Introduction
Acute myelocytic leukemia (AML) is a tumor derived
from the myeloid line of blood cells, in which abnormal cells in
the bone marrow and blood are rapidly generated (1). AML is characterized by the
proliferation and abnormal differentiation of cells of the
hematopoietic system in bone marrow, blood and other tissues
(2). The typical clinical symptoms
of patients with AML are shortness of breath, fatigue, increased
risk of infection, and easy bleeding and bruising (3). The risk factors of AML mainly include
smoking, benzene exposure, myelodysplastic syndrome, and previous
radiation therapy or chemotherapy (4,5). AML is
more frequent in elderly men, and it progresses rapidly without
immediate therapy (6). AML affects
approximately one million individuals and resulted in 147,000
deaths globally in 2015(7). The
pathogenesis of AML needs to be further investigated to decrease
the prevalence and mortality of AML.
Long non-coding RNAs (lncRNAs) are transcripts with
a length >200 nucleotides. lncRNAs have no protein coding
potential but are implicated in various biological functions,
including cell cycle, apoptosis, epigenetic regulation and
imprinting (8). lncRNA expression is
closely associated with recurrent mutations, and some lncRNAs play
important roles in the AML therapeutic response and clinical
survival (9). HOX transcript
antisense RNA expression is upregulated in de novo patients
with AML, which is strongly associated with the clinicopathological
prognosis of patients with AML (10,11).
Insulin-like growth factor type I receptor (IGF1R) antisense
imprinted non-protein coding RNA is associated with long range
IGF1R DNA interactions and upregulation of the IGF pathway in AML,
and is a potential tumor suppressor and therapeutic target for the
disease (12). lncRNA H19 (imprinted
maternally expressed transcript) is an independent prognostic
biomarker for patients with tumors, and its expression is
significantly upregulated in AML and may play a role in AML cell
proliferation (13). lncRNA RUNXOR
[runt related transcription factor 1 (RUNX1) overlapping
promoter-derived noncoding RNA) is located in the RUNX1 locus
(14). Overexpressed RUNXOR may
serve as a tumor suppressor through RUNX1 and scaffolding mediated
translocation of DNA correlated in AML (15). However, the lncRNAs associated with
the prognosis of AML have not been fully reported.
In the present study, prognosis-associated lncRNAs
were screened to construct a risk score system for AML.
Additionally, the potential functions of the key lncRNAs were
predicted. The data could be valuable in clarifying the prognostic
mechanisms of AML and could contribute to the prognostic prediction
of the disease.
Materials and methods
Data source
All available RNA-sequencing data of AML samples
were searched using The Cancer Genome Atlas (TCGA) database
(https://cancergenome.nih.gov/) on March
17, 2018. In total, 187 AML samples with RNA-sequencing data were
obtained from TCGA (16,17). According to the age information of
the samples, 111 samples from patients under the age of 14 years
were included. The 111 samples were randomly divided into two
groups (56 samples in the training set and 55 samples in the
validation set). The clinical information of the samples in each
group is presented in Table I.
 | Table IClinical information of the samples
in the training set, the validation set and the entire set. |
Table I
Clinical information of the samples
in the training set, the validation set and the entire set.
| Clinical
characteristics | Training set,
n=56 | Validation set,
n=55 | Entire set,
n=111 |
|---|
| Age, years, mean ±
SD | 5.99±4.26 | 5.81±4.32 | 5.91±4.27 |
| Sex,
male/female | 32/24 | 31/24 | 63/48 |
| WBC at diagnosis,
109/l, mean ± SD | 64.56±74.03 | 97.09±113.96 | 80.67±96.86 |
| Bone marrow
leukemic blast percentage, %, mean ± SD | 69.51±20.44 | 72.99±20.39 | 71.19±20.39 |
| Peripheral blasts,
%, mean ± SD | 49.61±28.76 | 57.7±30.09 | 53.62±29.58 |
| CNS disease,
yes/no | 3/53 | 4/51 | 7/104 |
| Chloroma,
yes/no/- | 3/53 | 6/48/1 | 9/101/1 |
| FAB Category,
M0/M1/M2/M3/M4/M5/M6/M7/- |
0/3/9/0/18/12/1/6/7 |
3/11/14/0/10/11/1/1/4 |
3/14/23/0/28/23/2/7/11 |
| FLT3/ITD mutation,
yes/no | 4/52 | 5/50 | 9/102 |
| NPM mutation,
yes/no/- | 0/52/4 | 5/50/0 | 5/4/102 |
| CEBPA mutation,
yes/no/- | 2/52/2 | 4/51 | 6/103/2 |
| WT1 mutation,
yes/no | 2/50/4 | 4/51 | 6/101/4 |
| Relapse,
yes/no | 37/19 | 40/15 | 77/34 |
| Relapse free
survival time, months, mean ± SD | 33.01±34.84 | 31.52±32.42 | 32.27±33.52 |
| Death,
dead/alive | 24/32 | 24/31 | 48/63 |
| Overall survival
time, months, mean ± SD | 54.13±34.76 | 55.97±36.08 | 55.04±35.27 |
Differential expression analysis
Among the 56 AML samples in the training set, the
median survival time was 54.13 months. It was assumed that samples
from patients with a short survival were high risk, and patients
with longer survival being indicative of relatively low risk.
Combining these assumptions with the median survival time
established 54.13±18 months as the cut-off value to classify
different prognostic samples. Samples with a survival time <36
months who had died were classified into the bad prognosis group
(n=20). Samples with a survival time >72 months who were still
alive were divided into the good prognosis group (n=21). The
differentially expressed lncRNAs (DE-lncRNAs) between the two
groups were selected using the DEseq package (version 1.30.0;
https://bioconductor.org/packages/release/bioc/html/DESeq.html)
(18) and edgeR package (version
3.20.9; http://bioconductor.org/packages/release/bioc/html/edgeR.html)
(19) in R, which were also used in
other previous studies (20-22).
The false discovery rate (FDR) <0.05 and log fold change (FC)
>1 were defined as the thresholds. By comparing the results of
the two screening methods, the overlapped DE-lncRNAs were taken as
the final DE-lncRNAs. Based on the expressions of the DE-lncRNAs in
the training set, the bidirectional hierarchical clustering using
the pheatmap package in R (version 1.0.8; https://cran.r-project.org/web/packages/pheatmap/index.html)
(23) was performed.
Identification of prognosis-associated
lncRNAs and clinical factors
Using the survival package in R (version 2.41.3;
https://cran.r-project.org/web/packages/survival/index.html)
(24), univariate and multivariate
Cox regression analysis was conducted for the 56 AML samples in the
training set to screen the prognosis-associated lncRNAs and
clinical factors. The log-rank P<0.05 was set as the threshold
for the significant results.
Construction and evaluation of risk
score system
Based on the prognosis-associated lncRNAs, the
optimal lncRNA combination was selected using the Cox-Proportional
Hazards (Cox-PH) model in R package penalized (version 0.9.50;
https://cran.r-project.org/web/packages/penalized/index.html)
(25). The optimal parameter ‘λ’ in
the Cox-PH model was obtained through the cross-validation
likelihood (cvl) for 1,000 times. The risk score system was
constructed for AML samples combined with the linear combination of
the lncRNA expression with regression coefficient weighting. The
formula for calculating the prognostic index (PI) of each sample
was as follows:
PI=β lncRNA1 x exprlncRNA1 + β lncRNA2 x exprlncRNA2
+ ··· + β lncRNAn x exprlncRNAn
The PI of each sample in the training set was
calculated based on the aforementioned formula, and the median PI
was utilized as the cut-off value of the good and bad prognosis
samples. A Kaplan-Meier (KM) curve (26) was used to calculate the survival time
in the two groups when stratified by different prognostic factors,
based on which to evaluate the prognosis difference between the
good and bad prognosis groups using a certain prognostic factor,
and an area under the receiver operating characteristic (AUROC)
curve (27) was applied to assess
the performance of prognostic prediction effectiveness using the
risk score system. Then, the risk score system was used to evaluate
the prognosis of the samples in the validation set.
Construction of the lncRNA-mRNA
expression correlation network
In the training set, the samples were divided into
the good and bad prognosis groups according to their PIs in the
predictive risk score system model. The differentially expressed
mRNAs (DE-mRNAs) between the good and bad prognosis groups were
analyzed using the edgeR package (19), with the thresholds of FDR <0.05
and log FC >1. Then, the Pearson correlation coefficients (PCCs)
(28) of the DE-mRNAs and the
lncRNAs involved in the risk score system were calculated. Finally,
the DE-mRNAs with PPC >0.5 were obtained for constructing the
lncRNA-mRNA expression correlation network.
Pathway enrichment analysis
Based on Gene Set Enrichment Analysis (GSEA;
http://software.broadinstitute.org/gsea/index.jsp)
(29), pathway enrichment analysis
for the mRNAs in the network was performed to identify the
significant pathways correlated with key lncRNAs. The pathways with
nominal P<0.05 were considered as significant results.
Results
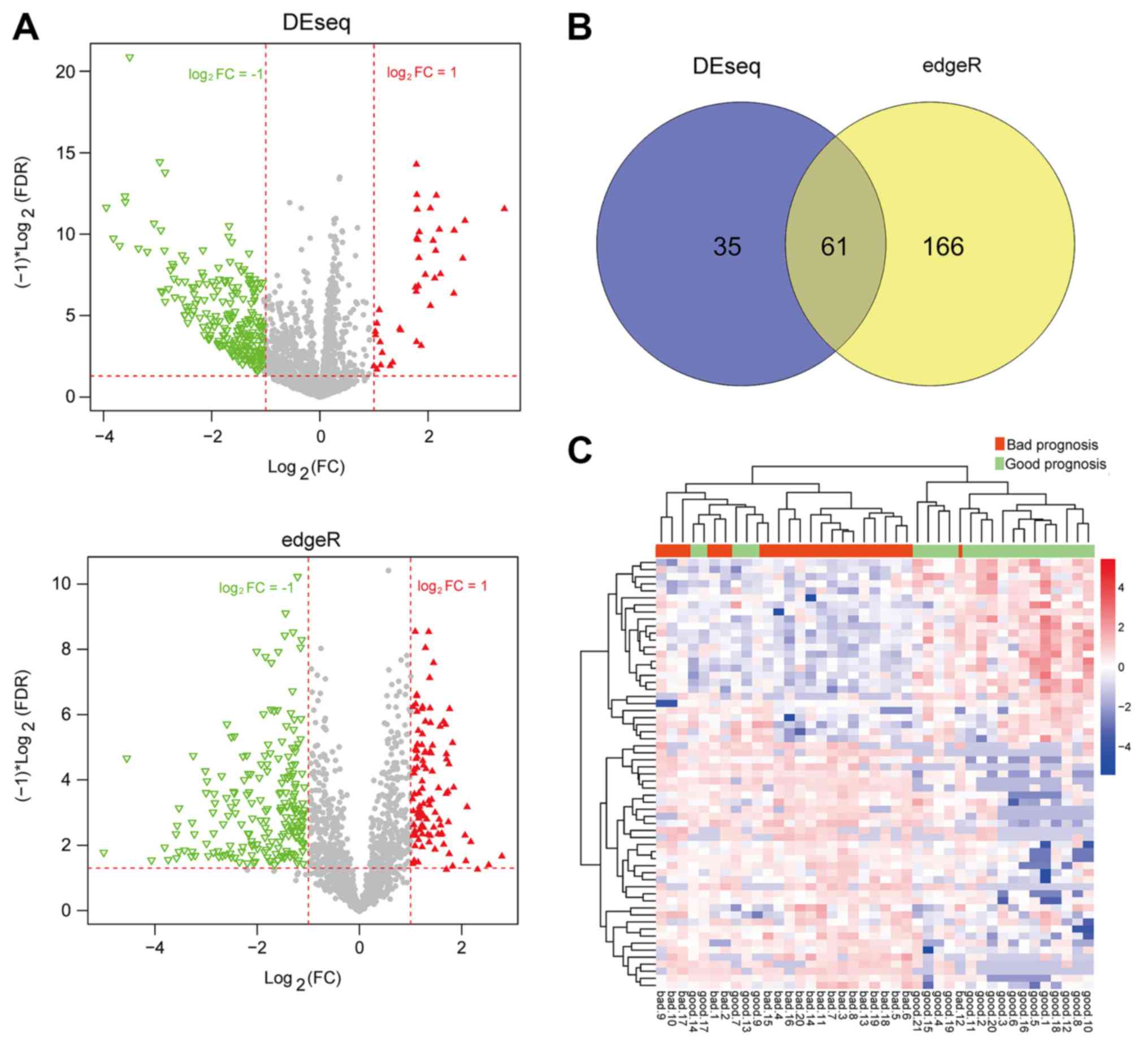
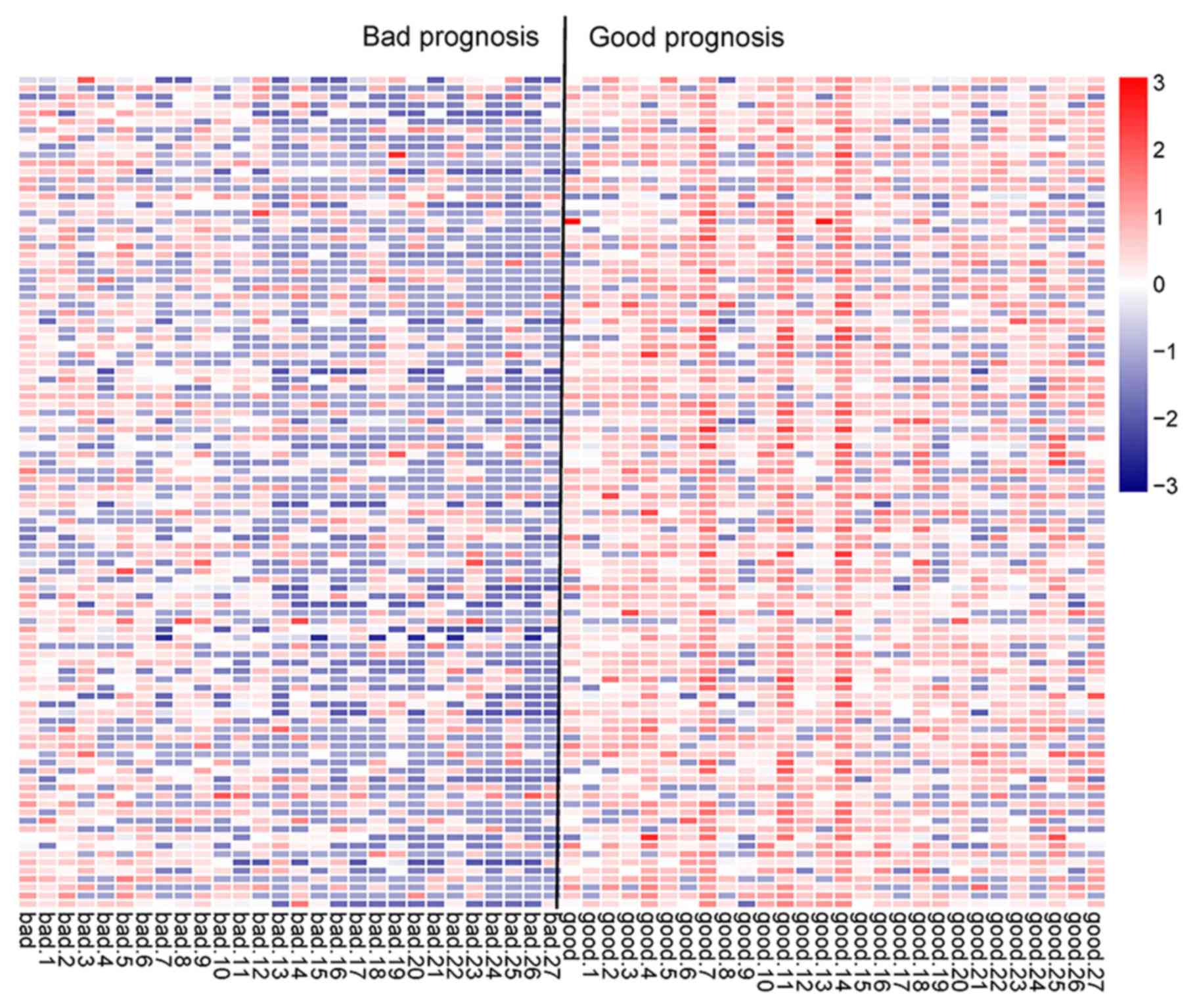
Differential expression analysis
After the AML samples in the training set were
classified into the bad and good prognosis groups, the DEseq
package and edgeR package were used for the selection of DE-lncRNAs
(Fig. 1A). A total of 61 overlapped
DE-lncRNAs were obtained by comparing the results of the two
screening methods (Fig. 1B). The
clustering heatmap of the 61 DE-lncRNAs suggested the samples could
be distinguished by these DE-lncRNAs (Fig. 1C).
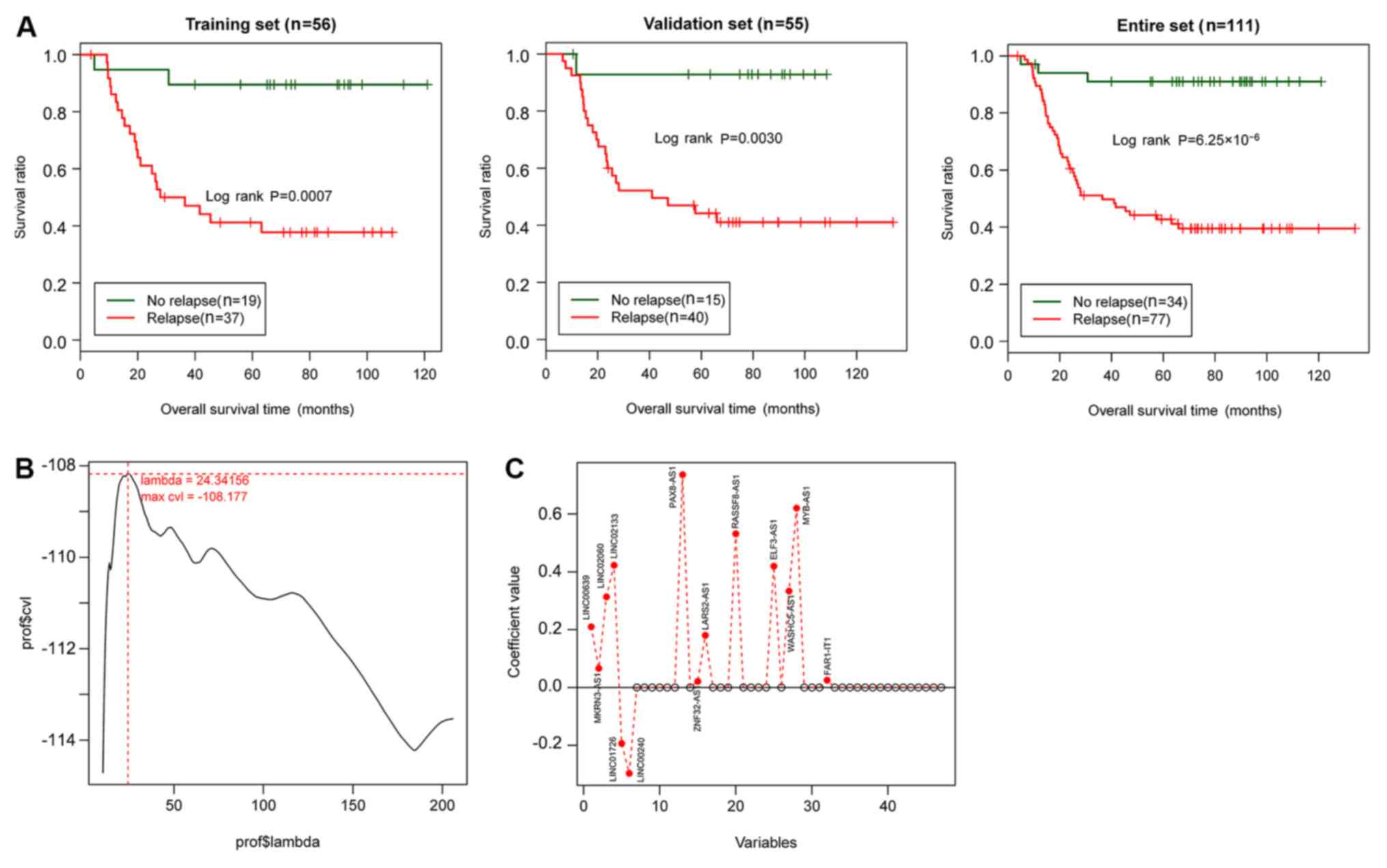
Identification of prognosis-associated
lncRNAs and clinical factors
In the training set, 61 lncRNAs were subjected to
univariate Cox regression analysis. A total of 48 lncRNAs were
significantly associated with prognosis (P<0.05; data not
shown). Univariate and multivariate Cox regression analysis
indicated that relapse was significantly associated with prognosis
in the training set, the validation set and the entire set (Table
II; P<0.05). Therefore, relapse was identified as an independent
clinical factor with prognostic significance. The KM curves of
relapse and the overall survival (OS) of the samples in the
training set, the validation set and the entire set are presented
in Fig. 2A. The results showed that
the AML samples without relapse had better clinical prognosis,
which was consistent with the actual situation.
Construction and evaluation of the
risk score system
Based on the expression matrix of the 48
prognosis-associated lncRNAs in the training set, the optimal
lncRNA combination was analyzed using the Cox-PH model using the
penalized package. After performing the 1,000 times cvl algorithm,
the maximum cvl value was-108.177 when the parameter ‘λ’ was set as
24.34156 (Fig. 2B). With the optimal
parameter, the optimal lncRNA combination involving 14 lncRNAs
[LINC00639, probable E3 ubiquitin-protein ligase makorin-3
(MKRN3)-antisense RNA 1 (AS1), LINC02060, LINC02133, LINC01726,
LINC00240, paired box protein Pax8 (PAX8)-AS1, zinc finger protein
32 (ZNF32)-AS1, probable leucine-tRNA ligase, mitochondrial
(LARS2)-AS1, Ras association domain family member 8 (RASSF8)-AS1,
E74-like factor 3 (ELF3)-AS1, WASH complex subunit 5-antisense RNA
1 (WASHC5)-AS1, MYB-AS1 and FAR1 intronic transcript 1 (FAR1-IT1)]
was screened (Fig. 2C; Table III) using the Cox-PH model.
| Table IIIInformation of the 14 optimal
lncRNAs. |
Table III
Information of the 14 optimal
lncRNAs.
| lncRNA | Coef | HR (95% CI) | P-value |
|---|
| LINC00639 | 0.20989291 | 1.075
(1.023-1.130) | 0.0025 |
| MKRN3-AS1 | 0.06638359 | 1.054
(1.015-1.096) | 0.0038 |
| LINC02060 | 0.3139191 | 1.069
(1.024-1.117) | 0.0014 |
| LINC02133 | 0.42299789 | 1.152
(1.061-1.250) | <0.0001 |
| LINC01726 | -0.19338239 | 0.938
(0.898-0.979) | 0.0023 |
| LINC00240 | -0.2969476 | 0.945
(0.897-0.996) | 0.0302 |
| PAX8-AS1 | 0.73636164 | 1.093
(1.012-1.179) | 0.0224 |
| ZNF32-AS1 | 0.02132678 | 1.070
(1.023-1.119) | 0.0010 |
| LARS2-AS1 | 0.1805946 | 1.075
(1.019-1.133) | 0.0030 |
| RASSF8-AS1 | 0.53198533 | 1.128
(1.023-1.244) | 0.0142 |
| ELF3-AS1 | 0.41975344 | 1.223
(1.045-1.431) | 0.0205 |
| WASHC5-AS1 | 0.33392756 | 1.195
(1.030-1.387) | 0.0076 |
| MYB-AS1 | 0.62016856 | 1.175
(1.038-1.330) | 0.0247 |
| FAR1-IT1 | 0.02508178 | 1.135
(1.017-1.267) | 0.0325 |
The risk score system was constructed and the
formula for calculating PIs was: PI=(0.20989291) x ExpLINC00639+
(0.06638359) x Exp MKRN3-AS1+(0.3139191) x ExpLINC02060
+(0.42299789) x ExpLINC02133+(-0.19338239) x ExpLINC01726
+(-0.2969476) x ExpLINC00240+(0.73636164) x ExpPAX8-AS1+
(0.02132678) x ExpZNF32-AS1+(0.1805946) x ExpLARS2-AS1+
(0.53198533) x ExpRASSF8-AS1+(0.41975344) x ExpELF3-AS1+
(0.33392756) x ExpWASHC5-AS1+(0.62016856) x ExpMYB-AS1+(0.02508178)
x Exp FAR1-IT1.
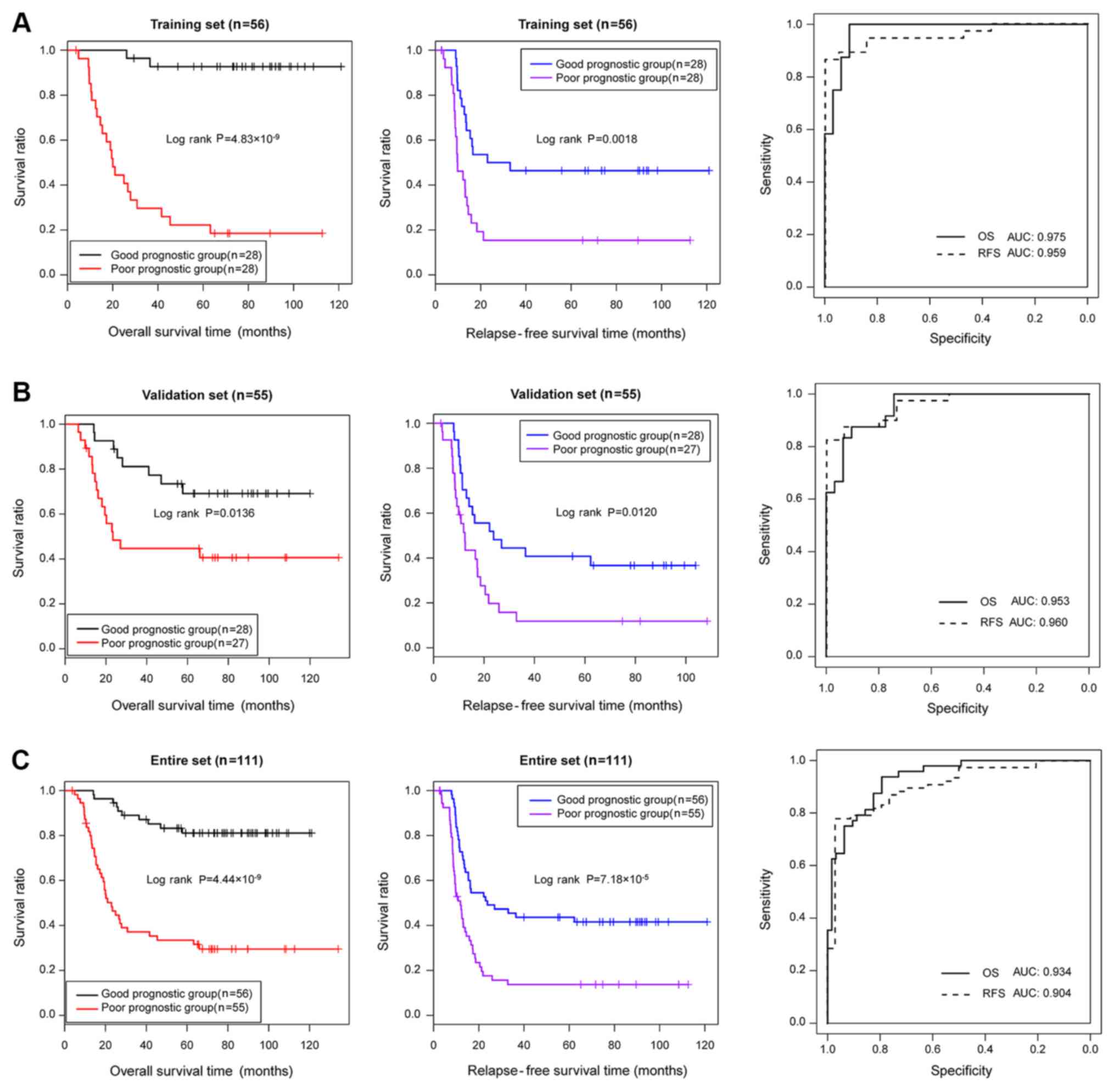
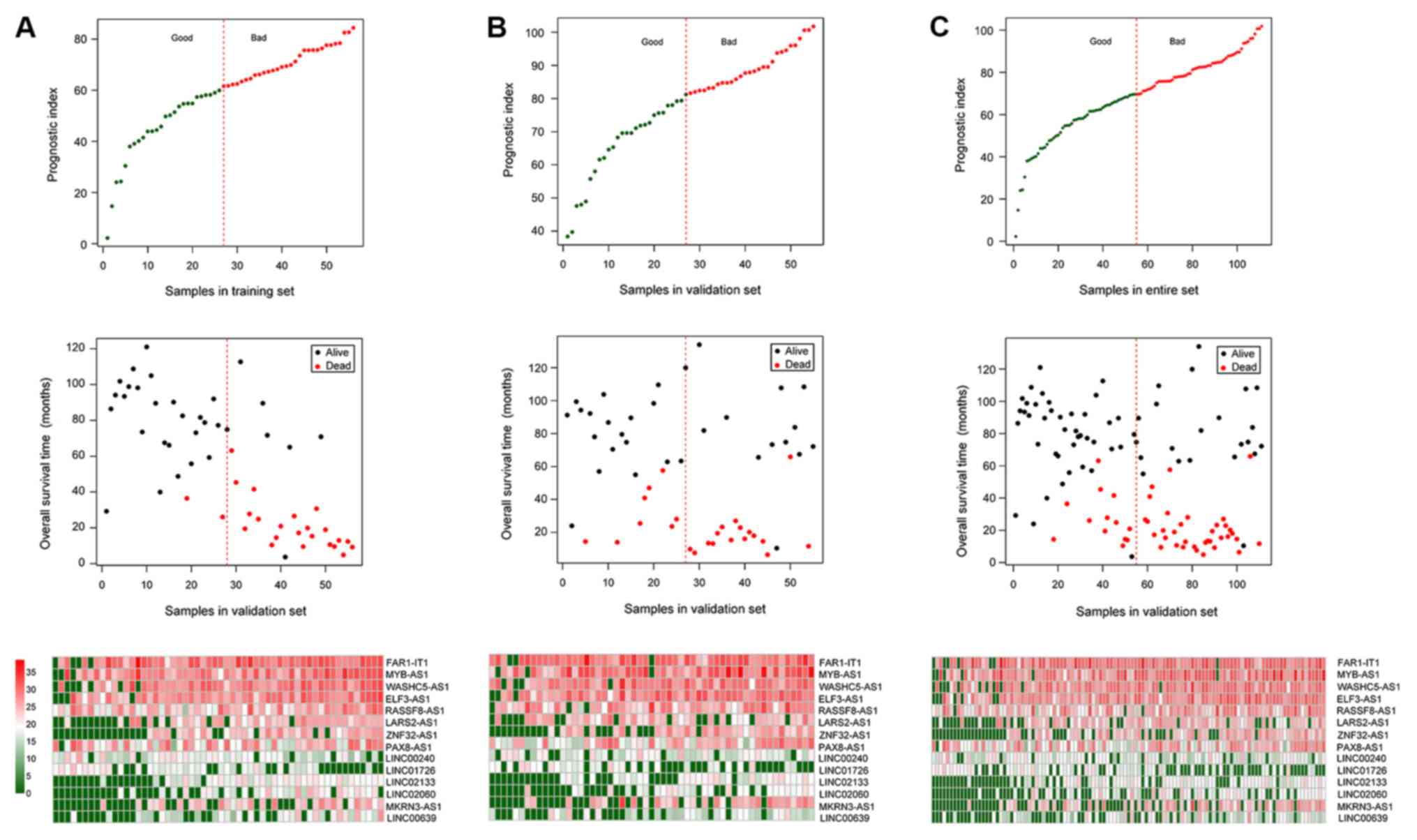
Subsequently, the performance of the prognostic
prediction of the risk score system for the samples in the training
set, the validation set and the entire set were evaluated using KM
curves and AUROC curves (Fig. 3).
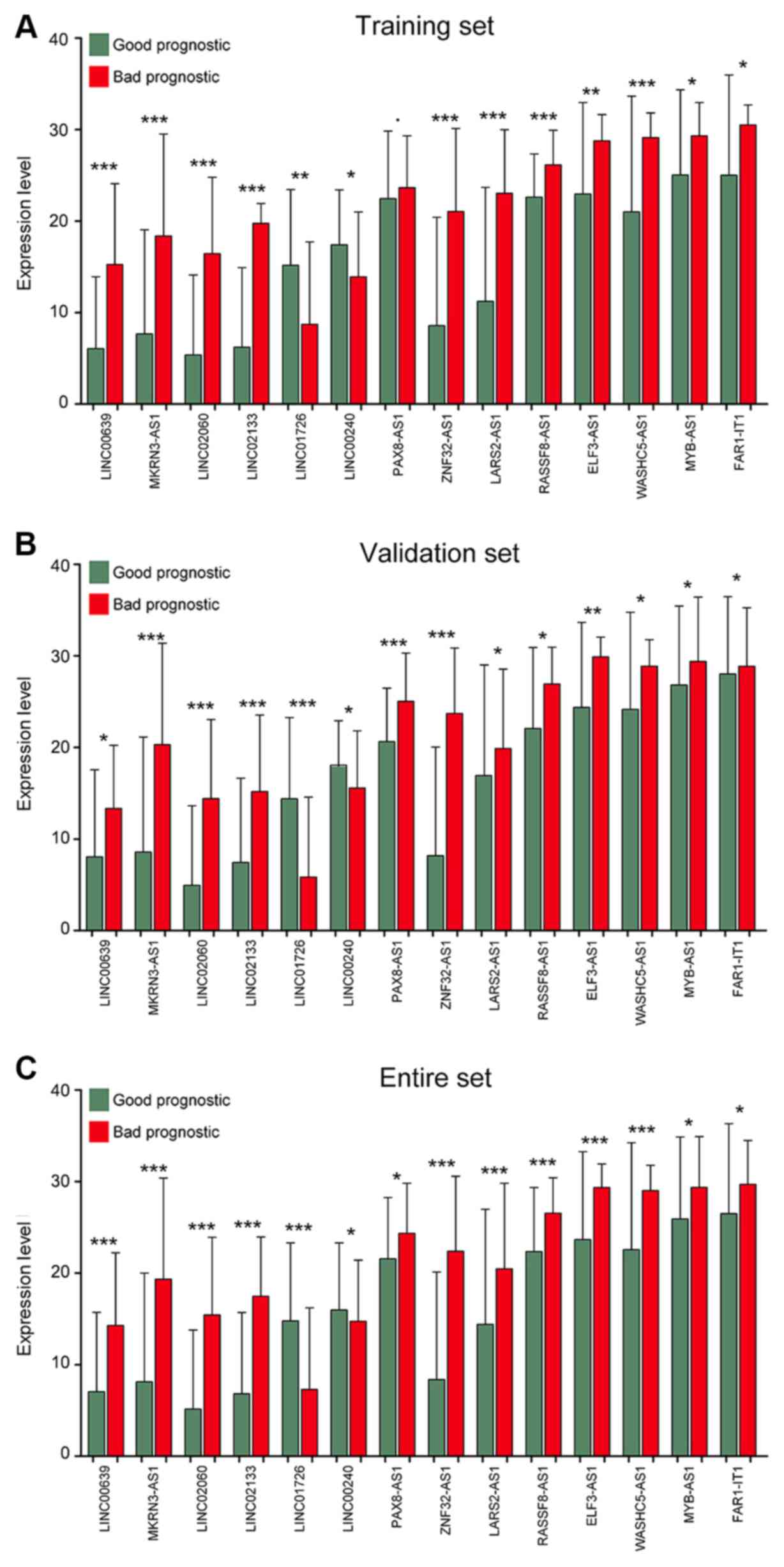
The expression levels of the 14 optimal lncRNAs in the
aforementioned sets downloaded from TCGA are presented in Fig. 4, where all 14 lncRNAs exhibited
distinct expression profiles between bad and good prognosis groups.
The PIs, OS and expression heatmap of the 14 lncRNAs in the
training set, the validation set and the entire set are presented
in Fig. 5.
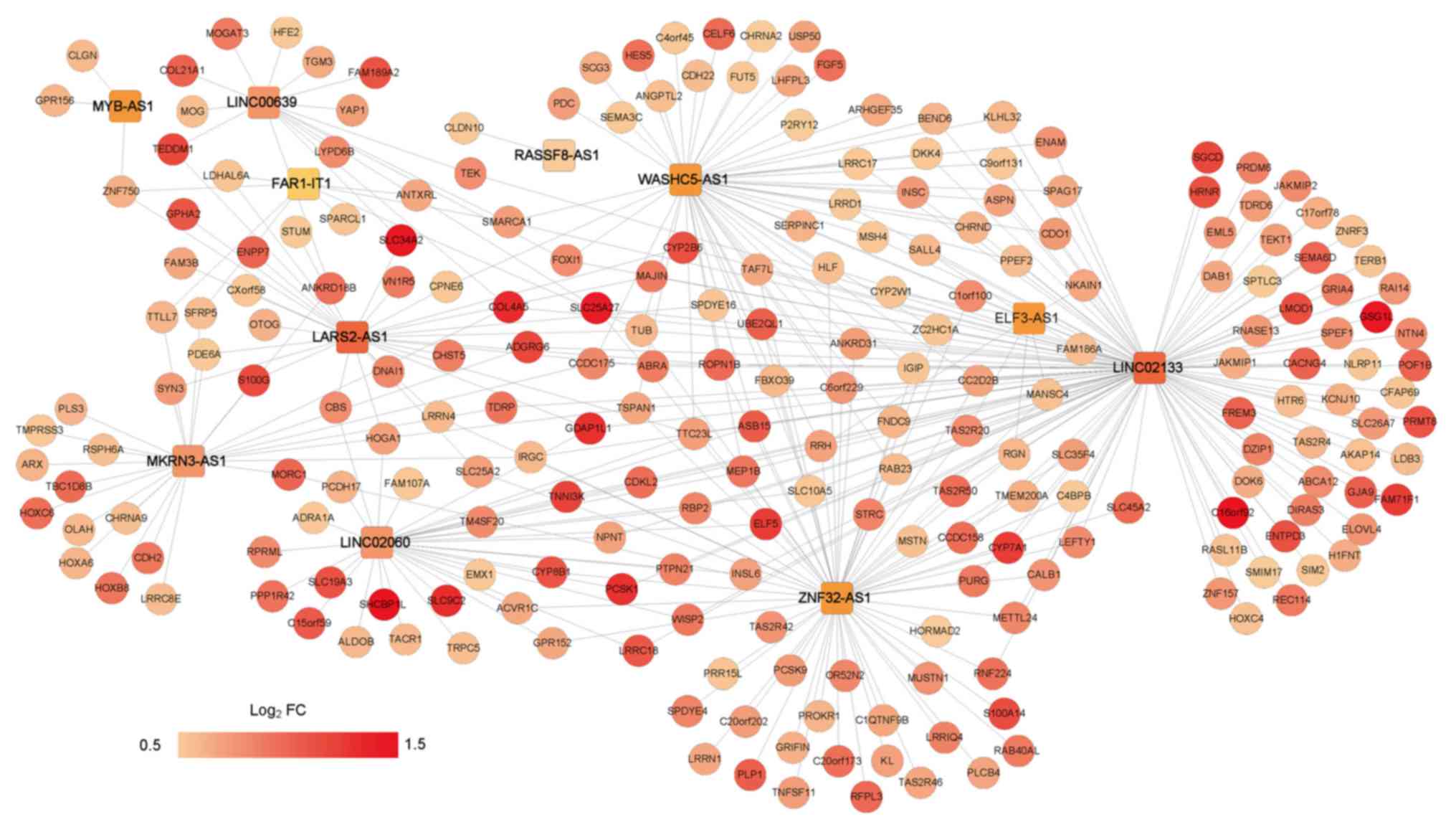
Construction of the lncRNA-mRNA
expression correlation network
Based on the PIs of the samples in the training set,
the samples were divided into the good and bad prognosis groups and
730 DE-mRNAs (718 upregulated and 12 downregulated) between the two
groups were identified. The PCC of the expression level of each
DE-mRNA and PI was calculated, and then the top 100 DE-mRNAs with
highest absolute values of PCC were used for sample clustering
(Fig. 6). Furthermore, the PCCs
between the DE-mRNAs and the 14 optimal lncRNAs were calculated.
The lncRNA-mRNA expression correlation network was constructed with
the criterion of PCC <0.5 (Fig.
7).
Pathway enrichment analysis
Based on GSEA pathway enrichment analysis, axon
guidance [involving semaphorin 3C (SEMA3C), semaphorin 6D and
netrin 4] and the Wnt signaling pathway [involving Dickkopf-related
protein 4, 1-phosphatidylinositol 4,5-biphosphate phosphodiesterase
β 4 and secreted frizzled-related protein 5 (SFRP5)] were
separately associated with the lncRNAs LARS2-AS1 and WASHC5-AS1,
respectively (Table IV).
| Table IVPathways significantly associated
with LARS2-AS1 and WASHC5-AS1. |
Table IV
Pathways significantly associated
with LARS2-AS1 and WASHC5-AS1.
| A, LARS2-AS1 |
|---|
| Name | ES | NES | NOM P-val | Genes |
|---|
| Axon guidance | 0.842324 | 1.775851 | 0.0081 | SEMA3C, SEMA6D,
NTN4 |
| B, WASHC5-AS1 |
| Name | ES | NES | NOM P-val | Genes |
| Wnt signaling
pathway | -0.6971 | -1.4588 | 0.0081 | DKK4, PLCB4,
SFRP5 |
Discussion
In recent decades, bioinformatics methods have
developed rapidly and are being applied for the analysis of gene
expression profiles to determine the pathogenesis of human diseases
(30). A total of six genes
(triggering receptor expressed on myeloid cells like 2, solute
carrier family 7 member 11, NLR family pyrin domain containing 2,
DNA damage inducible transcript 4, lymphocyte specific protein 1
and C-type lectin domain containing 11A) have been identified as
prognostic indicators of AML combining TCGA data with a Gene
Expression Omnibus dataset (31). In
total, two lncRNAs (NONHSAT027612.2 and NONHSAT134556.2) have been
implicated in the pathogenesis of childhood acute lymphoblastic
leukemia (ALL) (32). However, the
prognosis has not been investigated (32). In another previous study, although
both lncRNAs and their relationships with prognosis had been
examined in AML, the authors focused on the competing endogenous
RNAs regulation network (33). The
predictive role of lncRNAs on the prognosis of AML remains
obscure.
In the present study, 61 DE-lncRNAs between the good
and bad prognosis groups were obtained. Of these, 48 lncRNAs were
significantly associated with prognosis. Cox regression analysis
demonstrated that relapse was significantly associated with the
prognosis of patients with AML and was an independent prognostic
factor. From the 48 prognosis-associated lncRNAs, the optimal
lncRNA combination involving 14 lncRNAs was screened to construct
the risk score system. Subsequently, 730 DE-mRNAs were identified
for the good and bad prognosis groups divided by PIs, and the
lncRNA-mRNA expression correlation network was constructed.
The Wilms' tumor gene 1 (WT1) is overexpressed in
AML and functions as an oncogene (34). Therefore, it is a therapeutic target
of AML. PAX8 is a physiological regulator of WT1 that accounts for
its upregulation (35). The lncRNA
PAX8-AS1 located in the upstream region of the PAX8 gene regulates
the expression of the gene (36).
The rs4848320 and rs6726151 polymorphisms of PAX8-AS1 can
significantly increase the risk of ALL (37). Therefore, PAX8-AS1 may act as a risk
factor for childhood ALL (37).
CREB-binding protein/P300 coactivation derived by MYB is necessary
for ALL and AML, and MYB activity plays a critical role in
maintaining AML (38). MYB
expression is increased in T cell ALL, and the oncogene MYB could
be a therapeutic target for the disease (39,40).
These findings suggested that PAX8-AS1 and MYB-AS1 might be
associated with the pathogenesis of patients with AML.
LINC00240-Kruppel-like factor 3 crosstalk is
mediated by microRNA-26b-5p in esophageal squamous cell carcinoma
(ESCC) and its loss may be involved in the oncogenesis of ESCC
(41). ZNF32 is a transcription
factor closely associated with oxidative stress and protects tumor
cells against oxidative stress-induced cell death (42). Therefore, targeting ZNF32 may
suppress multidrug resistance during chemotherapy and improve the
clinical outcome of patients with lung adenocarcinoma (41,43).
RASSF8 expression is negatively associated with lymph node
metastasis and survival of patients with ESCC (44). Therefore, RASSF8 is a tumor
suppressor and a candidate therapeutic target for the disease
(44). The low expression of
E74-like factor 3 (ELF3) is significantly correlated with decreased
survival of patients with ovarian cancer, and ELF3 is a favorable
prognostic biomarker for the tumor (45). Although LINC00240, ZNF32-AS1,
RASSF8-AS1 and ELF3-AS1 have not been reported to be related to
AML, their roles in other tumors were identified. Based on the
findings of the present study, they could be prognostic factors for
AML. Therefore, LINC00240, ZNF32-AS1, RASSF8-AS1 and ELF3-AS1 might
also influence the prognosis of AML.
Dysregulated axon guidance molecules (AGMs) function
as tumor suppressors or oncogenes in breast cancer, which may be
promising targets for the prognosis and therapy of tumors (46). Some AGMs play roles in mediating cell
migration and apoptosis in tumorigenic tissues, suggesting that
promoting or inhibiting the activity of AGMs may be novel
approaches for treating tumors (47). Semaphorins, which are one type of
AGMs, serve as tumor suppressors (48). Of note, semaphorin 3B (SEMA3B) could
act as a tumor inhibitor in the early stage of breast cancer, and
could also promote metastasis during the late stage of the cancer
(49). The role of SEMA3C has not
been fully reported, but it belongs to the AGMs that promote tumor
metastasis (46). In the present
study, SEMA3C was significantly enriched in the axon guidance
pathway, and it was associated with the lncRNA LARS2-AS1,
suggesting that expression of SEMA3C may be regulated by LARS2-AS1,
and the alteration of the gene might disturb the pathway, which
might affect the pathogenesis of AML. However, these regulatory
regulations remain to be experimentally validated.
The Wnt signaling pathway is overexpressed in most
cases of AML and targeting the WNT/lymphoid enhancer-binding factor
1 signaling cascade may be used for developing novel therapies for
AML (50,51). The Wnt signaling pathway is directly
mediated by several secreted antagonist families, such as the SFRP
family (52). In AML, this pathway
is mediated by methylation of Wnt antagonists (53). In the present study, SFRP5 was
significantly associated with the Wnt signaling pathway and was
indirectly regulated by the lncRNA WASHC5-AS1 in the interaction
network, suggesting the interactions between them might influence
the signaling pathway, and regulate the development of AML.
Nonetheless, these interactions need to be experimentally
validated.
In the present study, relapse was identified as an
independent prognostic factor in both the training and validation
datasets, suggesting that relapse could affect the prognostic risk
using this 14-lncRNA predictive system. However, more clinical
evidence is required.
Although comprehensive bioinformatics analysis has
been conducted, there were several limitations of the present
study. The expressions of these lncRNAs and genes were not
validated in vitro and the interactions were not confirmed.
In addition, the predictions of prognostic roles of these lncRNAs
also need the support of further clinical data. More experiments
should be designed and conducted to support the present
results.
In conclusion, a 14-lncRNA (including PAX8-AS1 and
MYB-AS1) risk score system might be effective for the prediction of
AML prognosis. There were two pathways identified from the GSEA
pathway enrichment analysis; axon guidance, which might involve
SEMA3C and LARS2-AS1, and the Wnt signaling pathway, which might
include the interaction of SFRP5 and WASHC5-AS1. These two
identified pathways might be important factors that affect the
prognosis of childhood AML.
Acknowledgements
Not applicable.
Funding
The present study was supported by Medical
Scientific Research Project of Henan Province (grant no.
2018020905), Medical Scientific Research Project of Henan Province
(grant no. SB201903035), Medical and Health Program of Luoyang
Science & Technology Project (grant no. 1910014A).
Availability of data and materials
All data used in this study are available in TCGA
database (https://portal.gdc.cancer.gov/).
Authors' contributions
HRW designed this study; SLG, BL, XYX, WLW, SYW and
TL performed the data analysis. The final version of the manuscript
was read and approved by all authors.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Clarkson BD: Acute myelocytic leukemia in
adults. Cancer. 30:1572–1582. 2015.
|
|
2
|
Döhner H, Weisdorf DJ and Bloomfield CD:
Acute myeloid leukemia. N Engl J Med. 373:1136–1152.
2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kootte AM, Thompson J and Bruijn JA: Acute
myelocytic leukemia with acute aortic occlusion as presenting
symptom. Acta Haematol. 75:120–121. 2009.
|
|
4
|
Pyatt DW, Aylward LL and Hays SM: Is age
an independent risk factor for chemically induced acute myelogenous
leukemia in children? J Toxicol Environ Health B Crit Rev.
10:379–400. 2007.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Belson M, Kingsley B and Holmes A: Risk
factors for acute leukemia in children: A review. Environ Health
Perspect. 115:138–145. 2007.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Deschler B and Lübbert M: Acute myeloid
leukemia: Epidemiology and etiology. Cancer. 107:2099–2107.
2010.
|
|
7
|
GBD 2015 Mortality and Causes of Death
Collaborators: Global, regional, and national life expectancy,
all-cause mortality, and cause-specific mortality for 249 causes of
death, 1980-2015: A systematic analysis for the Global Burden of
Disease Study 2015. Lancet 388: 1459-1544, 2016.
|
|
8
|
Angrand PO, Vennin C, Le Bourhis X and
Adriaenssens E: The role of long non-coding RNAs in genome
formatting and expression. Front Genet. 6(165)2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Garzon R, Volinia S, Papaioannou D,
Nicolet D, Kohlschmidt J, Yan PS, Mrózek K, Bucci D, Carroll AJ,
Baer MR, et al: Expression and prognostic impact of lncRNAs in
acute myeloid leukemia. Proc Natl Acad Sci USA. 111:18679–18684.
2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Hao S and Shao Z: HOTAIR is upregulated in
acute myeloid leukemia and that indicates a poor prognosis. Int J
Clin Exp Pathol. 8:7223–7228. 2015.PubMed/NCBI
|
|
11
|
Wu S, Zheng C, Chen S, Cai X, Shi Y, Lin B
and Chen Y: Overexpression of long non-coding RNA HOTAIR predicts a
poor prognosis in patients with acute myeloid leukemia. Oncol Lett.
10:2410–2414. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sun J, Hu J, Li W and Sun Y: A novel
suppressive long noncoding RNA within the IGF1R Gene locus is
imprinted in acute myelocytic leukemia. Blood.
124(3592)2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Zhao TF, Jia HZ, Zhang ZZ, Zhao XS, Zou
YF, Zhang W, Wan J and Chen XF: LncRNA H19 regulates ID2 expression
through competitive binding to hsa-miR-19a/b in acute myelocytic
leukemia. Mol Med Rep. 16:3687–3693. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Nie Y, Zhou L, Wang H, Chen N, Jia L, Wang
C, Wang Y, Chen J, Wen X, Niu C, et al: Profiling the epigenetic
interplay of lncRNA RUNXOR and oncogenic RUNX1 in breast cancer
cells by gene in situ cis-activation. Am J Cancer Res. 9:1635–1649.
2019.PubMed/NCBI
|
|
15
|
Wang H, Guo R, Hoffman AR, Hu J and Li W:
Effect of long noncoding RNA RUNXOR on the epigenetic regulation of
RUNX1 in acute myelocytic leukemia. J Brazilian Soc Mechanical Sci
Engineering. 25:341–346. 2015.
|
|
16
|
Wang F, Tian X, Zhou J, Wang G, Yu W, Li
Z, Fan Z, Zhang W and Liang A: A three-lncRNA signature for
prognosis prediction of acute myeloid leukemia in patients. Mol Med
Rep. 18:1473–1484. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Lee SH, Chiu YC, Li YH, Lin CC, Hou HA,
Chou WC and Tien HF: High expression of dedicator of cytokinesis 1
(DOCK1) confers poor prognosis in acute myeloid leukemia.
Oncotarget. 8:72250–72259. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Anders S and Huber W: Differential
expression analysis for sequence count data. Genome Biol.
11(R106)2010.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Robinson M, Mccarthy D and Smyth GK:
edgeR: Differential expression analysis of digital gene expression
data. J Hospice Palliative Nurs. 4:206–207. 2010.
|
|
20
|
Chen LP, Wang H, Zhang Y, Chen QX, Lin TS,
Liu ZQ and Zhou YY: Robust analysis of novel mRNA-lncRNA cross talk
based on ceRNA hypothesis uncovers carcinogenic mechanism and
promotes diagnostic accuracy in esophageal cancer. Cancer Manag
Res. 11:347–358. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Perron AM, Girolimetti G, Procaccini M,
Marchio L, Livi A, Borghese G, Porcelli AM, De Iaco P and Gasparre
G: Potential for mitochondrial DNA sequencing in the differential
diagnosis of gynaecological malignancies. Int J Mol Sci. 19(pii:
E2048)2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
He JH, Han ZP, Zou MX, Wang L, Lv YB, Zhou
JB, Cao MR and Li YG: Analyzing the LncRNA, miRNA, and mRNA
regulatory Network in prostate cancer with Bioinformatics Software.
J Comput Biol. 25:146–157. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wang L, Cao C, Ma Q, Zeng Q, Wang H, Cheng
Z, Zhu G, Qi J, Ma H, Nian H and Wang Y: RNA-seq analyses of
multiple meristems of soybean: Novel and alternative transcripts,
evolutionary and functional implications. BMC Plant Biol.
14(169)2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wang P, Wang Y, Hang B, Zou X and Mao JH:
A novel gene expression-based prognostic scoring system to predict
survival in gastric cancer. Oncotarget. 7:55343–55351.
2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Goeman JJ: L1 penalized estimation in the
Cox proportional hazards model. Biom J. 52:70–84. 2010.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ranstam J and Cook JA: Kaplan-meier curve.
Br J Surg. 104(442)2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Kaushal B, Jain K and Sharma SK:
Estimation of area under receiver operating characteristic curve
for Bi-Pareto and Bi-Two parameter exponential models. Open J
Statistics. 4:1–10. 2014.
|
|
28
|
Hauke J and Kossowski T: Comparison of
values of Pearson's and Spearman's correlation coefficients on the
same sets of data. Quaestiones Geographicae. 30:87–93. 2011.
|
|
29
|
Tilford CA and Siemers NO: Gene set
enrichment analysis. Methods Mol Biol. 563:99–121. 2009.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Servant N, Roméjon J, Gestraud P, La Rosa
P, Lucotte G, Lair S, Bernard V, Zeitouni B, Coffin F,
Jules-Clément G, et al: Bioinformatics for precision medicine in
oncology: Principles and application to the SHIVA clinical trial.
Front Genet. 5(152)2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Zhao X, Li Y and Wu H: A novel scoring
system for acute myeloid leukemia risk assessment based on the
expression levels of six genes. Int J Mol Med. 42:1495–1507.
2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Li S, Bian H, Cao Y, Juan C, Cao Q, Zhou G
and Fang Y: Identification of novel lncRNAs involved in the
pathogenesis of childhood acute lymphoblastic leukemia. Oncol Lett.
17:2081–2090. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Yin X, Huang S, Zhu R, Fan F, Sun C and Hu
Y: Identification of long non-coding RNA competing interactions and
biological pathways associated with prognosis in pediatric and
adolescent cytogenetically normal acute myeloid leukemia. Cancer
Cell Int. 18(122)2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Rossi G, Minervini MM, Carella AM, Melillo
L and Cascavilla N: Wilms' tumor gene (WT1) expression and minimal
residual disease in acute myeloid leukemia. In: van den
Heuvel-Eibrink MM (ed), Wilms Tumor, Brisbane (AU), Codon
Publications, 2016.
|
|
35
|
Siehl JM, Thiel E, Heufelder K, Snarski E,
Schwartz S, Mailänder V and Keilholz U: Possible regulation of
Wilms' tumour gene 1 (WT1) expression by the paired box genes PAX2
and PAX8 and by the haematopoietic transcription factor GATA-1 in
human acute myeloid leukaemias. Br J Haematol. 123:235–242.
2003.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Han J, Zhou W, Jia M, Wen J, Jiang J, Shi
J, Zhang K, Ma H, Liu J, Ren J, et al: Expression quantitative
trait loci in long non-coding RNA PAX8-AS1 are associated with
decreased risk of cervical cancer. Mol Genet Genomics.
291:1743–1748. 2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Bahari G, Hashemi M, Naderi M,
Sadeghi-Bojd S and Taheri M: Long non-coding RNA PAX8-AS1
polymorphisms increase the risk of childhood acute lymphoblastic
leukemia. Biomed Rep. 8:184–190. 2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Ramaswamy K, Forbes L, Minuesa G, Gindin
T, Brown F, Kharas MG, Krivtsov AV, Armstrong SA, Still E, de
Stanchina E, et al: Peptidomimetic blockade of MYB in acute myeloid
leukemia. Nat Commun. 9(110)2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Lahortiga I, De Keersmaecker K, Van
Vlierberghe P, Graux C, Cauwelier B, Lambert F, Mentens N, Beverloo
HB, Pieters R, Speleman F, et al: Duplication of the MYB oncogene
in T cell acute lymphoblastic leukemia. Nat Genet. 39:593–595.
2007.PubMed/NCBI View
Article : Google Scholar
|
|
40
|
Mets E, Van der Meulen J, Van Peer G,
Boice M, Mestdagh P, Van de Walle I, Lammens T, Goossens S, De
Moerloose B, Benoit Y, et al: MicroRNA-193b-3p acts as a tumor
suppressor by targeting the MYB oncogene in T-cell acute
lymphoblastic leukemia. Leukemia. 29:798–806. 2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Shuang Y, Ning Q, Zhang G, Hong S, Zhen W
and Li Y: Construction of differential mRNA-lncRNA crosstalk
networks based on ceRNA hypothesis uncover key roles of lncRNAs
implicated in esophageal squamous cell carcinoma. Oncotarget.
7:85728–85740. 2016.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Li K, Gao B, Li J, Chen H, Li Y, Wei Y,
Gong D, Gao J, Zhang J, Tan W, et al: ZNF32 protects against
oxidative stress-induced apoptosis by modulating C1QBP
transcription. Oncotarget. 6:38107–38126. 2015.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Kai L, Bo G, Jun L, Chen H, Li Y, Wei Y,
Gong D, Gao J, Zhang J, Tan W, et al: ZNF32 protects against
oxidative stress-induced apoptosis by modulating C1QBP
transcription. Oncotarget. 6:38107–38126. 2015.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Zhang L, Wang JH, Liang RX, Huang ST, Xu
J, Yuan LJ, Huang L, Zhou Y, Yu XJ, Wu SY, et al: RASSF8
downregulation promotes lymphangiogenesis and metastasis in
esophageal squamous cell carcinoma. Oncotarget. 6:34510–34524.
2015.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Yeung TL, Leung CS, Wong KK,
Gutierrez-Hartmann A, Kwong J, Gershenson DM and Mok SC: ELF3 is a
negative regulator of epithelial-mesenchymal transition in ovarian
cancer cells. Oncotarget. 8:16951–16963. 2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Harburg GC and Hinck L: Navigating breast
cancer: Axon guidance molecules as breast cancer tumor suppressors
and oncogenes. J Mammary Gland Biol Neoplasia. 16:257–270.
2011.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Kerjan G and Moreaufauvarque C: The brain
within the tumor: New roles for axon guidance molecules in cancers.
Cell Death Differ. 12:1044–1056. 2005.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Neufeld G, Mumblat Y, Smolkin T, Toledano
S, Nir-Zvi I, Ziv K and Kessler O: The role of the semaphorins in
cancer. Cell Adh Migr. 10:652–674. 2016.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Rolny C, Capparuccia L, Casazza A, Mazzone
M, Vallario A, Cignetti A, Medico E, Carmeliet P, Comoglio PM and
Tamagnone L: The tumor suppressor semaphorin 3B triggers a
prometastatic program mediated by interleukin 8 and the tumor
microenvironment. J Exp Med. 205:1155–1171. 2008.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Minke KS, Staib P, Puetter A, Gehrke I,
Gandhirajan RK, Schlösser A, Schmitt EK, Hallek M and Kreuzer KA:
Small molecule inhibitors of WNT signaling effectively induce
apoptosis in acute myeloid leukemia cells. Eur J Haematol.
82:165–175. 2010.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Mikesch JH, Steffen B, Berdel WE, Serve H
and Müller-Tidow C: The emerging role of Wnt signaling in the
pathogenesis of acute myeloid leukemia. Leukemia. 21:1638–1647.
2007.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Pehlivan M, Çalışkan C, Yüce Z and Sercan
HO: Secreted Wnt antagonists in leukemia: A road yet to be paved.
Leuk Res. 69:24–30. 2018.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Valencia A, Romángómez J, Cervera J, Such
E, Barragán E, Bolufer P, Moscardó F, Sanz GF and Sanz MA: Wnt
signaling pathway is epigenetically regulated by methylation of Wnt
antagonists in acute myeloid leukemia. Leukemia. 23:1658–1666.
2009.PubMed/NCBI View Article : Google Scholar
|