Introduction
Lung cancer is a major cause of cancer-related
death, and the 5-year survival rate remains very low, as more than
half of patients are diagnosed too late for successful treatment
(1). Advances in molecular
translational research have resulted in greater understanding,
diagnosis and management of lung cancer, improving patient survival
rates (2). To date, efforts to
develop innovative treatments have focused on targeting key
signaling pathways involved in lung cancer growth and progression
(3). Although considerable progress
in targeted therapies has been achieved, further advances are
required to improve prognosis and increase the overall life
expectancy of patients with lung cancer (4). Distinct types of cancer result from the
abnormal proliferation of different cells in the organism, and
their properties and responses to treatment can vary substantially
(5). Among the hallmarks of cancer
cells, insensitivity to anti-growth signals, evading apoptosis and
sustained angiogenesis are common in most, if not all, types of
cancer (5,6). Accordingly, an effective strategy to
treat cancer is the termination of out-of-control cell growth
through activation of the intrinsic mechanisms of cell death
(7).
Panax ginseng, the most common species of
ginseng, is a herbal medicine that is widely applied in Asian
countries (8). P. ginseng has
been suggested to possess numerous beneficial properties, including
anti-inflammatory, antioxidant and anticancer activity (9). Ginsenosides, a form of triterpene
glycosides (saponins), are the major active components in ginseng
and have been extensively used in traditional Chinese medicine as
an anticancer agent (10). It has
been suggested that ginseng extract blocks the proliferation of
mammalian tumor cells by stimulating apoptosis (11). Ginsenoside Rh2 (Rh2) is characterized
by low toxicity, low molecular weight and exhibits good solubility
in lipids. Rh2 has been demonstrated to inhibit proliferation and
migration of tumor cells, as well as angiogenesis. In addition, its
inhibitory effect on angiogenesis in prostate cancer is mediated by
regulating the expression of the metal cation transporter
CNNM1(12). A previous study
suggested that, in liver cancer cells, Rh2 is able to regulate the
expression of a large number of non-coding RNAs (13), and an additional study in breast
cancer cells suggested that Rh2 inhibits proliferation via
epigenetic modifications of the cell-mediated immune pathway
(14). Rh2 has additionally been
suggested to inhibit the migration and invasion of lung cancer
cells by modulation of tumor-induced macrophages (15). Pseudo-Rh2 has also been reported to
induce apoptosis via the Ras/Raf/ERK/p53 pathway in the A549
adenocarcinoma cell line (16).
Together, these findings suggest that Rh2 may exert anticancer
activity through a range of diverse mechanisms.
Wnt signaling is essential during embryonic
development and has a crucial role in the maintenance of the
stem-like properties of tissue cells, including cancer cells
(17). Hedgehog (Hh) signaling
regulates diverse biological processes, among them the development
of invertebrate and vertebrate organisms (18). The canonical Wnt signaling pathway,
also known as the Wnt/β-catenin or β-catenin/T-cell factor pathway
(19), performs its regulatory
function by stabilizing the key transcription factor, β-catenin,
which activates downstream gene expression (20-22).
It is well documented that the activation of Wnt signaling is
closely associated with the development of cancer in numerous types
of tissue (23). Constitutive
activation of Hh signaling affects the development and progression
of cancer through several mechanisms (24). Aberrant activation of Hh signaling is
required for almost all basal cell carcinomas, rhabdomyosarcomas,
medulloblastomas and several other tumor types (18,25-27).
The binding of Hh and protein patched homolog 1 molecules results
in activation of the smoothened, frizzled class receptor (Smo)
protein (26,28), which subsequently upregulates the
expression of downstream transcriptional activator GLI-Kruppel
family transcription factors to stimulate Hh signaling (28). GLI family zinc finger (Gli)1 has been
demonstrated to function as a modulator of cancer cell properties
controlled by E-cadherin/β-catenin signaling. Gli1 activates
expression of the gel-forming mucin gene, MUC5AC, which in
turn inhibits E-cadherin-dependent cell-to-cell adhesion,
activating migration and invasion of pancreatic ductal
adenocarcinoma cells (15). Together
the available evidence indicates the involvement of Wnt and Hh
signaling in cancer cell proliferation. However, whether a
relationship exists between Rh2 and Wnt or Hh signaling remains to
be determined.
The present study aimed to investigate the impact of
Rh2 on the proliferation of A549 lung cancer cells, and on the
expression of Wnt and Hh signaling markers. The relationship
between the expression of β-catenin and Gli1 and the
proliferation of A549 cells in the presence or absence of Rh2 was
examined. The objective of this investigation was to establish the
mechanism by which Rh2 regulates Wnt and Hh signaling and
proliferation in A549 lung cancer cells.
Materials and methods
Cell culture and transfection
assays
The human lung adenocarcinoma cell line A549 was
obtained from the American Type Culture Collection. The cells were
grown in Dulbecco's modified Eagle's medium, supplemented with
glutamine, 10% fetal bovine serum, penicillin and streptomycin
(Gibco; Thermo Fisher Scientific, Inc.). cDNA encoding
β-catenin, α-catenin S641D and Gli1 were
synthesized by Sangon Biotech. Co., Ltd. and pcDNA3.1 (+)
(Invitrogen; Thermo Fisher Scientific, Inc.) was used to construct
overexpression (OX) vectors. Small interfering RNA (siRNA) for
β-catenin (ON-TARGETplus SMART pool; cat. no. L-004018),
siRNA for Gli1 (ON-TARGETplus SMART pool; cat. no.
J-041026-05) and ON-TARGETplus non-targeting pool (cat. no.
D-001810) were purchased from GE Healthcare Dharmacon, Inc. A total
of 1x106 cells were transfected with 2 µg
β-catenin, α-catenin S641D or Gli1 OX
plasmids, pcDNA3.1 (+) empty vector (OX control) and 30 nM siRNAs
on day 0 using Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) and Opti-MEM® I Reduced Serum
Medium (Gibco; Thermo Fisher Scientific, Inc.), according to the
manufacturers' protocol. Cells reached ~30% confluence 24 h
post-transfection and the transfection media were replaced with
full growth medium; cells were harvested for subsequent experiments
and 72 h after transfection.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR) analysis
Total RNA was extracted using TRIzol®
Reagent (Invitrogen; Thermo Fisher Scientific, Inc.) from A549
cells treated with Rh2 (cat. no. 209058; Sigma-Aldrich; Merck KGaA)
or transfected with β-catenin, α-catenin S641D and
Gli1 OX plasmids, or β-catenin and Gli1 siRNAs
for 72 h at 37˚C in an incubator with 5% CO2, according
to manufacturer's protocol. A total of 2 µg RNA was
reverse-transcribed using GoScript™ Reverse
Transcription kit at 42˚C for 1 h (Promega Corporation), according
to the manufacturer's instructions. SYBR® Green Master
Mix (Bio-Rad Laboratories, Inc.) was used to perform qPCR in an
Illumina Eco 3.0 Real-time PCR system (Illumina, Inc.). The
thermocycling conditions consisted of an initial denaturation at
95˚C for 3 min, followed by 40 cycles of denaturation for 30 sec at
95˚C, annealing for 30 sec at 58˚C and extension at 72˚C for 30
sec, ending with a final extension at 72˚C for 5 min. The
transcription levels were normalized against those of GAPDH
using the 2-ΔΔCq method (29). The sequences of primers used are
listed in Table I.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Primer | Sequences
(5'-3') |
|---|
| Wnt3 F |
ATCATAAGGGGCCGCCTGGCGAAGGCTGG |
| Wnt3 R |
CTTGCAGGTGTGCACGTCGTAGA |
| TCF7 F |
CTGCAGACCCCTGACCTCTCT |
| TCF7 R |
ATCCTTGATGCTAGGTTCTGGTGT |
| FZD8 F |
CTGGTGGAGATCCAGTGCTC |
| FZD8 R |
TTGTAGTCCATGCACAGCGT |
| Smo F |
ACCTATGCCTGGCACACTTC |
| Smo R |
AGGAAGTAGCCTCCCACGAT |
| Gli1 F |
CCAGAGTTCAAGAGCCTGG |
| Gli1 R |
CCTCGCTCCATAAGGCTCAG |
| Gli2 F |
GTTCCAAGGCCTACTCTCGCCTG |
| Gli2 R |
5'-CTTGAGCAGTGGAGCACGGACAT-3' |
| Gli3 F |
GGGTGAACAGCATCAAAATGGAG |
| Gli3 R |
CCGATAGCCATGTTGGTGG |
| β-catenin F |
TCGCCAGGATGATCCCAGC |
| β-catenin R |
GCCCATCCATGAGGTCCTG |
| GAPDH F |
GACCTGCCGTCTAGAAAAAC |
| GAPDH R |
CTGTAGCCAAATTCGTTGTC |
Western blot analysis
Both Rh2 treated and OX or siRNA construct
transfected A549 cells were harvested in ice-cold lysis buffer (7 M
urea, 2 M thiourea, 2% CHAPS, 40 mM Tris base, 40 mM dithiothreitol
and 1% protease inhibitor) to obtain whole-cell extracts. Protein
concentration was measured using a Bicinchoninic Acid assay kit
(Merck KGaA). The total proteins were separated on a 10% sodium
dodecyl sulfate polyacrylamide gel electrophoresis gel at 100 V for
2 h following extraction followed by transferal onto Immobilon-P
Transfer Membranes (EMD Millipore). The membranes were incubated in
TBS containing 5% skimmed milk and 0.05% Tween-20 (EMD Millipore)
at 25˚C for blocking for 1 h, followed by incubation with the
following primary antibodies at 25˚C for overnight: Anti-vimentin
(cat. no. ab193555; 1:1,000; Abcam), anti-E-cadherin (cat. no.
ab194982; 1:1,000; Abcam), anti-Smo (cat. no. ab8969; 1:1,000;
Abcam), anti-β-catenin (cat. no. ab16051; 1:2,000; Abcam),
anti-Gli1 (cat. no. ab49314; 1:2,000; Abcam), anti-α-catenin (cat.
no. 3240; 1:1,000; Cell Signaling Technology, Inc.),
anti-phosphorylated (p)-α-catenin Ser641 antibody (cat no. 11330,
1:1,000; Signalway Antibody LLC) and anti-GAPDH (cat. no. ab8245;
1:2,000; Abcam). The membranes were washed twice with PBS and
incubated with an anti-mouse or anti-rabbit horseradish
peroxidase-conjugated secondary antibody (cat. no. 7074 and 7076;
1:2,000; Cell Signaling Technology, Inc.) for 1 h at 25˚C.
Antigen-antibody complexes were visualized using an
electrochemiluminescence kit (Beijing BioTrand, Inc.). Protein
levels were normalized against GAPDH and protein expression was
analyzed using ImageJ2 version 2.0 software (National Institutes of
Health).
Phosphopeptide isolation and
analysis
Total protein was extracted from control and
Rh2-treated A549 cells and the protein concentration determined by
bicinchoninic acid assay. A total of 1 mg extracted protein was
digested overnight using trypsin (1:50 wt/wt) at 37˚C. Digested
peptides were extracted and incubated at 25˚C for 15 min in 25 mM
ammonium bicarbonate, and then for 15 min in 5% formic acid.
Samples were desalted on a C18 column (cat no. IQLAALGABXFANUMBBD,
Lancompare) according to the manufacturer's instructions and dried
using a SpeedVac. Phosphopeptides were enriched according to a
previously published protocol (30).
Prior to binding phosphopeptides, the TiO2 beads were
equilibrated with 200 µl 30 mg/ml 2,5-dihydroxybenzoic acid in 80%
acetonitrile and 0.1% TFA, and the pH of the digested peptide
lysate was adjusted to ≤1.9 with 1% TFA. The peptide mixture was
then added to a 2-ml reaction tube containing 10 mg TiO2
beads, and each batch was incubated for 30 min at 25˚C with
end-over-end rotation. Subsequently, the beads were spun down at
500 x g at 4˚C for 15 min and briefly washed once with 80%
acetonitrile and 0.1% TFA, and once with 10% acetonitrile and 0.1%
TFA. Finally, the bound peptides were eluted from the beads using
200 µl NH4OH in 30% acetonitrile (pH>10.0). The
eluate was immediately neutralized with 5% TFA and dried.
The TiO2-enriched
phosphopeptides (4 µl) were subjected to on-line nanoflow liquid
chromatography (LC) using the EASY-Nano LC system (Proxeon
Biosystems; Thermo Fisher Scientific, Inc.) with 10-cm capillary
columns of an internal diameter of 75 µm filled with 3 µm
Reprosil-Pur C18-A2 resin (Dr. Maisch HPLC GmbH). The sequence of
gradients consisted of 10-30% (v/v) CAN in 0.1% (v/v) formic acid
at the flow rate of 200 nl/min for 45 min, 30-100% (v/v) CAN in
0.1% (v/v) formic acid at a flow rate of 200 nl/min for 1 min, and
100% CAN in 0.1% formic acid at a flow rate of 200 nl/min for 10
min. The elution was electrosprayed using a Proxeon
nanoelectrospray ion source by electrospray ionization (ESI). The
ESI-tandem mass spectrometry (MS/MS) analysis was performed using a
Thermo Fisher LTQ Velos Pro (Thermo Fisher Scientific, Inc.) using
full ion scan mode over the m/z range of 200-1,800.
Collision-induced dissociation (CID) was performed in the linear
ion trap using a 4.0-Th isolation width and 35% normalized
collision energy with helium as the collision gas. Five independent
MS/MS scans were performed on each ion using dynamic exclusion.
Additionally, the precursor ion selected for CID was dynamically
excluded from further MS/MS analysis for 30 sec. The MS/MS spectra
were processed using Proteome Discoverer (Version 1.3; Thermo
Fisher Scientific, Inc.), and the database search was performed
using the Mascot search engine (Mascot 2.3; Matrix Science) against
a concatenated target decoy approach. The Uniprot-KB/Swiss-Prot
protein sequence database (release 54.5; https://web.expasy.org/docs/swiss-prot_guideline.html)
was searched, with corresponding taxonomy selection for different
samples. The following search parameters were applied: Mass error
tolerance for the precursor ions, 1 Da; mass error tolerance for
the fragment ions, 0.8 Da; fixed modifications,
carbamidomethylation (C); variable modifications, oxidation (M),
phosphorylation (S, T, Y); number of missed cleavages, 1;
significance threshold, P<0.05; type of instrument, ESI-TRAP.
Protein identifications were validated only if they met the
following three requirements: i) Their score was significant
(P<0.05) with cut-off criteria; ii) one peptide had a score
>15; iii) proteins were identified in at least two out of the
three runs. Proteins identified by a set or subset of peptides used
for identification of another protein were not considered.
Cell proliferation analysis
To determine cell proliferation rate, cells were
plated in a volume of 150 µl at a density of 2,000 cells/well in
96-well plates. Cell proliferation was analyzed following treatment
with 50 or 100 µM Rh2, or transfections with siRNAs and OX plasmids
using a CCK-8 kit (Dojindo Molecular Technologies, Inc.) as
previously described (31). At each
indicated time point (0, 24, 48, 72 and 96 h), MTT solution
(Beyotime Insitute of Biotechnology) was added to each well to a
final concentration of 5 mg/ml followed by incubation at 37˚C for 4
h. A total of 100 µl acidic isopropanol (10% SDS, 5% isopropanol
and 0.01 M HCl) was then added into each well to stop the reaction
and the plates were incubated at 37˚C overnight.
Cell invasion assay
The invasive properties of A549 cells treated with
100 µM Rh2 or transfected with α-catenin S641D were assessed
using a 96-well 3D spheroid cell invasion assay (cat. no.
3500-096-K; Trevigen Inc.; Bio-Techne), according to the
manufacturer's instructions.
Statistical analysis
Statistical analysis was performed with Prism 5
software package (GraphPad Software, Inc.). The data are presented
as the mean ± SEM with at least three experimental replicates.
Comparisons between two groups and the determination of statistical
significance was done by Student's t-test. Comparisons
between more than two groups were performed using one-way ANOVA,
followed by Bonferroni's multiple comparison test.
Results
Rh2 inhibits the proliferation of A549
lung cancer cells
Rh2 is well established as an anticancer molecule
and is widely used in cancer therapy in China, but its function in
lung cancer cells remains unclear (10,11).
Cell proliferation is a key process of spreading of cancer in human
tissues and is directly associated with the severity of the disease
(5). To analyze the function of Rh2
in lung cancer cells, the A549 cells were treated with Rh2 for 24,
48 and 72 h. In comparison with untreated cells, exposure to 50 or
100 µM Rh2 led to significant inhibition of the proliferation of
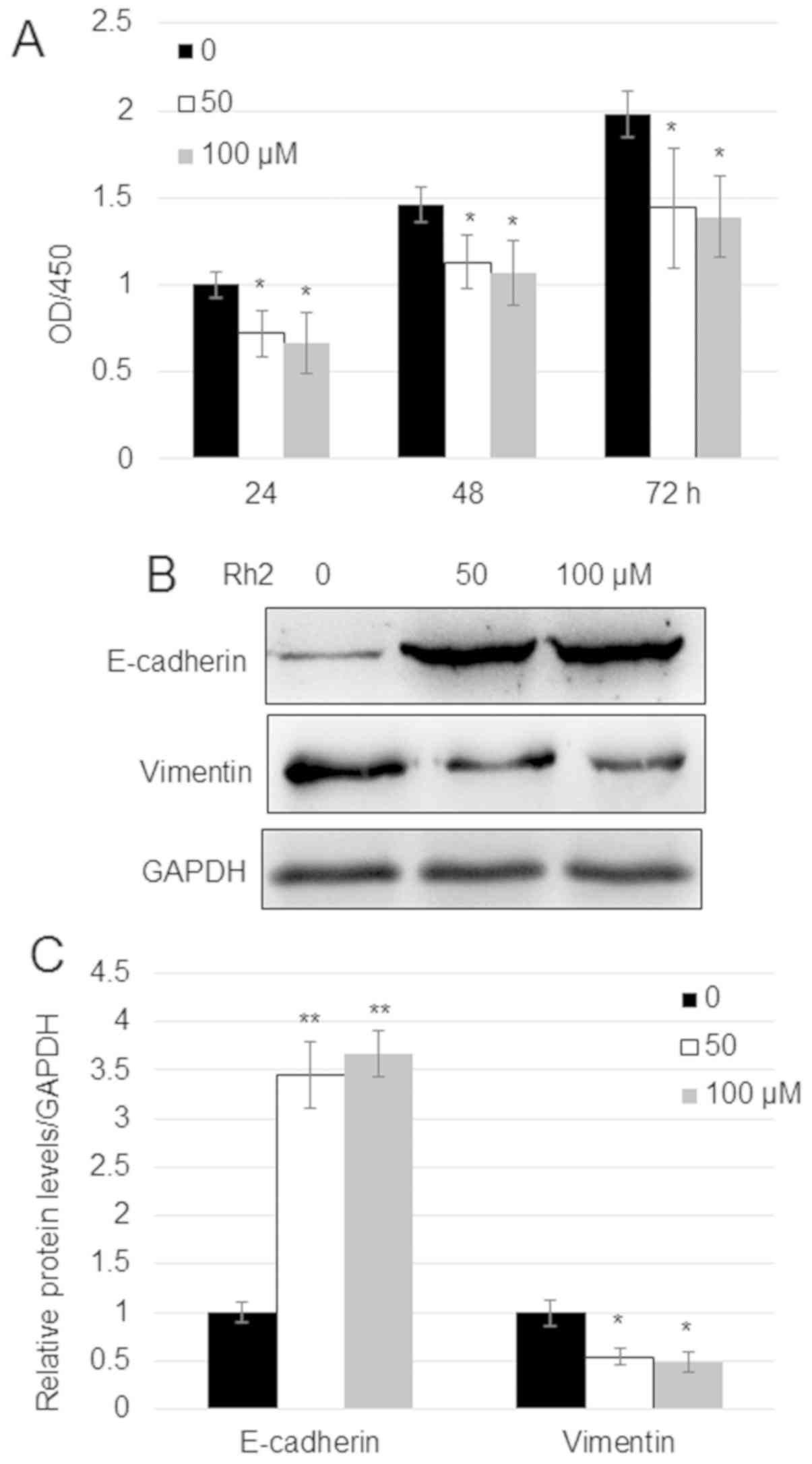
A549 cells (Fig. 1A). Since
E-cadherin and vimentin levels are related to the migration of
cancer cells, the impact of Rh2 on these proteins was analyzed.
Western blotting indicated that treatment with 50 and 100 µM Rh2
increased the expression levels of E-cadherin ~3.5-fold and reduced
the expression levels of vimentin by ~50% compared with untreated
cells (Fig. 1B and C).
Rh2 reduces the expression of Wnt and
Hh signaling genes in A549 lung cancer cells
Wnt and Hh signaling serve a critical role in the
proliferation of hepatocellular carcinoma (32). To analyze whether Rh2 regulates Wnt
and Hh signaling genes in A549 cells, the expression levels of key
genes implicated in the two signaling pathways were analyzed. The
mRNA expression levels of Wnt signaling genes [Wnt3,
transcription factor 7 (TCF7) and frizzled class receptor 8
(FZD8)] and Hh signaling genes (Smo, Gli1,
Gli2, and Gli3) were measured by RT-qPCR. The results
indicated that, in comparison with untreated cells, Rh2 reduced the
expression of both Wnt signaling genes (Wnt3, TCF7
and FZD8) and Hh signaling genes (Smo, Gli1,
Gli2 and Gli3) (Fig.
2A and B). In addition, the
protein expression levels of the key Wnt signaling regulator
β-catenin and Hh signaling regulator Smo, and the expression of
Gli1 were analyzed by western blotting. In agreement with the
results of RT-qPCR, the protein expression levels of β-catenin, Smo
and Gli1 were reduced with Rh2 treatment compared with untreated
cells (Fig. 2C and D).
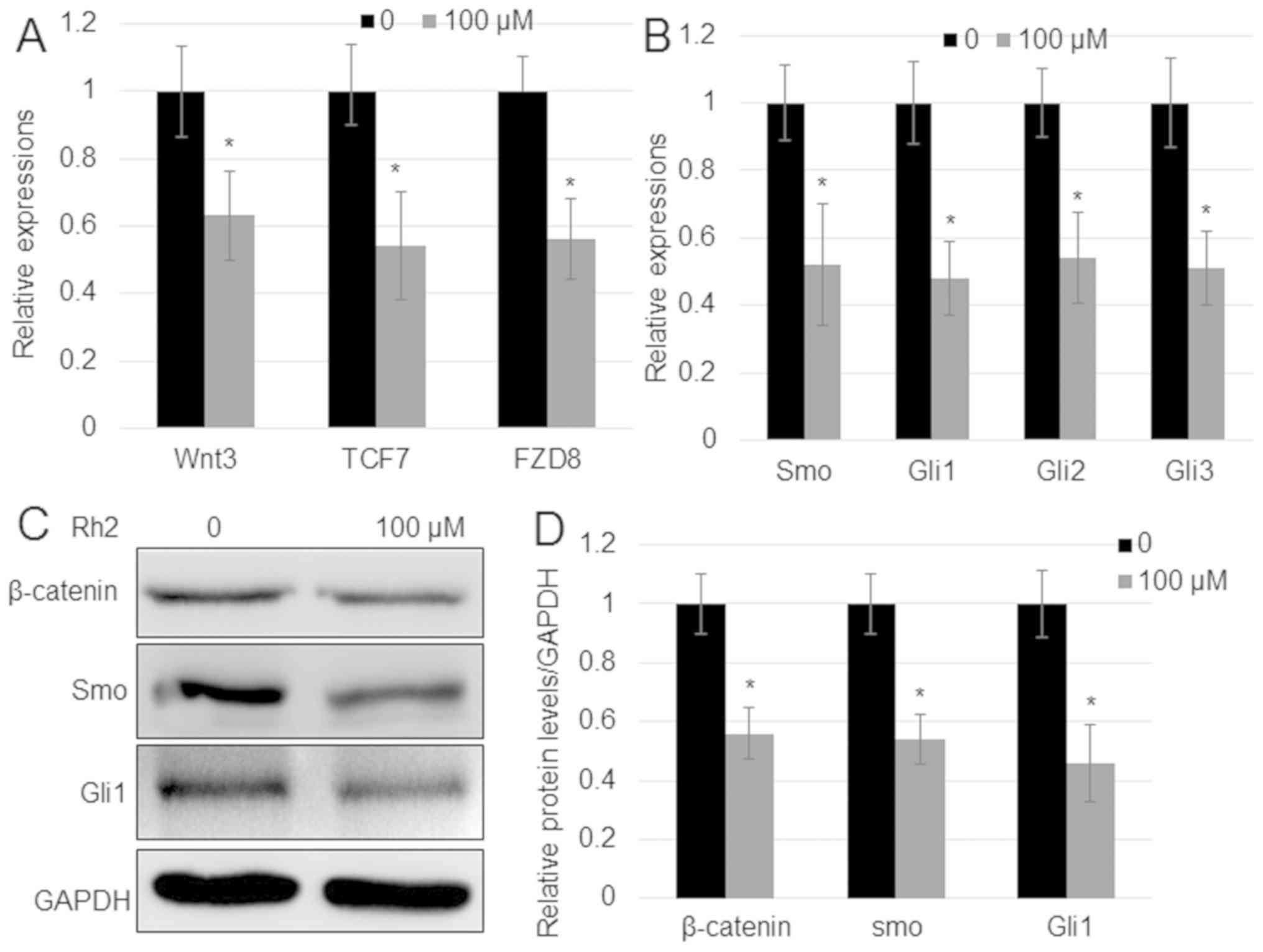 | Figure 2.Rh2 suppresses the expression of
genes implicated in Wnt and Hh signaling. (A) Expression of Wnt
signaling genes Wnt3, TCF7 and FZD8, and (B)
Hh signaling genes Smo, Gli1, Gli2 and
Gli3 in A549 cells after treatment with 100 µM Rh2. GAPDH
was used as an internal control to normalize expression levels.
Error bars indicate mean ± SE (n=3). (C) β-catenin, Smo and Gli1
protein levels in the presence or absence of 100 µM Rh2 examined by
western blot analysis. GAPDH was used as the loading control. (D)
Relative levels of proteins shown in (C). *P<0.05 vs.
Control. FZD8, frizzled class receptor 8; GLI, Gli family zinc
finger; Hh, hedgehog; Rh2, ginsenoside Rh2; Smo, smoothened,
frizzled class receptor; TCF7, transcription factor 7. |
Rh2 induces the phosphorylation of
α-catenin at S641
To further analyze the mechanisms implicated in the
effects of Rh2, phosphopeptides were analyzed using the
TiO2-enrichment method. Significant changes in the
levels of phosphorylation were identified in 14 phosphopeptides.
Among them, the phosphorylation level of five proteins was reduced,
whereas the phosphorylation of nine proteins was induced by Rh2
(Tables II and III). The five proteins displaying reduced
phosphorylation levels were ACTA, ITGA5, RACK1, ARHGEF6 and
FAM129B, while the nine with higher levels of phosphorylation were
PPP1R12A, FERMT2, α-catenin, CCDC6, LIMCH1, GRHPR, STEAP3, PPP3CA
and HS1BP3. Given the suppression of Wnt signaling by Rh2, the
phosphorylation of α-catenin was evaluated further. LC/MS data for
histogram of p-peptide showed that α-catenin at the S641 residue
exhibited the highest peak (691.70), suggesting that α-catenin
phosphorylation may be at the S641 residue (Fig. 3A). To verify further, the
phosphorylation level of α-catenin at the S641 residue, western
blotting using a specific p-α-catenin S641 antibody confirmed that
Rh2 treatment significantly increased the levels of p-α-catenin
S641 without changing the levels of total α-catenin (Fig. 3B). Expression of α-catenin
S641D, a phosphomimetic form of α-catenin S641, reduced the
accumulation of β-catenin and Gli1 in A549 cells (Fig. 3C). Additionally, RT-qPCR indicated
that the expression levels of Wnt signaling genes (Wnt3,
TCF7 and FZD8) and Hh signaling genes (Smo,
Gli1, Gli2 and Gli3) in A549 cells were
suppressed by the expression of α-catenin S641D (Fig. 3D). Furthermore, α-catenin
S641D expression significantly inhibited cell proliferation
(Fig. 3E). The results of a cell
invasion assay suggested that α-catenin S641D expression or
treatment with 100 µM Rh2 significantly reduced the invasiveness of
A549 cells. White dashed circles mark the borders of cell invasion
area in each cell (Fig. 3F and
G).
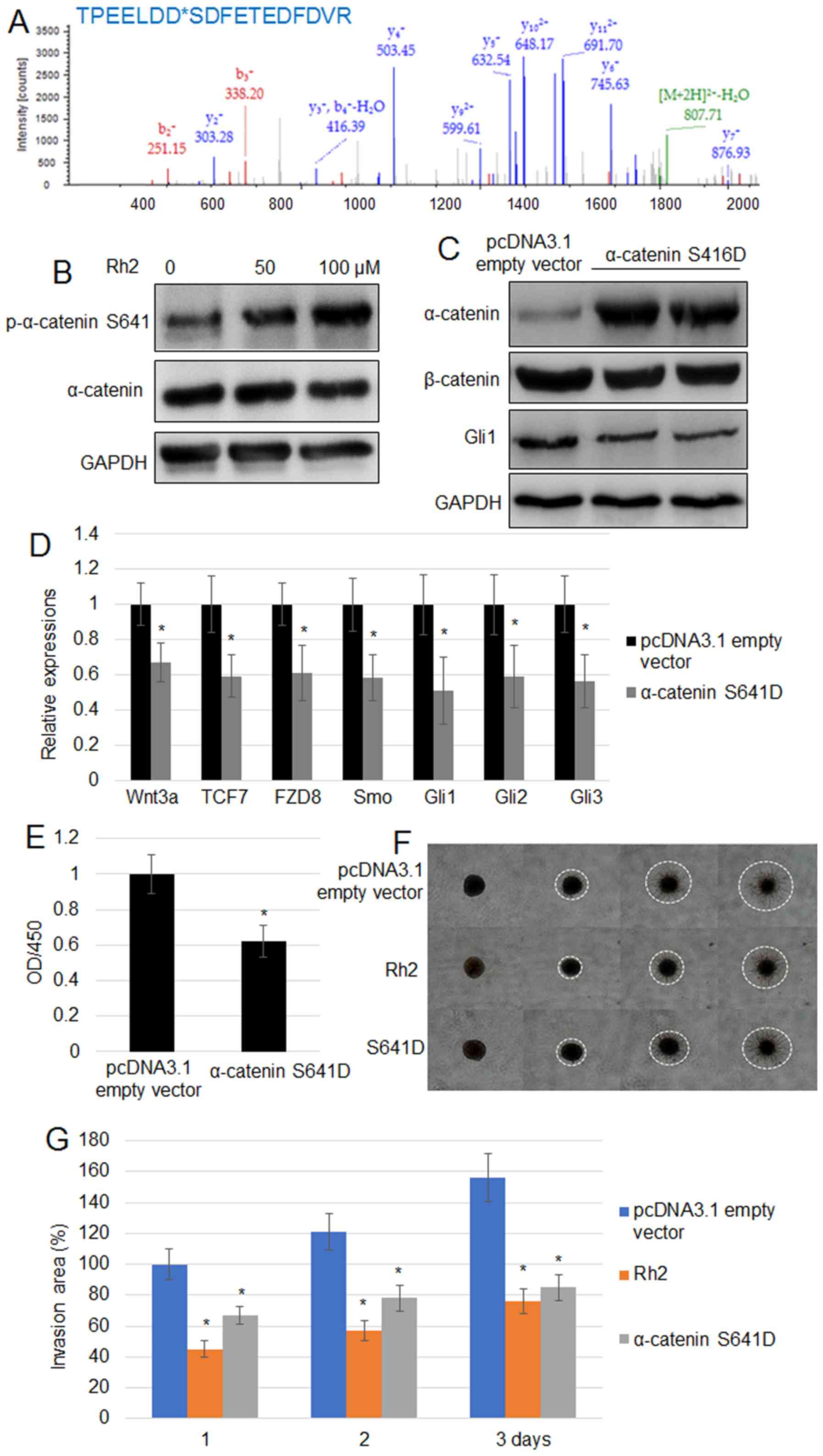 | Figure 3.Rh2 treatment increases the level of
p-α-catenin S641 in A549 cells. (A) Phosphopeptide diagram for
α-catenin showed phosphorylation of the S641 residue. (B)
p-α-catenin S641 and α-catenin levels were analyzed by western
blotting following treatment with 0, 50 and 100 µM Rh2. GAPDH was
used as the loading control. (C) α-catenin, β-catenin and GLI1
levels were examined by western blot analysis in pcDNA3.1 empty
vector transformed and α-catenin S641D-overexpressing A549 cells.
GAPDH was used as the internal control. (D) Expression levels of
Wnt signaling genes (Wnt3, TCF7 and FZD8) and
Hh signaling genes (Smo, Gli1, Gli2 and
Gli3) were assessed in the pcDNA3.1 empty vector transformed
and α-catenin S641D-expressing A549 cells. *P<0.05
vs. pcDNA3.1 empty vector. (E) Cell proliferation rate was measured
in the pcDNA3.1 empty vector transformed and α-catenin
S641D-expressing A541 cells. *P<0.05 vs. pcDNA3.1
empty vector. (F) Cell invasion assay using A549 cells transformed
with the pcDNA3.1 empty vector, expressing α-catenin S641D or
treated with Rh2 (100 µM) for 3 days. The images of cells invading
into the surrounding matrix were acquired at days 0, 1, 2 and 3.
White dashed circles indicate the area invaded by the cells. Bar,
500 µm. (G) Measurement of cell invasion areas depicted in (F).
Data are presented as the mean values ± SE of 10 replicates.
*P<0.05 vs. pcDNA3.1 empty vector. FZD8, frizzled
class receptor 8; GLI, GLI family zinc finger; OD, optical density;
p, phosphorylated; Rh2, ginsenoside Rh2; Smo, smoothened, frizzled
class receptor; TCF7, transcription factor 7. |
| Table IIProteins with phosphorylation
suppressed by Rh2. |
Table II
Proteins with phosphorylation
suppressed by Rh2.
| Accession
number | Gene name | Description | Mr | pI | Phosphorylated
peptides | Ion score | E-value | Ion precursor | Ion charge | Phosphosite |
|---|
| P68133 | ACTA | Actin, α skeletal
muscle | 42366 | 5.23 |
K.CDIDIRKDLYANNVMSGGTTMYPGIADR.M | 35 | 0.0072 | 1108.5046 | 3 | Y296 |
| P08648 | ITGA5 | Integrin α-5 | 115605 | 5.5 |
R.LLESSLSSSEGEEPVEYK.S | 72 | 7.60E-06 | 1032.2283 | 2 | S127 |
| P63244 | RACK1 | Guanine
nucleotide-binding protein subunit β-2-like 1 | 35511 | 7.6 | M.TEQMTLR.G | 35 | 0.034 | 528.0834 | 2 | T2, T6 |
| Q15052 | ARHGEF6 | Rho guanine
nucleotide exchange factor 6 | 88698 | 5.79 | R.MSGFIYQGK.I | 52 | 0.00074 | 555.6702 | 2 | S488 |
| Q96TA1 | FAM129B | Niban-like protein
1 | 84598 | 5.82 |
R.GLLAQGLRPESPPPAGPLLNGAPAGESPQPK.A | 48 | 0.0005 | 1033.0134 | 3 | S665, S652 |
| Table IIIProteins with phosphorylation
increased by Rh2. |
Table III
Proteins with phosphorylation
increased by Rh2.
| Accession
number | Gene name | Description | Mr | pI | Phosphorylated
peptides | Ion score | E-value | Ion precursor | Ion charge | Phosphosite |
|---|
| O14974 | PPP1R12A | Protein phosphatase
1 regulatory subunit 12A | 115610 | 5.31 |
R.RSTQGVTLTDLQEAEK.T | 64 | 8.4-5 | 928.7751 | 2 | T696 |
| Q96AC1 | FERMT2 | Fermitin family
homolog 2 | 78438 | 6.26 |
K.KLDDQSEDEALELEGPLITPGSG
SIYSSPGLYSK.T | 74 | 3.60E-05 | 1226.3796 | 3 | S159 |
| P35221 | CTNNA1 | Catenin α-1 | 100693 | 5.95 |
R.TPEELDDSDFETEDFDVR.S | 98 | 9.50E-09 | 1120.2859 | 2 | S641 |
| Q16204 | CCDC6 | Coiled-coil
domain-containing protein 6 | 53429 | 6.87 |
K.LDQPVSAPPSPR.D | 38 | 0.0045 | 712.4462 | 2 | S240, S244 |
| Q9UPQ0 | LIMCH1 | LIM and calponin
homology domains-containing protein 1 | 122818 | 6.1 |
K.SPEPEATLTFPFLDK.M | 52 | 0.0031 | 886.3337 | 2 | S718 |
| Q9UBQ7 | GRHPR | Glyoxylate
reductase/hydroxypyruvate reductase | 36045 | 7.01 |
R.GDVVNQDDLYQALASGK.I | 38 | 0.019 | 976.8146 | 2 | Y255 |
| Q658P3 | STEAP3 | Metalloreductase
STEAP3 | 55079 | 8.86 |
R.ESNAEYLASLFPTCTVVK.A | 36 | 0.0031 | 1095.3168 | 2 | S128, Y132 |
| Q08209 | PPP3CA |
Serine/threonine-protein phosphatase 2B
catalytic subunit α isoform | 59335 | 5.58 | R.IITEGASILR.Q | 31 | 0.023 | 617.1566 | 2 | T66, S70 |
| Q53T59 | HS1BP3 | HCLS1-binding
protein 3 | 42868 | 4.89 |
K.GEDAEESLEEEEALDPLGIMR.S | 30 | 0.015 | 1206.755 | 2 | S194 |
β-catenin and Gli1 positively regulate
lung cancer cell proliferation
As Rh2 treatment appeared to reduce the expression
of key genes involved in Wnt and Hh signaling, the role of these
pathways in A549 cell proliferation was analyzed. β-catenin
and Gli1 levels were reduced by siRNA or overexpressed
post-transfection with OX plasmids compared with control levels.
RT-qPCR results revealed that transfection with the siRNA duplex
and OX plasmid significantly suppressed or induced, respectively,
the expression of β-catenin (Fig.
4A) and Gli1 (Fig. 4C).
Comparison of cell proliferation among the control, siRNA and OX
groups indicated that β-catenin knockdown by siRNA inhibited
proliferation, whereas OX enhanced proliferation of A549 cells
(Fig. 4B). Similar results were
obtained with Gli1 knockdown and OX (Fig. 4D).
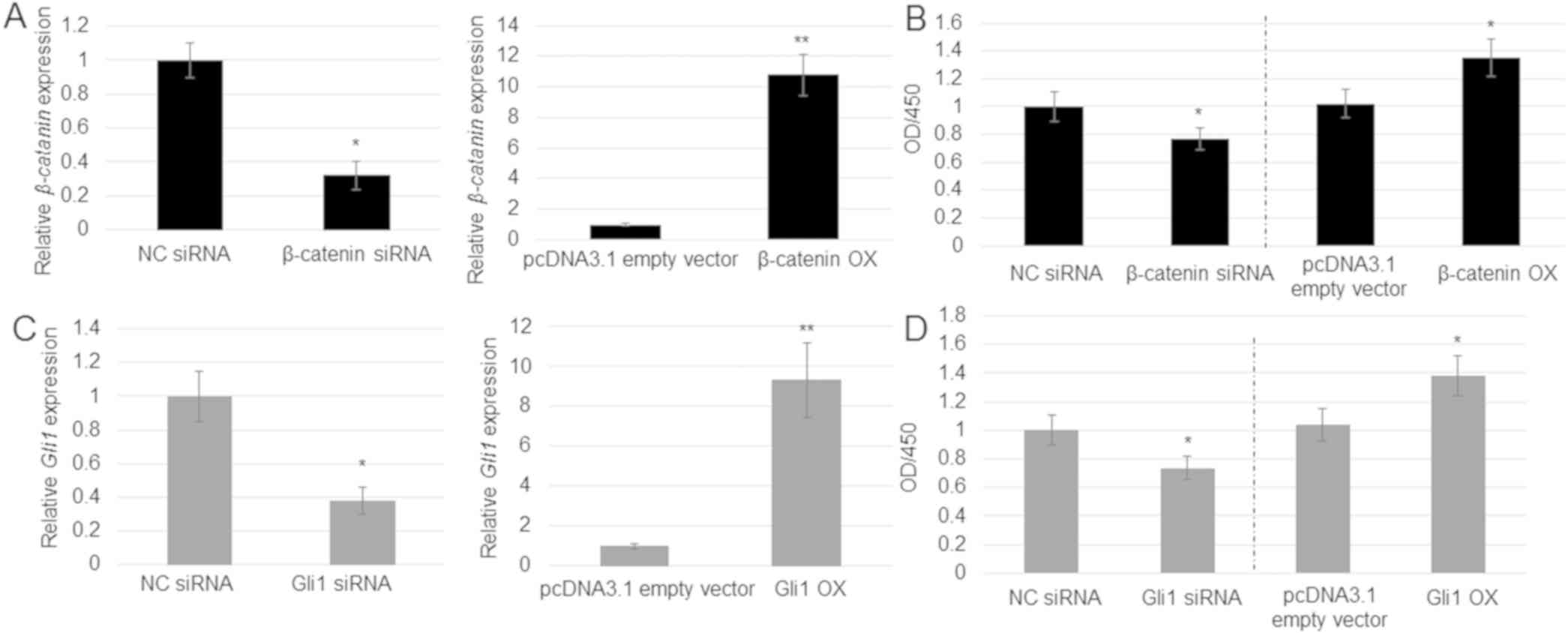 | Figure 4.Role of β-catenin and Gli1 in
A549 cell proliferation. (A) β-catenin expression was
analyzed in the pcDNA3.1 empty vector, NC siRNA, β-catenin
siRNA and β-catenin OX cells. Error bars indicate the mean ±
SE (n=3). *P<0.05 and
**P<0.01 vs. NC siRNA. (B) Cell
proliferation rate was analyzed in the pcDNA3.1 empty vector, NC
siRNA, β-catenin siRNA and β-catenin OX cells.
*P<0.05 vs. NC siRNA. (C) Gli1 expression was
analyzed in the pcDNA3.1 empty vector, NC siRNA, Gli1 siRNA
and Gli1 OX cells. Error bars indicate the mean ± SE (n=3).
*P<0.05 and **P<0.01 vs.
pcDNA3.1 empty vector. (D) Cell proliferation rate in the pcDNA3.1
empty vector, NC siRNA, Gli1 siRNA and Gli1 OX cells.
Data represent the mean ± SE of 6 replicates. *P<0.05
vs. pcDNA3.1 empty vector. GLI, GLI family zinc finger; NC,
non-targeting control; OD, optical density; OX, overexpression;
siRNA, small interfering RNA. |
β-catenin or Gli1 OX blocks the
inhibition of A549 cell proliferation by ginsenoside Rh2
As Rh2 reduced the expression of β-catenin and Gli1,
and levels of these proteins were positively associated with A549
cell proliferation, whether OX of β-catenin or Gli1
could counteract the inhibitory effect of Rh2 on A549 cell
proliferation was tested. RT-qPCR results suggested that
β-catenin and Gli1 were highly expressed in cells
transfected with OX plasmids compared with the control group
(Fig. 5A and C). The treatment with Rh2 inhibited the
rate of cell proliferation, whereas OX of β-catenin and
Gli1 had an opposite effect. Furthermore, OX of
β-catenin and Gli1 reversed the inhibitory effect of
Rh2, accelerating the proliferation of A549 cells (Fig. 5B and D).
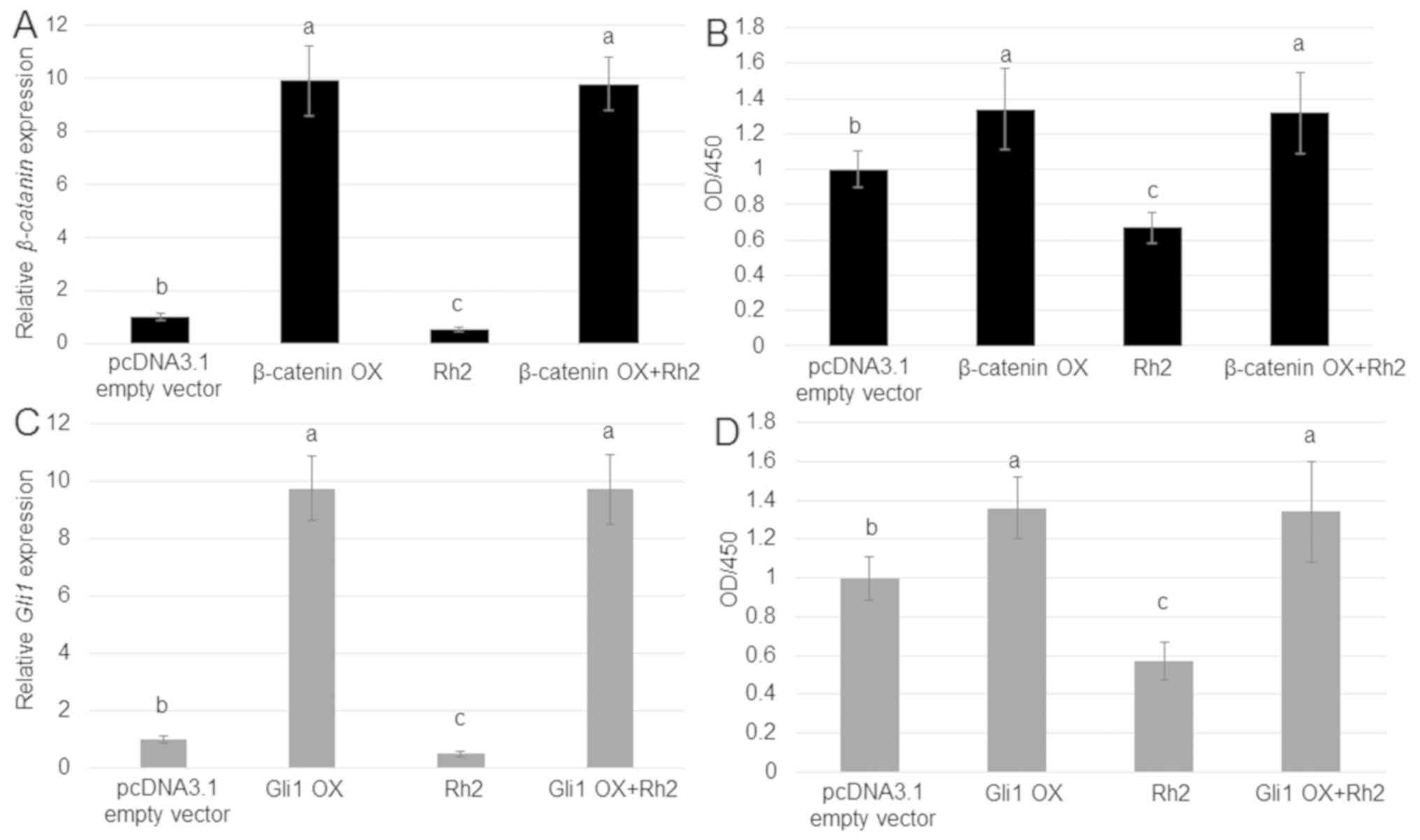 | Figure 5.Effects of β-catenin or Gli1 OX on
the impact of Rh2 on the proliferation of A549 cells. (A)
β-catenin expression was analyzed in A549 cells treated with
the β-catenin OX plasmid, 100 µM Rh2, or Rh2 together with
the β-catenin OX plasmid. pcDNA3.1 empty vector transformed
cells were used as control. (B) Cell proliferation in A549 cells
treated with the pcDNA3.1 empty vector, β-catenin OX
plasmid, 100 µM Rh2, or a combination of Rh2 and β-catenin
OX plasmid. (C) Gli1 expression was analyzed in A549 cells
treated with the Gli1 OX plasmid, 100 µM Rh2, or a combination of
Rh2 and the Gli1 OX plasmid. (D) Cell proliferation in A549 cells
treated with the pcDNA3.1 empty vector, 100 µM Rh2, Gli1 OX
plasmid, or a combination of Rh2 and Gli1 OX plasmid.
P<0.0.5 at 'a' vs. 'b', 'b' vs. 'c', and P<0.01 at 'a' vs.
'c' Data are presented as the GLI, GLI family zinc finger; OD,
optical density; OX, overexpression; Rh2, ginsenoside Rh2. |
Discussion
The majority of lung cancer cases worldwide, are
diagnosed as non-small-cell lung cancer; accounting for 85% of lung
cancer-related deaths (33). The
identification of an effective therapeutic approach to lung cancer
is therefore an urgent issue. Rh2 has been suggested to act as an
anticancer molecule that affects diverse types of cancer cells.
However, the mechanism by which it regulates the proliferation of
lung cancer cells is not yet known.
The present study sought to determine whether Rh2
could regulate the proliferation of A549 cells and to identify the
molecular mechanism involved. Treatment with Rh2 significantly
inhibited cell proliferation, upregulated E-cadherin expression and
downregulated vimentin compared with in untreated cells, suggesting
that this molecule affects the behavior of A549 lung cancer cells.
Subsequent experiments elucidated the role of Wnt and Hh signaling
in mediating the effects of Rh2. This was an essential question,
since the Wnt (23) and Hh signaling
pathways are known to be associated with cancer development and
progression (24). Rh2 treatment
suppressed key Wnt signaling genes, Wnt3, TCF7 and
FZD8, and Hh signaling genes, Smo, Gli1,
Gli2 and Gli3. Moreover, it decreased the protein
expression levels of β-catenin (Wnt signaling), Smo (Hh signaling)
and GLI1 (Hh signaling) in A549 cells, suggesting that Rh2
suppresses both signaling pathways. The phosphoproteomic study
revealed that the phosphorylation of 14 distinct peptides was
significantly altered by treatment with Rh2. Among them,
phosphorylation levels of five peptides were suppressed, and
phosphorylation levels of nine peptides were increased. Further
analysis demonstrated that phosphorylation of the α-catenin S641
residue was significantly increased and that the expression of
α-catenin S641D, a phosphomimetic of α-catenin S641,
markedly reduced the levels of β-catenin and Gli1. α-catenin
S641D expression additionally significantly suppressed key Wnt
signaling genes, Wnt3, TCF7 and FZD8, as well
as Hh signaling genes, Smo, Gli1, Gli2 and
Gli3. These findings indicated that the expression of
α-catenin S641D may inhibit Wnt and Hh signaling in A549 cells.
Previously, α-catenin was reported as a tumor suppressor acting as
an inhibitor of the inflammatory response in breast cancer cells
(34). However, α-catenin
phosphorylation signaling has not been extensively investigated.
The present results indicated that Rh2 stimuli may activate the
phosphorylation of α-catenin, which, in turn, inhibits the
accumulation of β-catenin and Gli1. In addition, expression of
α-catenin S641D severely attenuated the proliferation and
invasion of A549 cells, resembling the effect of Rh2 on lung cancer
cells. The roles of Wnt and Hh signaling in A549 cell proliferation
were also investigated. The data suggested that suppression of the
expression of β-catenin and Gli1 by siRNAs inhibited
cell proliferation, whereas OX of these genes promoted cell
proliferation, indicating that Wnt and Hh signaling cascades exert
positive control on the proliferation rate of A549 cells. To test
the possibility that Rh2 might suppress Wnt and Hh signaling to
inhibit lung cancer cell proliferation, β-catenin and
Gli1 were overexpressed in A549 cells. Administration of Rh2
did not change β-catenin and Gli1 levels in
overexpressing cells but downregulated the expression of these
genes in untransfected A549 cells. This finding suggested that Rh2
might control activities of the promoters of β-catenin and
Gli1 genes. It will be of interest to further dissect
the regulatory mechanism by which Rh2 affects the transcription of
β-catenin and Gli1. These data indicated that
β-catenin and Gli1 promoted proliferation not only of
control cells, but also of cells treated with Rh2, suggesting that
β-catenin and Gli1 may function downstream of Rh2
signaling in the control of lung cancer cell proliferation.
The obtained results indicated that Rh2 suppressed
both Wnt and Hh signaling in the A549 lung cancer cell line. A
previous study on skin fibroblasts reported that β-catenin complex
directly activates two Hh signaling genes, Smo and
Gli1, via promoter binding (35). Thus, it may be hypothesized that, in
lung cancer cells, Rh2 regulates Wnt directly and it is the
modification of Wnt signaling that affects the activity of genes
involved in Hh signaling. It is apparent that further experiments
are needed to define the regulatory model. In conclusion, the
present study documented that Rh2 may suppress Wnt and Hh signaling
via the activation of α-catenin S641 phosphorylation, inhibiting
lung cancer cell proliferation and invasion. These findings expand
our understanding of the regulatory mechanism controlled by Rh2 and
its anticancer activity.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GZ and ZM designed the experiments. GZ, LH, JC and
BX performed the experiments. GZ, LH, JC, BX and ZM analyzed the
data. GZ and ZM wrote the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent for
publication
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Chan BA and Hughes BG: Targeted therapy
for non-small cell lung cancer: Current standards and the promise
of the future. Transl Lung Cancer Res. 4:36–54. 2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Roberts PJ, Stinchcombe TE, Der CJ and
Socinski MA: Personalized medicine in non-small-cell lung cancer:
Is KRAS a useful marker in selecting patients for epidermal growth
factor receptor-targeted therapy? J Clin Oncol. 28:4769–4777.
2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
de Looff M, de Jong S and Kruyt FA: The
role of Src in TRAIL signaling in non-small cell lung cancer cells.
Proceedings: AACR Annual Meeting 2018. April 14-18, 2018. Cancer
Res 78: 4377, 2018.
|
|
5
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Arbiser JL, Bonner MY and Gilbert LC:
Targeting the duality of cancer. NPJ Precision Oncol 1: pii: 23,
017.
|
|
7
|
Pfeffer CM and Singh ATK: Apoptosis: A
target for anticancer therapy. Int J Mol Sci.
19(448)2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Rastogi V, Santiago-Moreno J and Doré S:
Ginseng: A promising neuroprotective strategy in stroke. Front Cell
Neurosci. 8(457)2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kiefer D and Pantuso T: Panax ginseng. Am
Fam Physician. 68:1539–1542. 2003.PubMed/NCBI
|
|
10
|
Helms S: Cancer prevention and
therapeutics: Panax ginseng. Altern Med Rev. 9:259–274.
2004.PubMed/NCBI
|
|
11
|
Fei XF, Wang BX, Tashiro S, Li TJ, Ma JS
and Ikejima T: Apoptotic effects of ginsenoside Rh2 on human
malignant melanoma A375-S2 cells. Acta Pharmacol Sin. 23:315–322.
2002.PubMed/NCBI
|
|
12
|
Huang Y, Huang H, Han Z, Li W, Mai Z and
Yuan R: Ginsenoside Rh2 inhibits angiogenesis in prostate cancer by
targeting CNNM1. J Nanosci Nanotechnol. 19:1942–1950.
2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Chen WW, Huang YF, Hu ZB, Liu YM, Xiao HX,
Liu DB and Zhuang YZ: Microarray analysis of altered long
non-coding RNA expression profile in liver cancer cells treated by
ginsenoside Rh2. J Asian Nat Prod Res. 21:742–753. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lee H, Lee S, Jeong D and Kim SJ:
Ginsenoside Rh2 epigenetically regulates cell-mediated immune
pathway to inhibit proliferation of MCF-7 breast cancer cells. J
Ginseng Res. 42:455–462. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Li H, Huang N, Zhu W, Wu J, Yang X, Teng
W, Tian J, Fang Z, Luo Y, Chen M and Li Y: Modulation the crosstalk
between tumor-associated macrophages and non-small cell lung cancer
to inhibit tumor migration and invasion by ginsenoside Rh2. BMC
Cancer. 18(579)2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wang Y, Xu H, Lu Z, Yu X, Lv C, Tian Y and
Sui D: Pseudo-ginsenoside Rh2 induces A549 cells apoptosis via the
Ras/Raf/ERK/p53 pathway. Exp Ther Med. 15:4916–4924.
2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhan T, Rindtorff N and Boutros M: Wnt
signaling in cancer. Oncogene. 36:1461–1473. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Bushman W: Hedgehog signaling in
development and cancer. In: Prostate cancer Springer, pp107-118,
2007.
|
|
19
|
Shtutman M, Zhurinsky J, Simcha I,
Albanese C, D'Amico M, Pestell R and Ben-Ze'ev A: The cyclin D1
gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad
Sci USA. 96:5522–5527. 1999.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Moon RT, Kohn AD, De Ferrari GV and Kaykas
A: WNT and beta-catenin signalling: Diseases and therapies. Nat Rev
Genet. 5:691–701. 2004.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Clevers H: Wnt/β-catenin signaling in
development and disease. Cell. 127:469–480. 2006.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ,
Keller C and Rando TA: Increased Wnt signaling during aging alters
muscle stem cell fate and increases fibrosis. Science. 317:807–810.
2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Reya T and Clevers H: Wnt signalling in
stem cells and cancer. Nature. 434:843–850. 2005.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Xin M, Ji X, De La Cruz LK, Thareja S and
Wang B: Strategies to target the Hedgehog signaling pathway for
cancer therapy. Med Res Rev. 38:870–913. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Beauchamp EM, Ringer L, Bulut G, Sajwan
KP, Hall MD, Lee YC, Peaceman D, Ozdemirli M, Rodriguez O,
Macdonald TJ, et al: Arsenic trioxide inhibits human cancer cell
growth and tumor development in mice by blocking Hedgehog/GLI
pathway. J Clin Invest. 121:148–160. 2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wang K, Pan L, Che X, Cui D and Li C:
Sonic Hedgehog/GLI1 signaling pathway inhibition restricts cell
migration and invasion in human gliomas. Neurol Res. 32:975–980.
2010.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Huangfu D and Anderson KV: Signaling from
Smo to Ci/Gli: Conservation and divergence of Hedgehog pathways
from Drosophila to vertebrates. Development. 133:3–14.
2006.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Rohatgi R, Milenkovic L and Scott MP:
Patched1 regulates hedgehog signaling at the primary cilium.
Science. 317:372–376. 2007.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Larsen MR, Thigholm TE, Jensen ON,
Roepstorff P and Jørgensen TJ: Highly selective enrichment of
phosphorylated peptides from peptide mixtures using titanium
dioxide microcolumns. Mol Cell Proteomics. 4:873–886.
2005.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Xuan YH, Huang BB, Tian HS, Chi LS, Duan
YM, Wang X, Zhu ZX, Cai WH, Zhu YT, Wei TM, et al: High-glucose
inhibits human fibroblast cell migration in wound healing via
repression of bFGF-regulating JNK phosphorylation. PLoS One.
9(e108182)2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Tripathy A, Thakurela S, Sahu MK,
Uthanasingh K, Behera M, Ajay AK and Kumari R: The molecular
connection of histopathological heterogeneity in hepatocellular
carcinoma: A role of Wnt and Hedgehog signaling pathways. PLoS One.
13(e0208194)2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Neal RD, Hamilton W and Rogers TK: Lung
cancer. BMJ. 349(g6560)2014.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Piao HL, Yuan Y, Wang M, Sun Y, Liang H
and Ma L: α-catenin acts as a tumour suppressor in
E-cadherin-negative basal-like breast cancer by inhibiting NF-κB
signalling. Nat Cell Biol. 16:245–254. 2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Wang Y, Lin P, Wang Q, Zheng M and Pang L:
Wnt3a-regulated TCF4/β-catenin complex directly activates the key
Hedgehog signalling genes Smo and GLI1. Exp Ther Med. 16:2101–2107.
2018.PubMed/NCBI View Article : Google Scholar
|