examinations. J
Clinic Exp Ophthalmol. S5(006)2012. View Article : Google Scholar
|
|
31
|
Castro NG, de Mello MC, de Mello FG and
Aracava Y: Direct inhibition of the N-methyl-D-aspartate receptor
channel by dopamine and (+)-SKF38393. Br J Pharmacol.
126:1847–1855. 1999.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wu Y, Pearl SM, Zigmond MJ and Michael AC:
Inhibitory glutamatergic regulation of evoked dopamine release in
striatum. Neuroscience. 96:65–72. 2000.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Kaneko M, Sugawara T and Tazawa Y:
Electrical responses from the inner retina of rats with
streptozotocin-induced early diabetes mellitus. Nippon Ganka Gakkai
Zasshi. 104:775–778. 2000.(In Japanese). PubMed/NCBI
|
|
34
|
Lešták J and Rozsíval P: The influence of
corneal thickness on progression of hypertensive glaucoma. J Clin
Exp Ophthalmol. 3(245)2012. View Article : Google Scholar
|
|
35
|
Javitt DC and Zukin SR: Recent advances in
the phencyclidine model of schizophrenia. Amer J Psychiatry.
148:1301–1308. 1991.PubMed/NCBI View Article : Google Scholar
|