Introduction
Inflammation, an important process of host immune
response, defends against various injurious insults, including
pathogens and their products, damaged cells or noxious stimuli
(1). An increasing number of studies
suggested that macrophages are critical components of the
inflammatory response by secreting various enzymes, cytokines,
chemokines and prostaglandins via the activation of several
signaling cascades, including nuclear factor (NF) κ-B and
mitogen-activated protein kinases (MAPKs) (2,3).
Although pro-inflammatory mediators and cytokines produced by
macrophages can help to defend against various injurious insults,
including pathogens and their products, damaged cells or noxious
stimuli, the pro-inflammatory mediators and cytokines, such as
nitric oxide (NO), prostaglandin E2, tumor necrosis
factor-α (TNF-α), interleukin (IL)-1β and IL-6, which are produced
by macrophages, can result in metabolic stress of cells and severe
damage within the tissues (4).
Notably, misdirected inflammation has been proven to be associated
with the pathogenesis of numerous diseases, including rheumatoid
arthritis (RA), cancer, diabetes, cardiovascular and
cerebrovascular diseases (5-8).
Thus, the regulation of activated macrophages followed by the
reduction of the aforementioned inflammatory mediators and
cytokines may aid in the development of novel treatment strategies
for alleviation of inflammatory responses and its related
diseases.
Carboxyamidotriazole (CAI) was originally developed
as a non-cytotoxic anti-cancer drug; however, multiple in
vitro and in vivo preclinical research studies have
revealed its anti-proliferative, -angiogenic, and -migratory
activities (9-12).
Most side effects of CAI were minor, identified from a series of
clinical trials, indicating that CAI is typically well tolerated,
although the drug exhibits only mild anti-cancer action in a
clinical setting (13-15).
Therefore, the current primary application for the anti-tumor use
of CAI is to combine it with other drugs to synergize the efficacy
according to its underlying mechanism (16). Recent studies published by authors of
the present study in several animal inflammatory models, such as
croton oil-induced ear edema, cotton-induced granuloma, adjuvant
arthritis and colitis, revealed significant anti-inflammatory
potency of CAI (17-20).
In addition, CAI-associated amelioration of inflammatory symptoms
were associated with decreasing levels of cytokines and
phosphorylated inhibitor of nuclear factor-κBα (IκBα), as well as
blocking IκBα degradation in inflammatory tissues (18-20).
Moreover, inhibition of TNF-α production in tumor-associated
macrophages was found to be another important mechanism for the
anti-cancer action of CAI (16).
Based on this, the combination of CAI with a low dose of
dexamethasone, an effective anti-inflammatory drug, strengthened
the suppression of CAI on the proliferation and invasion of tumor
cells co-cultured with macrophages (16). Therefore, the clarification of the
effect of CAI on macrophages will promote further understanding of
its anti-inflammatory and anti-cancer actions. However, the
detailed mechanisms in macrophages and the signaling cascade
pathways influenced by CAI have yet to be elucidated.
In the present study, the effects of CAI on
LPS-stimulated RAW264.7 cells, which is a commonly used model of
macrophage inflammation, was investigated. The results revealed the
potency of CAI against inflammatory responses in RAW264.7
macrophages via the inhibition of NO and cytokines. In addition,
the influences of CAI on cyclooxygenase (COX) activity and NF-κB
and MAPK signal transduction pathways to reveal its molecular
mechanism was determined.
Materials and methods
Reagents
CAI was synthesized by the Institute of Materia
Medica, Chinese Academy of Medical Sciences and dissolved in
dimethyl sulfoxide (DMSO; Sigma-Aldrich; Merck KGaA) as a 40 mM
stock solution. Cell culture reagents were purchased from Gibco
(Thermo Fisher Scientific, Inc.) and the COX inhibitor screening
assay kit, sc-560 and DuP697 were purchased from Cayman Chemical
Company. Cell Counting Kit-8 (CCK-8) was obtained from Dojindo
Molecular Technologies, Inc. LPS (Escherichia coli 055:B5)
was obtained from Sigma-Aldrich (Merck KGaA). TRIzol®
reagent and SYBRGreen dye were purchased from Invitrogen (Thermo
Fisher Scientific, Inc.). The NO assay kit (cat. no. A013-2-1) was
purchased from Nanjing Jiancheng Bioengineering Institute. TNF-α
(cat. no. EM008), IL-1β (cat. no. EM001) and IL-6 (cat. no. EM004)
enzyme-linked immunosorbent assay (ELISA) kits were purchased from
Shanghai ExCell Biology, Inc.. The TransScript II First-Strand cDNA
Synthesis SuperMIX was purchased from Transgen Biotech Co., Ltd.
The TransAM NF-κB p65 transcription factor assay kit (cat. no.
40096) and nuclear extract kit (cat. no. 40010) were purchased from
Active Motif Inc.. Primary antibodies against inducible nitric
oxide synthase (iNOS, cat. no. 13120), phosphorylated (p)-MAPKs
(p-MAPK family antibody sampler kit; cat. no. 9910), MAPKs (MAPK
family antibody sampler kit; cat. no. 9926), p-IκB (cat. no. 2859),
IκB (cat. no. 4814), p-p65 (cat. no. 3033) and p65 (cat. no. 8242)
were purchased from Cell Signaling Technology Inc., while
anti-β-actin primary antibody (cat. no. sc-47778) and horseradish
peroxidase (HRP)-conjugated secondary antibodies (cat. nos. sc-2004
and sc-2005) were purchased from Santa Cruz Biotechnology, Inc..
The chemiluminescent reagent was purchased from EMD Millipore. The
immunofluorescence staining kit (cat. no. P0176), containing goat
anti-rabbit immunoglobulin G (IgG) conjugated with Alexa Fluor 488
was purchased from Beyotime Institute of Biotechnology. Other
chemical reagents including Tris, NaCl, glycerol, Triton X-100,
egtazic acid, sodium orthovanadate and sodium fluoride were of
analytical grade.
COX activity assay
The COX inhibitor screening kit, which can directly
detect prostaglandin F2α (PGF2α) produced by
tin chloride reduction of COX-derived prostaglandin H2
generated in the COX reaction, was used to evaluate the effects of
CAI on COX-1 and COX-2 activity. The assay was performed according
to the manufacturer's instructions. Heme and the reaction buffer
(Tris-HCl buffer containing 5 mM EDTA and 2 mM phenol, pH 8.0) were
added to test tubes. Following addition of CAI and COX-1 or COX-2,
the tubes were incubated at 37˚C for 10 min. Subsequently, the
substrate (arachidonic acid) was added and the tubes were incubated
at 37˚C for a further 2 min. The amount of PGF2α
produced was determined by utilizing ELISA contained within the
aforementioned kit. The absorbance was quantified using
spectrophotometry at a wavelength of 405 nm and PGF2α
content was determined from a standard curve. CAI was dissolved in
DMSO at final concentrations of 1.56, 3.125, 6.25, 12.5, 25, 50 and
100 µM. DMSO alone was placed in the control tubes. Sc-560 and
DuP697, COX-1 and COX-2 specific inhibitors, respectively, were
used at final concentrations of 20 nM as positive controls for 10
min at 37˚C. All measurements were performed in triplicate.
Cell culture
The RAW264.7 mouse macrophage cell line was provided
by the Chinese National Infrastructure of Cell Line Resource. Cells
were cultured in Dulbecco's modified Eagle medium (DMEM) containing
100 U/ml penicillin, 100 µg/ml streptomycin, 10 mM HEPES, 2 mM
L-glutamine and 10% fetal bovine serum at 37˚C in a humidified
incubator with 5% CO2.
Cell viability assay
The CCK-8 assay was used to measure cell viability.
In brief, RAW264.7 cells were plated at a density of
2x104 cells/well in a 96-well plate, and either
untreated (control), or pretreated with vehicle (0.1% DMSO) or
various concentrations of CAI (10, 20 and 40 µM) for 2 h in a 37˚C
incubator, and subsequently stimulated for 22 h with 1 µg/ml LPS at
37˚C. CCK-8 solution (20 µl/well) was then added to the cells and
incubated for an additional 2 h at 37˚C. Subsequently, the
absorbance was determined at a wavelength of 450 nm to measure cell
viability.
Measurement of cytokine and NO
levels
RAW264.7 cells were cultured at a density of
2x105 cells/well in 24-well plates, and either untreated
(control), or pretreated with vehicle (0.1% DMSO) or various
concentrations of CAI (10, 20 and 40 µM) for 2 h in a 37˚C
incubator, and subsequently stimulated with 1 µg/ml LPS at 37˚C.
The culture supernatants were collected after 6 h of LPS
stimulation (for detection of cytokine levels) or 24 h (for
detection of NO levels) (21). The
levels of TNF-α, IL-1β and IL-6 in the supernatants were determined
using corresponding ELISA kits, and NO levels were measured using
an NO assay kit according to the manufacturer's instructions.
Reverse transcription-quantitative PCR
(RT-qPCR)
Following either untreated (control), pretreatment
with vehicle (0.1% DMSO) or various concentrations of CAI (10, 20
and 40 µM) for 2 h in a 37˚C incubator, RAW264.7 cells were
stimulated for 6 h with 1 µg/ml LPS at 37˚C. Total mRNA was
extracted from the cells using TRIzol® reagent and RNA
concentration was determined using a NanoDrop 2000
spectrophotometer (Thermo Fisher Scientific, Inc.). Subsequently,
total RNA from each sample was reverse transcribed into cDNA using
the TransScript II First-Strand cDNA Synthesis SuperMIX according
to the manufacturer's instructions. RT-qPCR was performed using the
CFX Connect Real-Time PCR Detection system (Bio-Rad Laboratories,
Inc.) and SYBRGreen dye. The sequences of the primers used are
shown in Table I. The following
thermocycling conditions were used for the qPCR: Initial
denaturation at 94˚C for 3 min; 40 cycles of 94˚C for 30 sec, 60˚C
for 40 sec, and a final extension at 72˚C for 1 min. mRNA levels
were quantified using the 2-ΔΔCq method (22). β-actin was used as the endogenous
control.
 | Table IPrimer sequences used for the
qPCR. |
Table I
Primer sequences used for the
qPCR.
| Gene | Primer | Sequence |
|---|
| iNOS | Forward |
5'-CAGCTGGGCTGTACAAACCTT-3' |
| | Reverse |
5'-CATTGGAAGTGAAGCGTTTCG-3' |
| TNF-α | Forward |
5'-GCCTCCCTCTCATCAGTTCTA-3' |
| | Reverse |
5'-GGCAGCCTTGTCCCTTG-3' |
| IL-1β | Forward |
5'-GGGCTGCTTCCAAACCTTTG-3' |
| | Reverse |
5'-GCTTGGGATCCACACTCTCC-3' |
| IL-6 | Forward |
5'-AGTTGTGCAATGGCAATTCTGA-3' |
| | Reverse |
5'-AGGACTCTGGCTTTGTCTTTCT-3' |
| β-actin | Forward |
5'-TGCTGTCCCTGTATGCCTCT-3' |
| | Reverse |
5'-TTTGATGTCACGCACGATTT-3' |
Western blot analysis
Western blot analysis was used to analyze the
protein expression levels of corresponding proteins. For the
detection of iNOS, cells were either untreated (control), or
preincubated with vehicle (0.1% DMSO) or various concentrations of
CAI (10, 20 and 40 µM) for 2 h in a 37˚C incubator and subsequently
stimulated for 6 h with 1 µg/ml LPS at 37˚C. Since inflammatory
pathways, including NF-κB and MAPKs, are activated quickly
following LPS stimulation, the time of LPS incubation for analysis
of the two pathways was set to 1 h following CAI pretreatment, as
previously described (23,24). Briefly, RAW264.7 cells were washed
twice with phosphate-buffered saline (PBS) and lysed on ice for 30
min with ice-cold lysis buffer (10 mM Tris, 150 mM NaCl, 10%
glycerol, 1% Triton X-100, 1 mM egtazic acid, 1 mM sodium
orthovanadate, 10 mM sodium fluoride and 1% protease inhibitor mix;
pH 7.5), followed by centrifugation for 30 min at 12,000 x g (4˚C).
The supernatants were collected, and protein concentrations were
determined using a Bradford assay. Equivalent amounts of protein
(60 µg/lane) from the supernatant were transferred onto
polyvinylidene difluoride membranes following separation on 10%
SDS-PAGE. The membranes were subsequently blocked with
Tris-buffered saline solution containing 0.1% Tween-20 (TBS-T),
supplemented with 5% skimmed milk for 2 h at room temperature.
Membranes were then probed overnight (4˚C) with primary antibodies
against iNOS (1:500), IκB (1:1,000), p-IκB (1:1,000), p65
(1:1,000); p-p65 (1:1,000), β-actin (1:1,000), p-p38 (1:500), p38
(1:500), p-JNK (1:500), JNK (1:800), p-ERK (1:1,000) and ERK
(1:1,000). Following washing with TBS-T three times, the membranes
were incubated for 2 h at room temperature with HRP-labeled
secondary antibodies at a dilution of 1:5,000. Protein bands were
visualized using an enhanced chemiluminescent reagent and the band
density was quantified using Kodak 1D software (version 3.5;
Kodak). β-actin served as the internal control and total levels of
the proteins were used in the evaluation of the phosphorylated
forms of the proteins.
DNA-binding activity of NF-κB p65
RAW264.7 cells were either untreated (control), or
preincubated with vehicle (0.1% DMSO) or various concentrations of
CAI (10, 20 and 40 µM) for 2 h in a 37˚C incubator and then
stimulated for 1 h with 1 µg/ml LPS at 37˚C. Cellular nuclear
protein was extracted using a nuclear extraction kit and analyzed
using the NF-κB p65 transcription factor assay kit, which uses an
ELISA-based assay to determine NF-κB p65 activity. Briefly, the kit
contained a 96-well microtiter plate to which an oligonucleotide
containing the NF-κB consensus binding site (5'-GGGACTTTCC-3') was
immobilized. The nuclear extracts were added to the 96-well plate
and incubated for 1 h at room temperature, allowing the active form
of p65 contained in the extracts to bind to the oligonucleotide.
Following sufficient washing with the wash buffer contained in the
kit, anti-NF-κB p65 primary antibody (1:1,000) was added and
incubated for 1 h at room temperature. The plate was washed
extensively, and subsequently incubated with HRP-conjugated
secondary antibody (1:1,000). Finally, tetramethyl benzidine
substrate was added for color development and the absorbance was
quantified using spectrophotometry at a wavelength of 450 nm.
Immunofluorescence
RAW264.7 cells were cultured on coverslips and
either untreated (control), or preincubated with vehicle (0.1%
DMSO) or various concentrations of CAI (10, 20 and 40 µM) for 2 h
in a 37˚C incubator followed by stimulation for 1 h with 1 µg/ml
LPS at 37˚C. Following washing with PBS, cells were fixed for 20
min with 4% paraformaldehyde at room temperature and permeabilized
for 10 min using 0.1% Triton X-100 on ice. Subsequently, the cells
were treated for 1 h with blocking solution (1% bovine serum
albumin) and incubated overnight with a primary antibody against
p65 (1:400) at 4˚C. Subsequently, Alexa Fluor 488-conjugated
anti-rabbit IgG secondary antibody (1:1,000) was added to the cells
for 30 min at 37˚C. The nuclei were stained with DAPI at room
temperature for 5 min. Images were acquired using a Zeiss LSM 780
laser scanning confocal microscope (Zeiss AG) and a Leica DM4000
upright fluorescence microscope (Leica Microsystems GmbH) under
x400 magnification. The pixel intensities of p65 in the nuclear
area were measured as the percent area of immunoreactivity using
ImageJ software (version 1.52u; National Institutes of Health)
(25,26).
Statistical analysis
SPSS software (version 20.0; IBM Corp.) was used for
statistical analysis. Data are presented as mean ± SD and were
analyzed using one-way ANOVA followed by Bonferroni's multiple
comparisons test. P<0.05 was considered to indicate a
statistically significant difference.
Results
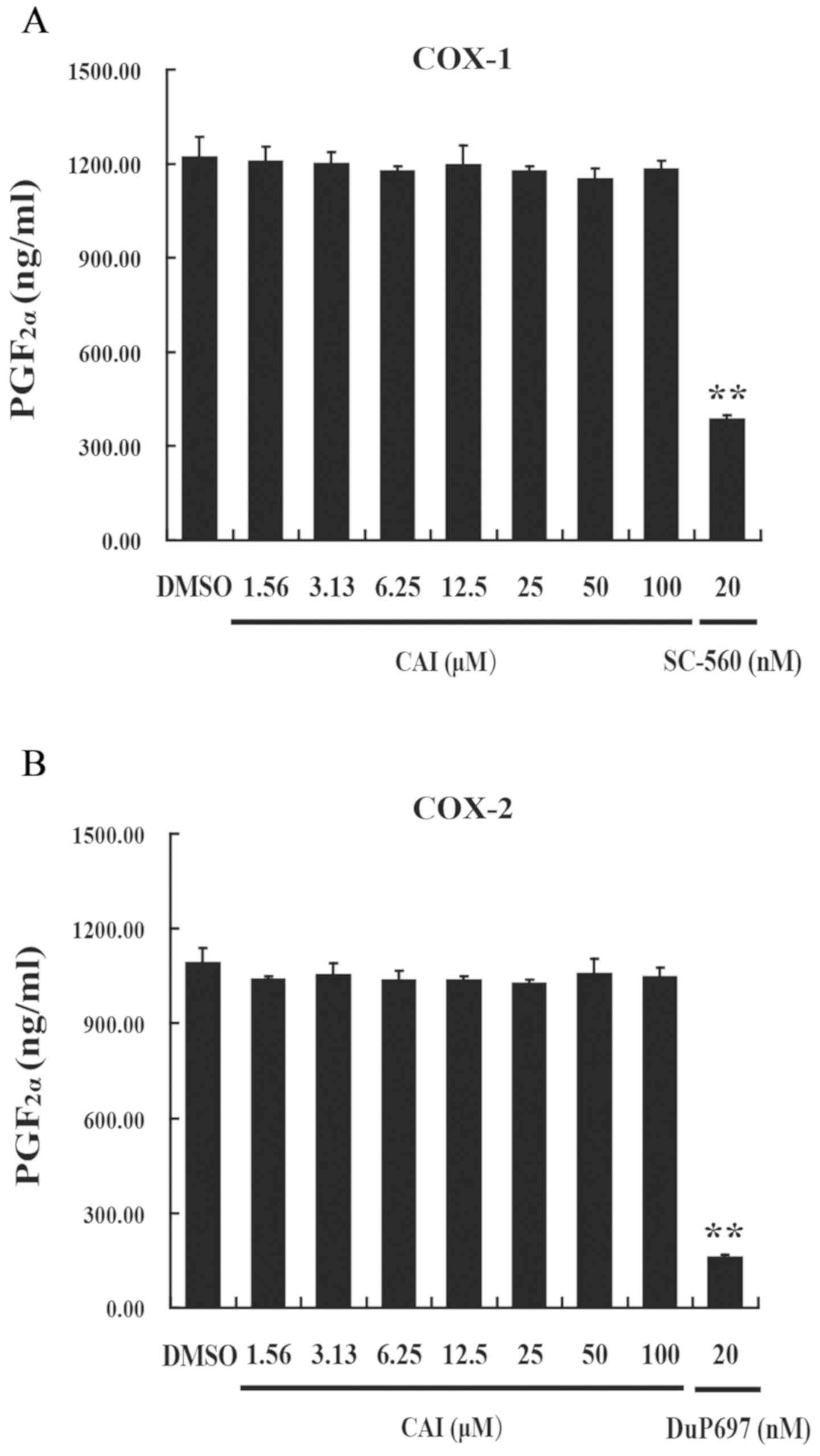
CAI has no effect on COX-1 and COX-2
activities
Non-steroidal anti-inflammatory drugs (NSAIDs) are
the main drugs used for inflammation-related diseases, which have
been known to possess anti-inflammatory action via inhibiting COX
activity and numerous arachidonic acid-related pro-inflammatory
factors, such as prostaglandins and leukotrienes (27). By contrast, CAI did not affect the
enzyme activities of both COX-1 and COX-2, at concentrations
ranging from 1.56 to 100 µM. Furthermore, sc-560 (COX-1-specific
inhibitor) and DuP697 (COX-2-specific inhibitor), significantly
suppressed COX-1 and COX-2 activity, respectively, compared with
control cells (Fig. 1). These
results suggested that CAI exerted its anti-inflammatory action via
a different mechanism to COX-inhibiting NSAIDs.
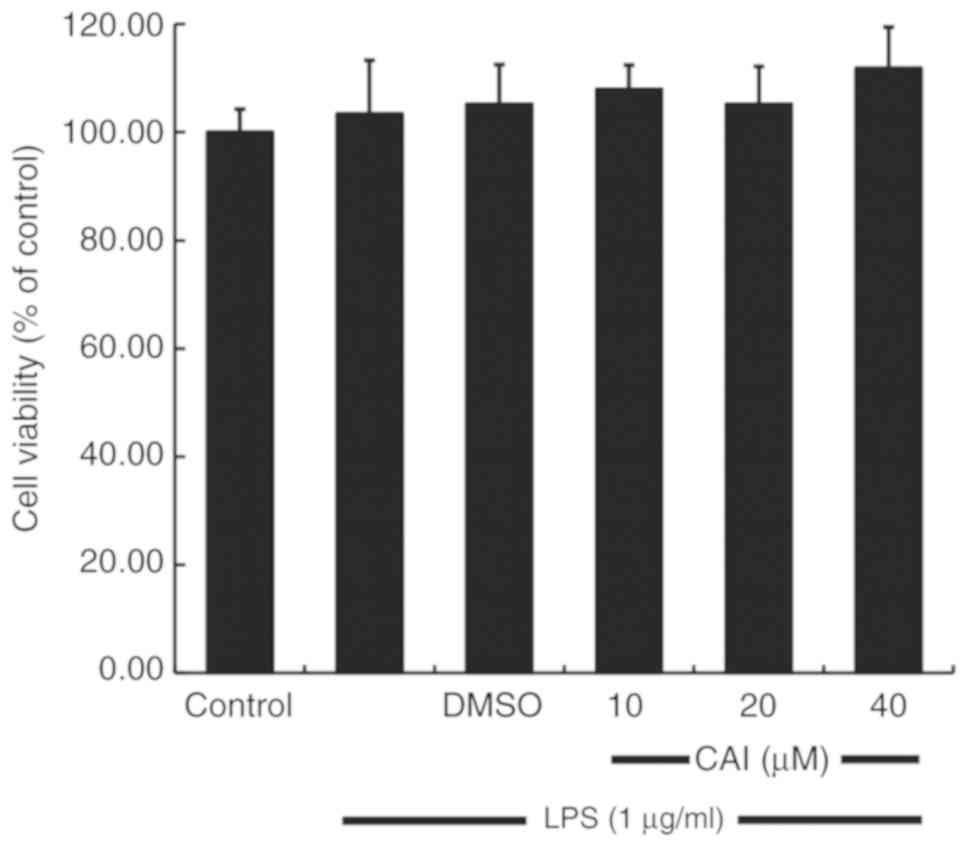
Effect of CAI on cell viability of
RAW264.7 macrophages
The anti-inflammatory effects and mechanism of CAI
on RAW264.7 macrophages was investigated. To rule out the
possibility that decreased cellular inflammatory responses were
caused by direct toxicity of CAI to the cells, the potential
cytotoxicity of CAI was evaluated using a CCK-8 assay. As shown in
Fig. 2, CAI did not affect cell
viability at all the concentrations tested compared with controls,
which was subsequently found to inhibit LPS-induced inflammatory
responses. Therefore, the effect of CAI on RAW264.7 macrophages was
not due to cytotoxicity.
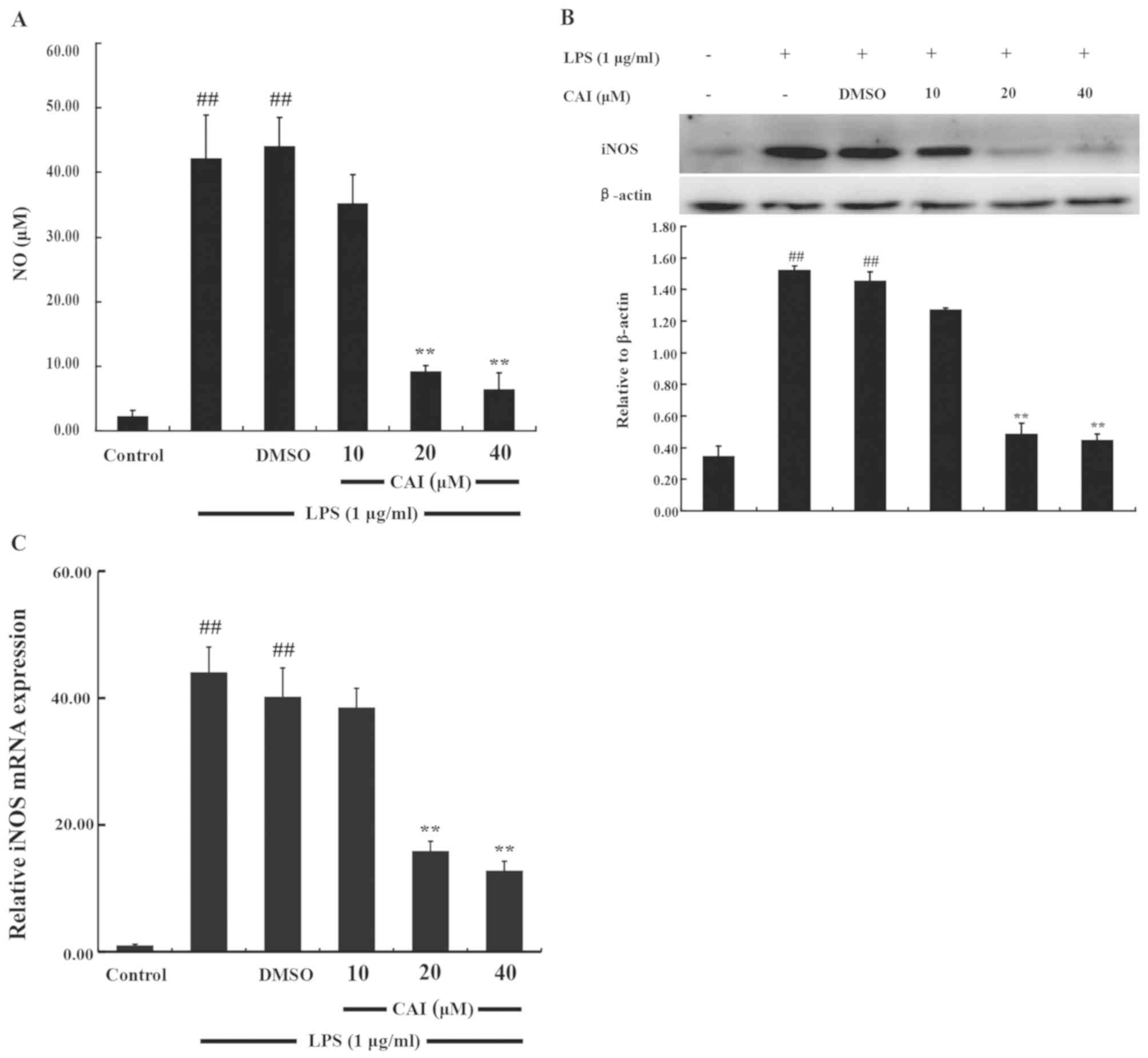
CAI decreases NO production and iNOS
expression in LPS-induced RAW264.7 macrophages
The anti-inflammatory action of CAI in macrophages
was firstly investigated by determining the levels of NO production
following stimulation of RAW 264.7 cells with LPS. NO production
significantly increased following LPS stimulation compared with
controls. However, cells pretreated with CAI (20 and 40 µM) showed
a significant downregulation in NO levels compared with the LPS +
DMSO group (Fig. 3A).
Since iNOS is an important NO-generating enzyme
(4), western blot analysis was
performed to further analyze the protein expression levels of iNOS.
iNOS expression was almost undetectable in the absence of LPS but
was significantly increased following LPS stimulation compared with
controls. However, CAI pretreatment (20 and 40 µM) significantly
suppressed the increased protein expression of iNOS in LPS-induced
RAW264.7 cells compared with the LPS + DMSO group (Fig. 3B). In addition, LPS-induced mRNA
expression levels of iNOS were also significantly inhibited by
pretreatment with CAI (20 and 40 µM) compared with the LPS + DMSO
group, as shown from the RT-qPCR analysis (Fig. 3C).
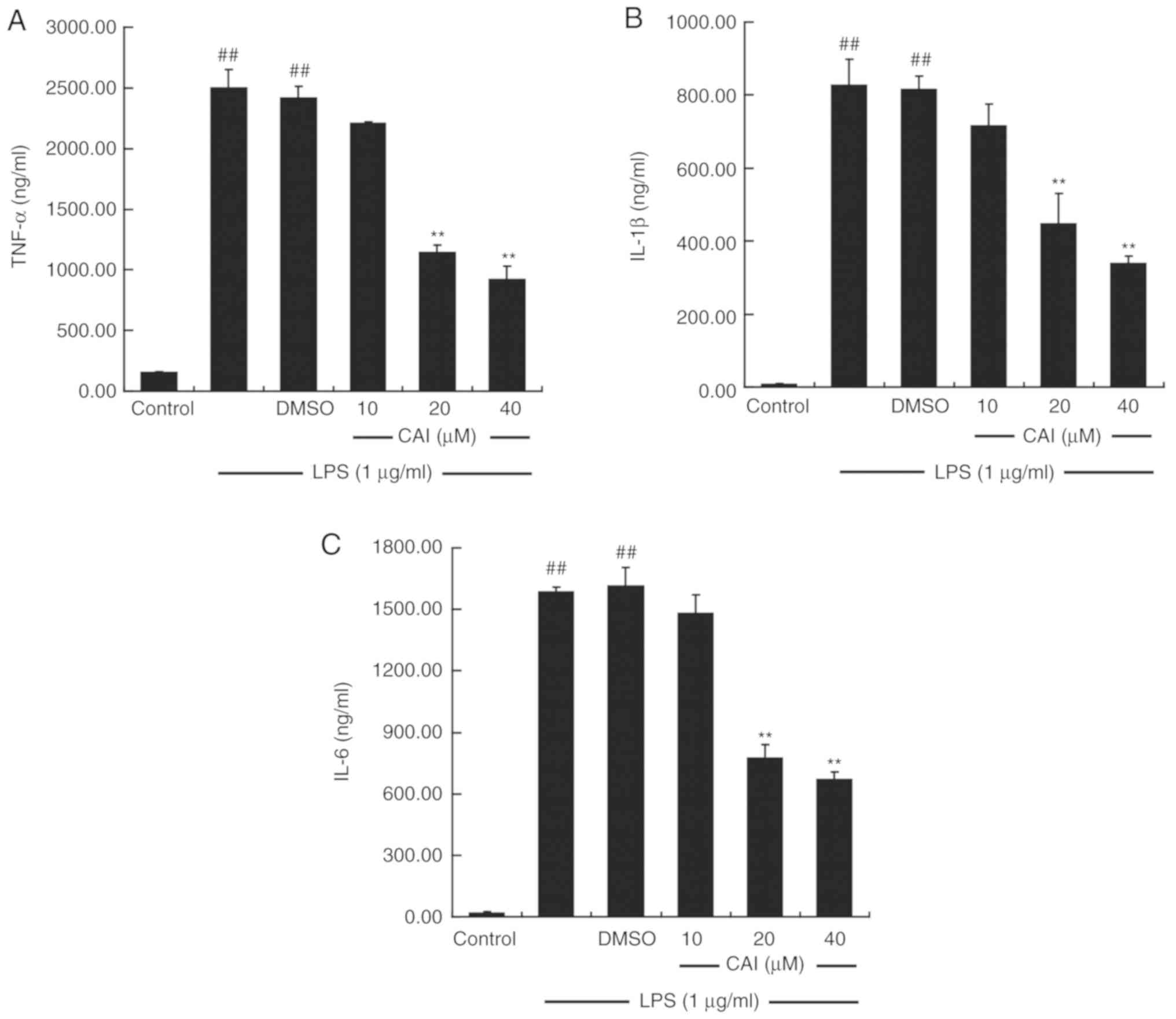
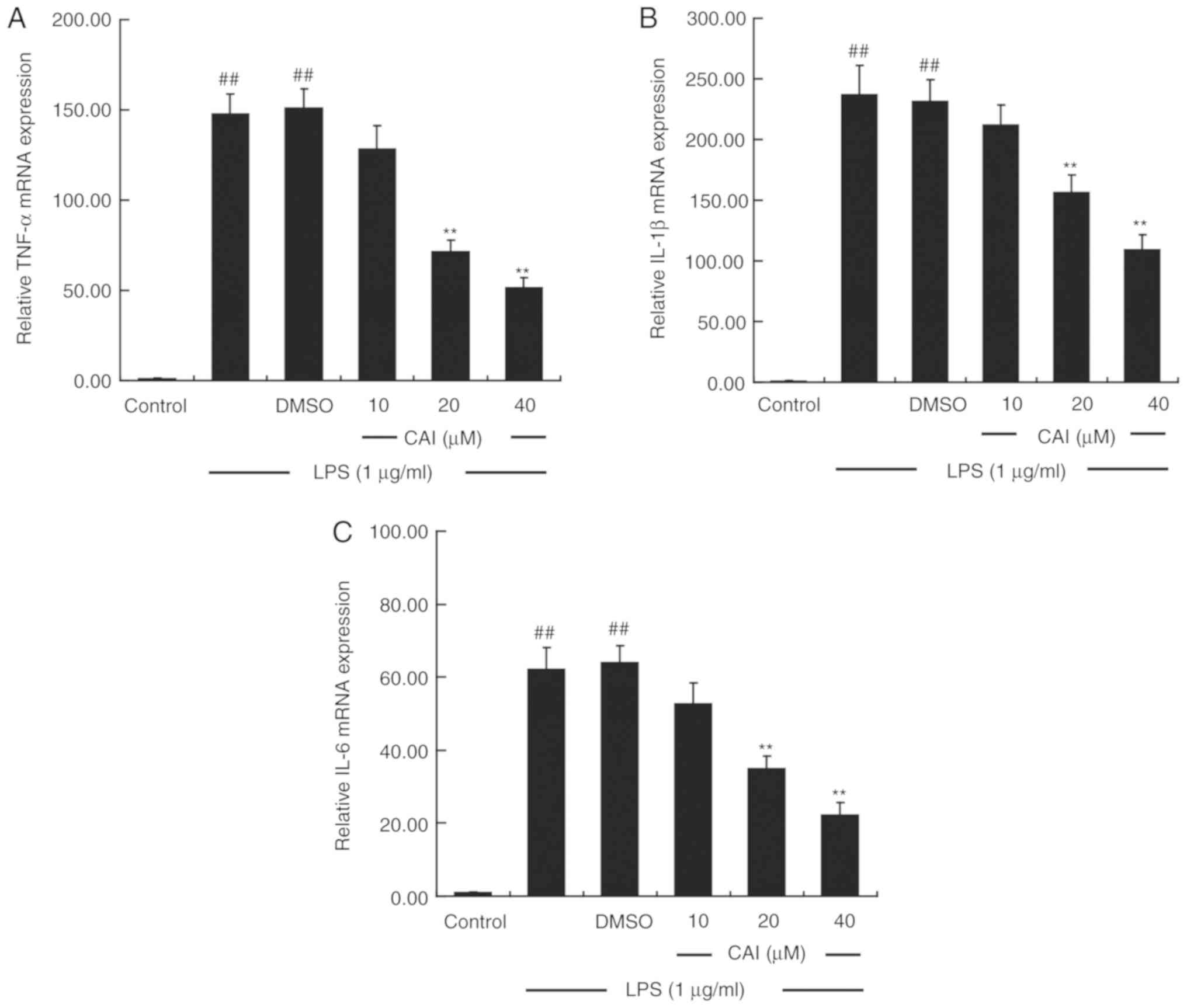
CAI downregulates the levels of
pro-inflammatory cytokines in LPS-induced RAW264.7 macrophages
The production of pro-inflammatory cytokines was
measured using ELISA. Pretreating the cells with LPS alone
significantly upregulated the levels of TNF-α, IL-1β and IL-6 in
the supernatant compared with controls, while CAI (20 and 40 µM)
prior to LPS treatment inhibited the elevated secretion of these
cytokines compared with the LPS + DMSO group (Fig. 4). RT-qPCR analysis further confirmed
that treatment with LPS increased the mRNA expression levels of the
aforementioned cytokines, which were attenuated by CAI at 20 and 40
µM (Fig. 5).
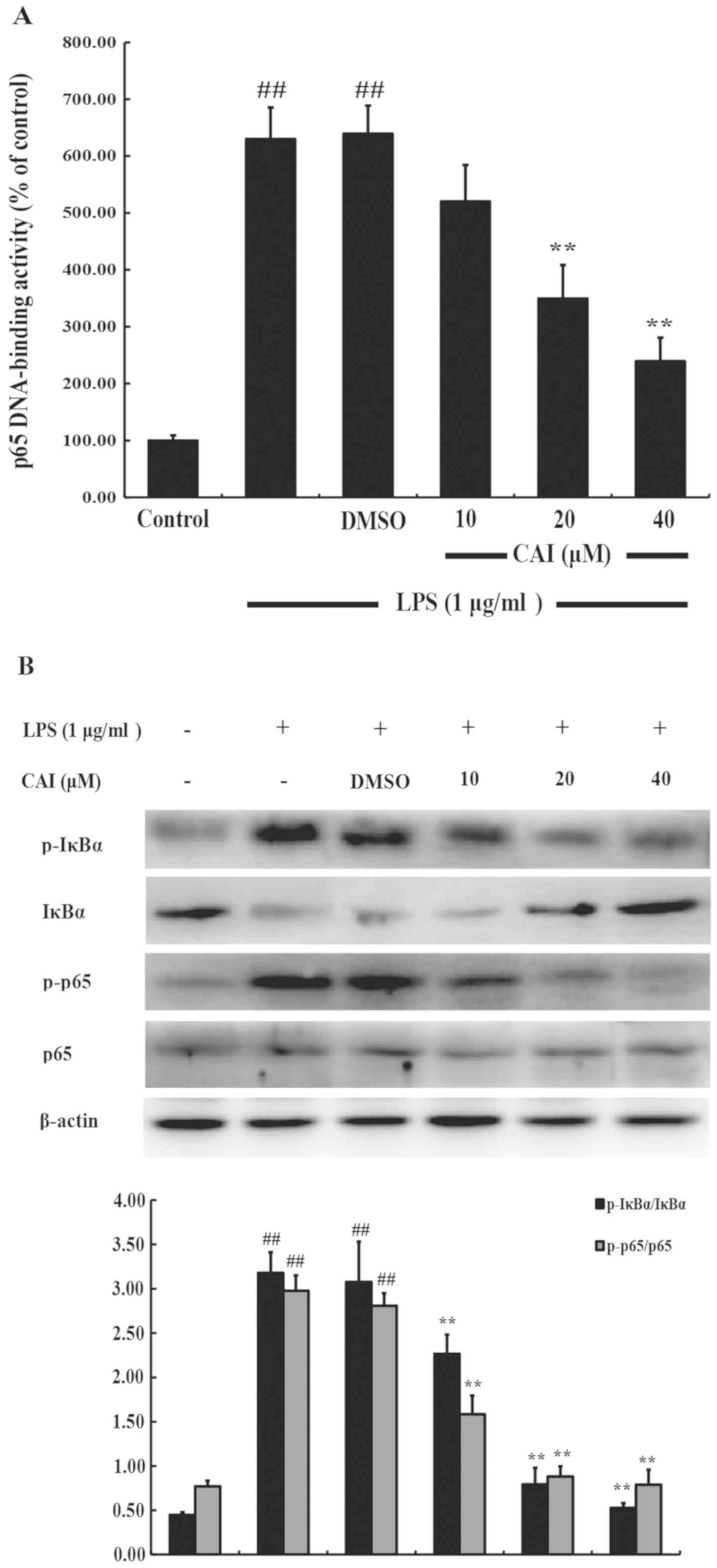
CAI inhibits NF-κB activation in
LPS-induced RAW264.7 macrophages
The transcription factor NF-κB is known to be
implicated in the regulation of numerous genes that code for the
mediators of inflammatory and immune responses (28). To explore the potential mechanism
involved in the anti-inflammatory action of CAI in the macrophages,
a DNA-binding assay was performed to determine the effects of CAI
on LPS-induced NF-κB activation. The DNA-binding activity of NF-κB
p65 in nuclear extracts was significantly increased in response to
LPS treatment compared with controls, and this increase was
significantly reduced by pretreatment with CAI (20 and 40 µM)
compared with the LPS + DMSO group (Fig.
6A).
Since the activation of NF-κB is associated with the
sequential transduction cascade, including IκBα phosphorylation and
degradation, and translocation of activated NF-κB, which is
facilitated by the phosphorylation of the p65 subunit (28), protein extracts of RAW264.7 cells
were probed for IκBα, p-IκBα, p-65 and p-p65. The results showed
that the phosphorylation of IκBα and p65 in RAW264.7 cells
increased after LPS stimulation compared with controls, but was
significantly inhibited by CAI pretreatment (10, 20 and 40 µM)
(Fig. 6B).
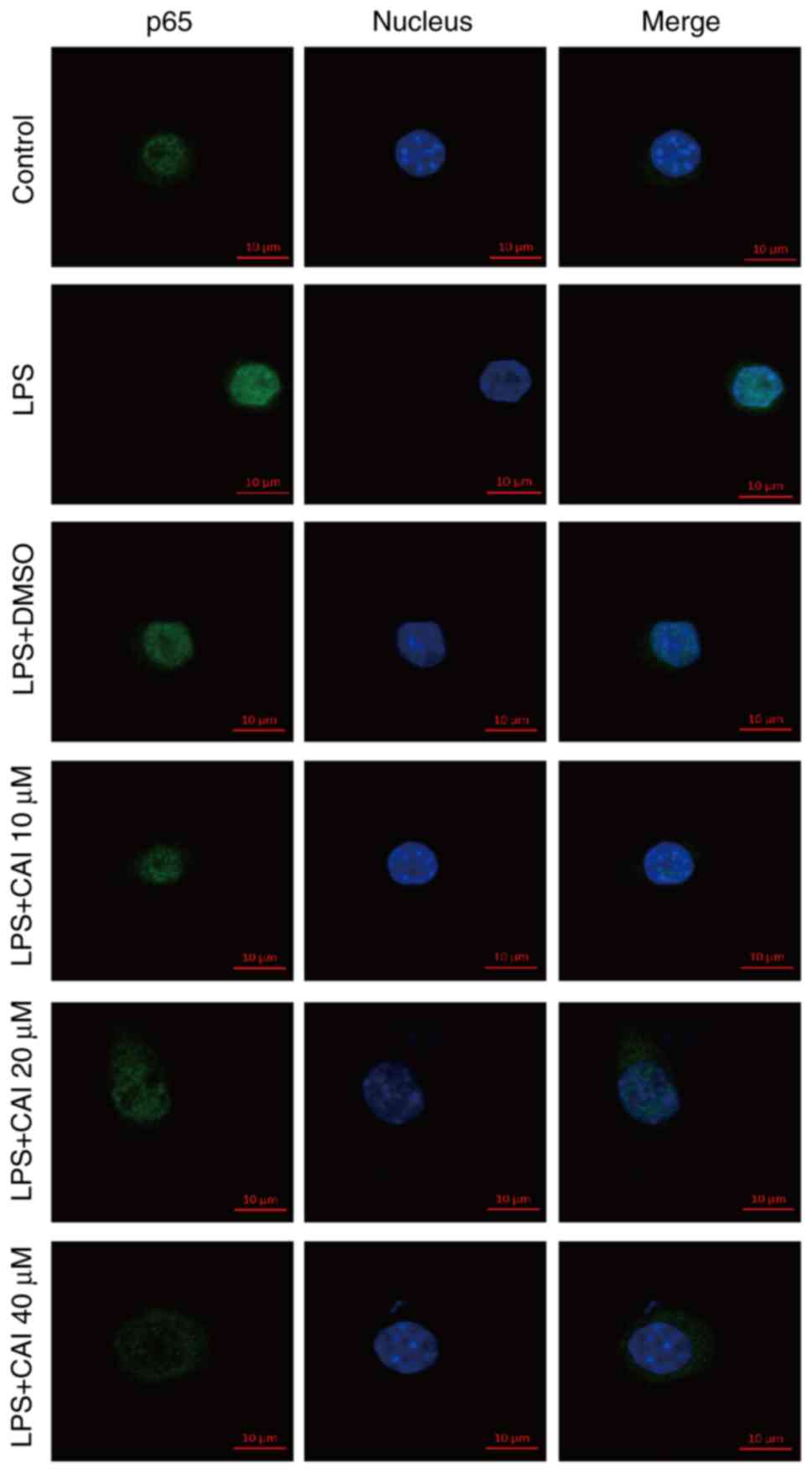
NF-κB nuclear translocation is considered as a
hallmark of activation, therefore it was subsequently visualized
using immunofluorescence of p65. The expression level of p65 was
low in unstimulated cells. Following LPS stimulation, the
overlapping of p65-Alexa Fluor 488 green fluorescence with DAPI
blue staining was increased, suggesting the enhanced nuclear
expression of p65. However, p65 staining was weakened in the
nucleus and mainly located in the cytoplasm by pretreatment with
CAI (10, 20 and 40 µM), which indicated the inhibitory nuclear
translocation of p65 (Figs. 7 and
S1, S2). These findings indicated that CAI
could inhibit NF-κB activation in LPS-stimulated RAW264.7
cells.
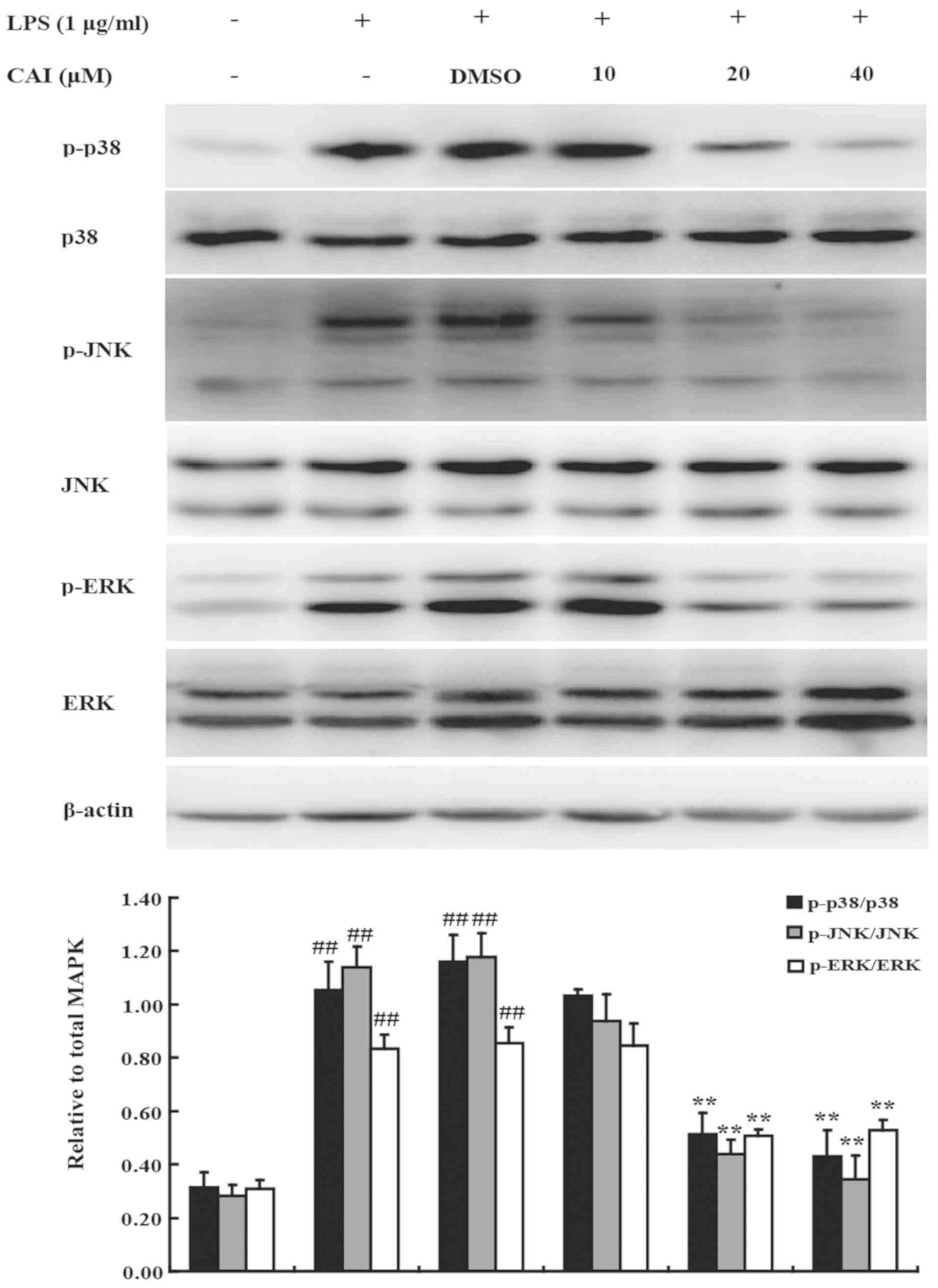
CAI blocks MAPK activation in
LPS-induced RAW264.7 macrophages
MAPKs play critical roles in the induction of
pro-inflammatory gene expression (29). Moreover, the activation of NF-κB is
regulated by p38, JNK and ERK (8,30). To
further characterize the mechanism underlying the effects of CAI,
the MAPK signaling pathway was examined. The phosphorylation of
p38, JNK and ERK was significantly increased in RAW264.7 cells
stimulated with LPS compared with controls; however, CAI treatment
significantly reduced protein levels compared with the LPS + DMSO
groups (Fig. 8). However, the total
protein expression levels of p38, JNK and ERK were not altered
following LPS stimulation or treatment with CAI and LPS. The
aforementioned results suggested that CAI inactivated the MAPK
signaling pathway to exert its inhibition on the inflammatory
responses in macrophages stimulated by LPS.
Discussion
The anti-inflammatory effect of CAI in several
animal inflammatory models were revealed in previous studies;
however, the underlying mechanisms involved has not been fully
determined (18-20).
Since COX-2 is an important enzyme regulating the inflammation
process (31), the effect of CAI on
COX activity was initially determined to investigate whether the
anti-inflammatory mechanism of CAI is similar to NSAIDs. However,
either COX-1 or COX-2 activities were not affected by CAI up to 100
µM, which is much higher compared with the effective concentration
(10-40 µM) in anti-inflammatory application observed in the present
study and in our previous report (32). The results suggested that CAI exerted
its anti-inflammatory action via a different mechanism compared
with COX-inhibiting NSAIDs. Thus, further investigation was
required.
Activated macrophages play pivotal roles in the
initiation and amplification of the inflammatory response and the
immune reaction, where one of the mechanisms is via release of a
variety of pro-inflammatory mediators (2,3). The
regulation of activated macrophages can restrain or control the
inflammatory process and may be developed as a promising
therapeutic target for various inflammatory diseases (33). LPS, a prototypic endotoxin, is a
common inducer of inflammation (4).
Therefore, the model mostly used for investigating inflammation and
evaluating anti-inflammatory candidates with their mechanisms of
action is by stimulating macrophages with LPS (7,8). NO,
which is endogenously catalyzed by one of three NOS isoenzymes from
L-arginine, is one of the most important inflammatory mediators
produced by LPS-stimulated macrophages (34). In contrast to the constitutively
expressed neuronal and endothelial NOS, exposure to microbial
components, such as LPS or cytokines (for example, IL-1β and TNF-α)
causes the induced increase of iNOS in the macrophage, which
produces large amounts of NO consistently (35). The NO produced by iNOS is cytotoxic
to pathogens and causes host cell and tissue injury, either by
activating the NF-κB pathway to release cytokines or directly
causing peroxynitrate formation (36-38).
In the present study, CAI was found to reduce NO production via
suppressing mRNA and protein expression levels of iNOS. In
addition, the inhibition of CAI on LPS-induced increases of
pro-inflammatory cytokines, including TNF-α, IL-1β and IL-6 also
provided supportive evidence for the anti-inflammatory effect of
CAI, since these cytokines are widely accepted to be involved in
inflammatory induction and perpetuation, and their excessive
production leads to multiple tissue damage and organ dysfunction
(39). Moreover, this repression was
regulated by CAI at the transcriptional level by decreasing the
gene expression of the aforementioned cytokines. Our previous study
reported that CAI downregulated the levels of the aforementioned
cytokines in the serum and at the inflammatory sites of
experimental inflammation animal models (18,20).
Taken together, the decrease in the aforementioned inflammatory
mediators confirmed the anti-inflammatory action of CAI in
macrophages, which partially explains the mechanisms underlying the
therapeutic effect of CAI on inflammatory animal models observed in
our previous studies (18-20).
One of the most important cascades for LPS
activation in macrophages is the NF-κB pathway (40,41).
NF-κB is a protein complex consisting of five subunits (RelA/p65,
RelB, c-Rel, p50 and p52) that form hetero- or homodimers,
typically a dimer of p50 and p65(28). In the unstimulated state, NF-κB
dimers are sequestered by the inhibitory IκB protein and remain
inactive in the cytoplasm (28).
When the cell is induced by a variety of different stimuli,
including pathogen-derived molecules (for example, LPS), cytokines
and oxidants, IκB is rapidly phosphorylated and undergoes
polyubiquitination and proteasomal degradation (28). This leads to the release of NF-κB
dimers and allows them to consequently translocate into the nucleus
where they bind κB motifs in the promoter, contributing to the
transcription of a number of genes associated with inflammatory
activation, such as iNOS, chemokines, adhesion molecules and
cytokines (28). The positive
feedback between the inflammatory molecules and NF-κB resulted in
the persistence of inflammation and is involved in the pathogenesis
of several inflammatory diseases including RA and inflammatory
bowel disease (42,43). Consistent with this, suppressing
NF-κB was demonstrated to be beneficial for the control of
inflammation-related diseases (44,45).
Accordingly, the present study discovered that CAI inhibited the
increased LPS-induced DNA-binding activity of NF-κB p65. To explore
the mechanisms by which CAI inhibited NF-κB activity, the effects
of CAI on NF-κB activation pathways was investigated. CAI
suppressed the LPS-induced phosphorylation and degradation of IκBα,
as well as decreasing the phosphorylated levels and the subsequent
nuclear translocation of p65. Similarly, our previous research
revealed that CAI could block IκBα phosphorylation and degradation
in the inflammatory tissues of arthritic or colitis animal models
(18,20). Based on these results, the
anti-inflammatory mechanism of CAI is possibly due to its ability
to break down the integrating feedback loop between the
inflammatory mediators and the NF-κB pathway.
In addition to NF-κB, the MAPK signaling pathway is
another important signaling transduction cascade regulating
LPS-induced inflammatory responses in macrophages (23,24).
MAPKs, which primarily include p38, JNK, and ERK, are a family of
serine/threonine kinases (29).
Under a variety of extracellular signaling molecules, p38, JNK, and
ERK are phosphorylated by consecutive multilevel cascade, which
subsequently causes the phosphorylation and activation of various
intracellular targets, including additional protein kinases,
phospholipases, transcription factors and cytoskeletal proteins
(29). The significance of MAPKs in
the regulation of cell growth, differentiation, stress adaptation
to the environment, inflammatory responses and additional important
cellular physiological or pathological processes has identified
them as a focus for research into numerous human diseases (46,47). In
particular, previous reports revealed that the MAPK signaling
pathway exerted a key role in the induction of inflammatory genes
expression, including cytokines as well as iNOS. Additionally,
MAPKs could increase the expression of these inflammatory genes by
regulating the activation of the NF-κB pathway (8,30).
Therefore, MAPKs are considered as an important target for the
development of novel drugs against inflammation (29,48,49). In
the present study, MAPKs, including p38, JNK and ERK were
significantly activated by LPS in RAW264.7 macrophages via
elevating their phosphorylation levels, which was in line with the
results of previous studies (8,23,24).
However, CAI suppressed the increased phosphorylated levels of p38,
JNK, and ERK stimulated by LPS, but had no effect on their total
levels, suggesting the inhibition of CAI on MAPK activation but not
on their biosynthesis. To the best of our knowledge, this is the
first study that identified the involvement of CAI in the
perturbation of the MAPK signaling pathway resulting in
anti-inflammatory activity.
However, there are certain limitations to the
present study. COX-2-mediated PGE2 is also a key
pro-inflammatory mediator. Further experiments are required to
determine the effects of CAI on PGE2 levels and COX-2
expression. In addition, it is essential to set up a positive
control in the experiments, which is preferably similar to the
experimental drug in terms of its mechanism of action. However, the
present study lacks a positive control since the exact
anti-inflammatory mechanisms of CAI was not clear at the beginning
of the study. With the development of research and understanding of
the mechanisms of CAI, an appropriate positive control is clearly
required in future studies.
In summary, findings from the present study revealed
that CAI has no direct effect on COX activity, which differs from
COX-inhibiting NSAIDs. Further investigation demonstrated that the
inhibitory effects of CAI on cytokine and iNOS-dependent NO
production was via inactivation of NF-κB and MAPK pathways
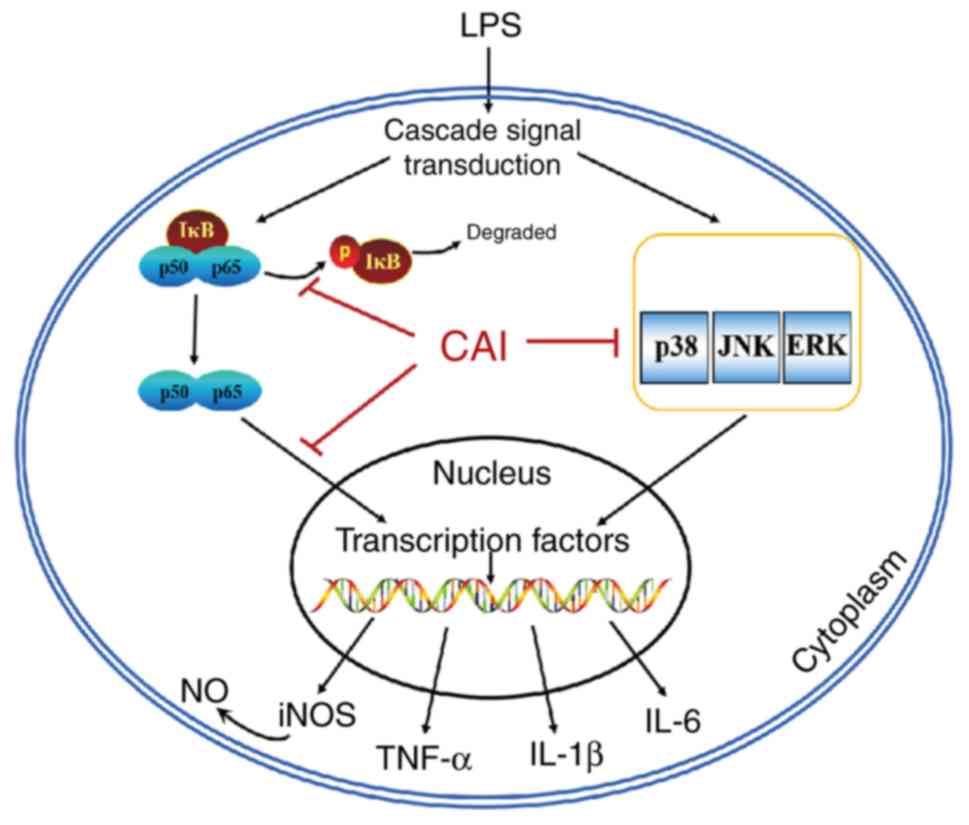
LPS-stimulated macrophages (Fig. 9).
These results therefore identified a novel perspective for
understanding the anti-inflammatory mechanism of CAI and provided a
basis for modeling the therapeutic effects of CAI in animal models.
Further work may warrant the application of CAI in the near future
against inflammatory diseases related to the excessive activation
of macrophages.
Supplementary Material
Fluorescence microscope images of
NF-κB p65 in RAW264.7 macrophages. Cells were pretreated with CAI
(10, 20 and 40 μM) or vehicle (0.1% DMSO) for 2 h prior to
stimulation by 1 μg/ml LPS for 1 h. They were then fixed and
processed as described in section Materials and methods. Samples in
each group were triplicate. Images were obtained using a Leica
DM4000 upright fluorescence microscope and representative images
from different group were shown. CAI, carboxyamidotriazole; NF,
nuclear factor.
Immunofluorescence quantification of
NF-κB p65 in Fig. S1. The pixel
intensity of the immunostained p65 in the nucleus area was
quantified using ImageJ software to assess the effect of CAI on p65
nuclear translocation. The values were normalized to 100% for the
expression of control group. Data are presented as the mean ± SD of
three samples in each group. ##P<0.01 versus control
group; *P<0.05, **P<0.01 versus
LPS+DMSO group. LPS, lipopolysaccharide; CAI, carboxyamidotriazole;
NF, nuclear factor.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81102454); CAMS Innovation
Fund for Medical Sciences (grant no. 2016-I2M-1-002); and Fund of
Medical Epigenetics Research Center, Chinese Academy of Medical
Sciences (grant. nos. 2019PT310017, 2018PT31015 and
2017PT31035).
Availability of data and materials
The datasets used and/or analyzed in this study are
available from the corresponding author on reasonable request.
Authors' contributions
LZ, DZ and CY designed the study. SL, MD, ZG, XZ and
YZ conducted the experiments and performed data entry. YCW, DWW, RJ
and JL performed statistical analysis and data interpretation. SL,
MD, ZG, XZ and LZ wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Krishnamoorthy S and Honn KV: Inflammation
and disease progression. Cancer Metastasis Rev. 25:481–491.
2006.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kawai T and Akira S: TLR signaling. Cell
Death Differ. 13:816–825. 2006.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Siebert S, Tsoukas A, Robertson J and
McInnes I: Cytokines as therapeutic targets in rheumatoid arthritis
and other inflammatory diseases. Pharmacol Rev. 67:280–309.
2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Jang KJ, Choi SH, Yu GJ, Hong SH, Chung
YH, Kim CH, Yoon HM, Kim GY, Kim BW and Choi YH: Anti-inflammatory
potential of total saponins derived from the roots of panax ginseng
in lipopolysaccharide-activated RAW 264.7 macrophages. Exp Ther
Med. 11:1109–1115. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wei X, Song H, Yin L, Rizzo MG, Sidhu R,
Covey DF, Ory DS and Semenkovich CF: Fatty acid synthesis
configures the plasma membrane for inflammation in diabetes.
Nature. 539:294–298. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zhu L, Wei W, Zheng YQ and Jia XY: Effects
and mechanisms of total glucosides of paeony on joint damage in rat
collagen-induced arthritis. Inflamm Res. 54:211–220.
2005.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Diakos CI, Charles KA, McMillan DC and
Clarke SJ: Cancer-related inflammation and treatment effectiveness.
Lancet Oncol. 15:e493–e503. 2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ferrucci L and Fabbri E: Inflammageing:
Chronic inflammation in ageing, cardiovascular disease, and
frailty. Nat Rev Cardiol. 15:505–522. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kohn EC, Sandeen MA and Liotta LA: In vivo
efficacy of a novel inhibitor of selected signal transduction
pathways including calcium, arachidonate, and inositol phosphates.
Cancer Res. 52:3208–3212. 1992.PubMed/NCBI
|
|
10
|
Kohn EC, Felder CC, Jacobs W, Holmes KA,
Day A, Freer R and Liotta LA: Structure-function analysis of signal
and growth inhibition by carboxyamido-triazole, CAI. Cancer Res.
54:935–942. 1994.PubMed/NCBI
|
|
11
|
Wasilenko WJ, Palad AJ, Somers KD,
Blackmore PF, Kohn EC, Rhim JS, Wright GL Jr and Schellhammer PF:
Effects of the calcium influx inhibitor carboxyamido-triazole on
the proliferation and invasiveness of human prostate tumor cell
lines. Int J Cancer. 68:259–264. 1996.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Moody TW, Chiles J, Moody E, Sieczkiewicz
GJ and Kohn EC: CAI inhibits the growth of small cell lung cancer
cells. Lung Cancer. 39:279–288. 2003.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hussain MM, Kotz H, Minasian L, Premkumar
A, Sarosy G, Reed E, Zhai S, Steinberg SM, Raggio M, Oliver VK, et
al: Phase II trial of carboxyamidotriazole in patients with
relapsed epithelial ovarian cancer. J Clin Oncol. 21:4356–4363.
2003.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Dutcher JP, Leon L, Manola J, Friedland
DM, Roth B and Wilding G: Eastern Cooperative Oncology Group. Phase
II study of carboxyamidotriazole in patients with advanced renal
cell carcinoma refractory to immunotherapy: E4896, an Eastern
cooperative oncology group study. Cancer. 104:2392–2399.
2005.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Mikkelsen T, Lush R, Grossman SA, Carson
KA, Fisher JD, Alavi JB and Rosenfeld S: Phase II clinical and
pharmacologic study of radiation therapy and carboxyamido-triazole
(CAI) in adults with newly diagnosed glioblastoma multiforme.
Invest New Drugs. 25:259–263. 2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ju R, Wu D, Guo L, Li J, Ye C and Zhang D:
Inhibition of pro-inflammatory cytokines in tumour associated
macrophages is a potential anti-cancer mechanism of
carboxyamidotriazole. Eur J Cancer. 48:1085–1095. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Guo L, Ye C, Chen W, Ye H, Zheng R, Li J,
Yang H, Yu X and Zhang D: Anti-inflammatory and analgesic potency
of carboxyamidotriazole, a tumorostatic agent. J Pharmacol Exp
Ther. 325:10–16. 2008.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zhu L, Li J, Guo L, Yu X, Wu D, Luo L, Zhu
L, Chen W, Chen C, Ye C and Zhang D: Activation of NALP1
inflammasomes in rats with adjuvant arthritis; a novel therapeutic
target of carboxyamidotriazole in a model of rheumatoid arthritis.
Br J Pharmacol. 172:3446–3459. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhu L, Li J, Guo L, Yu XL, Wu DW, Chen W,
Chen C, Du XW, Zhang DC and Ye CY: Therapeutic effect of
carboxyamidotriazole on adjuvant arthritis in rats. Zhongguo Yi Xue
Ke Xue Yuan Xue Bao. 38:49–54. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Du X, Chen W, Wang Y, Chen C, Guo L, Ju R,
Li J, Zhang D, Zhu L and Ye C: Therapeutic efficacy of
carboxyamidotriazole on 2,4,6-trinitrobenzene sulfonic acid-induced
colitis model is associated with the inhibition of NLRP3
inflammasome and NF-κB activation. Int Immunopharmacol. 45:16–25.
2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Jeon HL, Yoo JM, Lee BD, Lee SJ, Sohn EJ
and Kim MR: Anti-inflammatory and antioxidant actions of
N-arachidonoyl serotonin in RAW264.7 cells. Pharmacology.
97:195–206. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Guo T, Lin Q, Li X, Nie Y, Wang L, Shi L,
Xu W, Hu T, Guo T and Luo F: Octacosanol attenuates inflammation in
both RAW264.7 macrophages and a mouse model of colitis. J Agric
Food Chem. 65:3647–3658. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Fengyang L, Yunhe F, Bo L, Zhicheng L,
Depeng L, Dejie L, Wen Z, Yongguo C, Naisheng Z, Xichen Z and
Zhengtao Y: Stevioside suppressed inflammatory cytokine secretion
by downregulation of NF-κB and MAPK signaling pathways in
LPS-stimulated RAW264.7 cells. Inflammation. 35:1669–1675.
2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Choi WS, Shin PG, Lee JH and Kim GD: The
regulatory effect of veratric acid on NO production in
LPS-stimulated RAW264.7 macrophage cells. Cell Immunol.
280:164–170. 2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Choi WS, Seo YB, Shin PG, Kim WY, Lee SY,
Choi YJ and Kim GD: Veratric acid inhibits iNOS expression through
the regulation of PI3K activation and histone acetylation in
LPS-stimulated RAW264.7 cells. Int J Mol Med. 35:202–210.
2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Bacchi S, Palumbo P, Sponta A and
Coppolino MF: Clinical pharmacology of non-steroidal
anti-inflammatory drugs: A review. Antiinflamm Antiallergy Agents
Med Chem. 11:52–64. 2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Viatour P, Merville MP, Bours V and
Chariot A: Phosphorylation of NF-kappaB and IkappaB proteins:
Implications in cancer and inflammation. Trends Biochem Sci.
30:43–52. 2005.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Seger R and Krebs EG: The MAPK signaling
cascade. FASEB J. 9:726–735. 1995.PubMed/NCBI
|
|
30
|
Nemoto S, DiDonato JA and Lin A:
Coordinate regulation of IkappaB kinases by mitogen-activated
protein kinase kinase kinase 1 and NF-kappaB-inducing kinase. Mol
Cell Biol. 18:7336–7343. 1998.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Seibert K, Zhang Y, Leahy K, Hauser S,
Masferrer J, Perkins W, Lee L and Isakson P: Pharmacological and
biochemical demonstration of the role of cyclooxygenase 2 in
inflammation and pain. Proc Natl Acad Sci USA. 91:12013–12017.
1994.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zhu L, Duan MY, Zhang XJ, Li J, JU R, Guo
L, Lu S and Ye CY: Inhibitory effects of carboxyamidotriazole on
cytokines production by peritoneal macrophages from adjuvant
arthritis rats. Basic Clin Med. 38:798–802. 2018.
|
|
33
|
Zhang X and Mosser DM: Macrophage
activation by endogenous danger signals. J Pathol. 214:161–178.
2008.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Rim HK, Cho W, Sung SH and Lee KT:
Nodakenin suppresses lipopolysaccharide-induced inflammatory
responses in macrophage cells by inhibiting tumor necrosis factor
receptor-associated factor 6 and nuclear factor-κB pathways and
protects mice from lethal endotoxin shock. J Pharmacol Exp Ther.
342:654–664. 2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Bogdan C: Nitric oxide and the immune
response. Nat Immunol. 2:907–916. 2001.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Davis KL, Martin E, Turko IV and Murad F:
Novel effects of nitric oxide. Annu Rev Pharmacol Toxicol.
41:203–236. 2001.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Aktan F: iNOS-mediated nitric oxide
production and its regulation. Life Sci. 75:639–653.
2004.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Farlik M, Reutterer B, Schindler C, Greten
F, Vogl C, Müller M and Decker T: Nonconventional initiation
complex assembly by STAT and NF-kappaB transcription factors
regulates nitric oxide synthase expression. Immunity. 33:25–34.
2010.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kalliolias GD and Ivashkiv LB: TNF
biology, pathogenic mechanisms and emerging therapeutic strategies.
Nat Rev Rheumatol. 12:49–62. 2016.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Ulevitch RJ and Tobias PS: Recognition of
gram-negative bacteria and endotoxin by the innate immune system.
Curr Opin Immunol. 11:19–22. 1999.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Aderem A and Ulevitch RJ: Toll-like
receptors in the induction of the innate immune response. Nature.
406:782–787. 2000.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Bonizzi G and Karin M: The two NF-kappaB
activation pathways and their role in innate and adaptive immunity.
Trends Immunol. 25:280–288. 2004.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Oeckinghaus A, Hayden MS and Ghosh S:
Crosstalk in NF-κB signaling pathways. Nat Immunol. 12:695–708.
2011.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Li Q and Verma IM: NF-kappaB regulation in
the immune system. Nat Rev Immunol. 2:725–734. 2002.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Shibata W, Maeda S, Hikiba Y, Yanai A,
Ohmae T, Sakamoto K, Nakagawa H, Ogura K and Omata M: Cutting edge:
The IkappaB kinase (IKK) inhibitor, NEMO-binding domain peptide,
blocks inflammatory injury in murine colitis. J Immunol.
179:2681–2685. 2007.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Strnisková M, Barancík M and Ravingerová
T: Mitogen-activated protein kinases and their role in regulation
of cellular processes. Gen Physiol Biophys. 21:231–255.
2002.PubMed/NCBI
|
|
48
|
Kristof AS, Marks-Konczalik J and Moss J:
Mitogen-activated protein kinases mediate activator
protein-1-dependent human inducible nitric-oxide synthase promoter
activation. J Biol Chem. 276:8445–8452. 2001.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Tanoue T and Nishida E: Docking
interactions in the mitogen-activated protein kinase cascades.
Pharmacol Ther. 93:193–202. 2002.PubMed/NCBI View Article : Google Scholar
|