Introduction
Status epilepticus (SE) is one of the most common
types of neurological disorders, and can cause patient disability
or mortality if not diagnosed and treated early (1). SE can have long-term consequences
following 30 min (tonic-clonic SE) or 60 min (focal SE with
impaired consciousness), including neuronal death, neuronal injury
and alteration of neuronal networks (2). However, the underlying mechanism of SE
is yet to be elucidated.
The mTOR signaling pathway is an important signaling
mechanism in epileptogenic processes (3). AKT substrate of 40 kDa (PRAS40) is a
novel downstream factor of the PI3K/AKT signaling pathway and was
first identified by Kovacina et al (4) in 2003. When the PI3K/AKT signaling
pathway is activated, PRAS40 can be phosphorylated (p-) by p-AKT at
Thr246 site and promote the signal downstream to the mTOR signaling
pathway (5). Moreover, PRAS40 is a
subunit of the mTOR complex 1 (mTORC1). PRAS40 inhibits mTORC1
autophosphorylation (6) and prevents
mTORC1 binding to downstream ribosomal protein S6 kinase 1 (P70S6K)
and eukaryotic initiation factor 4E binding protein 1(7), in order to inhibit the activation of
the mTOR signaling pathway. In addition, p-PRAS40 binds to the
14-3-3 scaffold protein and separates from mTORC1, and does not
inhibit the mTOR signaling pathway (8). PRAS40 also participates in the mTOR
signaling pathway by regulating neural development, circuit
formation and synaptic plasticity (9).
Abnormal activation of the mTOR pathway in SE has
been reported by previous studies. For instance, San et al
(10) revealed the elevation of the
p-mTOR/mTOR and p-P70S6K/P70S6K ratios in the posttraumatic
amnesia-induced SE rat model. Furthermore, Brewster et al
(11) showed that mTORC1
hyperactivation was associated with SE-induced memory deficits and
dendritic dysregulation, which could be partially reversed by
rapamycin, an inhibitor of the mTOR pathway. In a study by Wang
et al (12), it was
demonstrated that hyperactivation of the mTOR pathway could lead to
increased levels of NF-κB, as well as the promotion of inflammation
in the brain early after SE. However, to the best of our knowledge,
there have been few reports focused on the association between
PRAS40 and SE.
Autophagy is one of the most important methods to
eliminate and recycle intracellular materials in eukaryotic cells
(13). Under conditions of nutrient
sufficiency, the mTOR pathway is activated when mTORC1 inhibits
autophagy by phosphorylating the Unc-51 like autophagy activating
kinase 1 complex, which is a promoter of autophagy (14). However, under nutrient starvation,
opposite events lead to the elevation of autophagy levels and
cytoplasmic contents are eliminated to generate energy for
essential cellular activities (10).
There has been controversy regarding autophagy-associated
alterations following SE. For example, some previous studies
reported an abnormal elevation in autophagy levels following SE
(15,16), but other researchers have
hypothesized that autophagy-associated activities induce dynamic
changes during this cellular event (17-19).
As mTOR is one of major pathways that regulates the onset of
autophagy, it was hypothesized that PRAS40 may participate in this
regulation by influencing the activity of the mTOR signaling
pathway.
The aim of the present study was to investigate the
role of PRAS40 in SE and the associated mechanism, as well as how
PRAS40 participates in the mTOR associated regulation of autophagy
following SE.
Materials and methods
Antibody and reagents
Primary antibodies were purchased as follows:
P-PRAS40 (Thr246; cat. no. 2997; Cell Signaling Technology, Inc.),
PRAS40 (cat. no. 2691; Cell Signaling Technology, Inc.), p-mTOR
(Ser2448; cat. no. 5536; Cell Signaling Technology, Inc.), mTOR
(cat. no. 2983; Cell Signaling Technology, Inc.), p-AKT (Ser473;
cat. no. 4060; Cell Signaling Technology, Inc.), AKT (cat. no.
4685; Cell Signaling Technology, Inc.), p-P70S6K (Thr389; cat. no.
9234; Cell Signaling Technology, Inc.), P70S6K (cat. no. 14130;
Cell Signaling Technology, Inc.), Light chain 3-I/II (LC3-I/II;
cat. no. 12741; Cell Signaling Technology, Inc.), 14-3-3 (cat. no.
8312; Cell Signaling Technology, Inc.), P62 (cat. no. 18420-1-AP;
ProteinTech Group, Inc.) and GAPDH (cat. no. sc-365062; 1:2,000;
Santa Cruz Biotechnology, Inc.). Goat anti-rabbit horseradish
peroxidase (HRP)-conjugated secondary antibody (cat. no. sc-2054;
Santa Cruz Biotechnology, Inc.). The following drugs were obtained
from commercial sources as follows: Pilocarpine (cat. no. S4231;
Selleck Chemicals), lithium chloride (cat. no. L9650;
Sigma-Aldrich; Merck KGaA), Scopolamine (cat. no. S2508; Selleck
Chemicals) and LY3023414 (cat. no. S8322; Selleck Chemicals).
Animals and drug treatment
50 adult male Sprague Dawley rats which provided by
Shanghai SLAC Laboratory Animal Co. Ltd. (weight, 200-250 g; age, 5
weeks old) were used in the present study and the protocols used
were approved by the Experimental Animal Ethics Committee of the
Basic Medical College of Fudan University (approval no.
20170223-066). Rats were housed in standard cages with free access
to food and water on a 12-h light/dark cycle in a
temperature-controlled room (26˚C, the humidity was 40-60%) and
were allowed a 3 days period to acclimatize prior to treatment with
drugs. Lithium chloride [127 mg/kg; intraperitoneal (i.p.)] was
administered 18 h prior to pilocarpine administration. Pilocarpine
(30 mg/kg; i.p.) was administered 1 h following pre-treatment with
scopolamine (1 mg/kg; i.p.; to prevent the effects of peripheral
cholinergic stimulation) to induce seizures. The
electroencephalograms (EEG) was acquired and analyzed with a
RM6240C multichannel physiological signal acquisition and
processing system (Chengdu Instrument Factory). EEG was used to
confirm epileptic seizures. Seizures were graded using the Racine
scale (20). Seizure latency and
duration were also recorded. Rats that survived following 2 h of
continued grade 4 or greater seizures (SE model establishment) were
included in the present study. Pentobarbital sodium (40 mg/kg;
i.p.) was administered to stop seizures. Following SE, rats were
kept warm and fed with glucose saline through a gastric tube. Rats
in the normal control group (n=8) received sodium chloride (0.9%;
i.p.) instead of pilocarpine, and rats in the SE + inhibitor (SE +
inh) group (n=6) were pretreated with the mTOR inhibitor LY3023414
(10 mg/kg; i.p.) before seizures. Rats were sacrificed with
pentobarbital sodium (400 mg/kg; i.p.) at 3 h (SE-3 h group, n=9),
6 h (SE-6 h group, n=9), 1 day (SE-1d group, n=9) and 3 days (SE-3d
group, n=9) following SE.
Histology and immunohistochemistry
(IHC)
After rats were sacrificed with lethal pentobarbital
sodium, the brains were perfused with 0.9% sodium chloride via
intramyocardial injection, in order to excrete blood from the brain
of rats. Then, the brains were perfused with 4% paraformaldehyde at
room temperature for ~5 min, after which the brains were harvested
and post-fixed in 4% paraformaldehyde overnight at 4˚C. Brains were
embedded in paraffin and sectioned in the coronal plane in 4-µm
thick slides. The sections containing the hippocampus were selected
for IHC. Sections were heated to 98˚C for 10 min using a microwave
in citrate buffer (pH, 6.0) and treated with 3%
H2O2 for 15 min at room temperature to
abolish endogenous peroxidase activity. After blocking with 3%
BSA-PBS (cat. no. ST023; Beyotime Institute of Biotechnology) for
30 min in 37˚C, sections were incubated with the primary antibodies
(p-PRAS40; dilution 1:100) at 4˚C overnight and then with
corresponding HRP-conjugated secondary antibodies for 30 min at
37˚C. The sections were incubated with 3,3-diaminobenzidine
solution for 2 min at room temperature for visualization and were
subsequently dehydrated and mounted using neutral balsam. The
sections were observed using a light microscope (magnification
400x, Zeiss observer A1; Zeiss AG).
Western blot analysis
After sacrifice, the rat hippocampus was isolated
and total proteins were extracted using RIPA buffer (cat. no.
P0013; Beyotime Institute of Biotechnology) supplemented with
serine protease inhibitor, PMSF. The protein was measured using a
BCA Protein Assay kit (cat. no. P0010; Beyotime Institute of
Biotechnology). A total of 30 µg protein samples were resolved
using 10% SDS-PAGE, then transferred to PVDF membranes. Then, the
membranes were blocked with 5% non-fat dry milk in Tris-buffered
saline/0.1% Tween-20 for 1 h at 37˚C, followed by incubation with
primary antibodies (dilution 1:1,000) at 4˚C overnight. After 1 h
incubation with the corresponding HRP-conjugated secondary
antibodies (dilution 1:1,000) at room temperature, signals were
detected using ECL reagents (cat. no. 34096; Thermo Fisher
Scientific, Inc.). Signal intensities were quantified using ImageJ
v1.28 program (National Institutes of Health) and normalized to
GAPDH expression.
Co-immunoprecipitation (Co-IP)
Total proteins from the rat hippocampi were
extracted for Co-IP using the Pierce Co-IP kit (cat. no. 26149;
Thermo Fisher Scientific, Inc.) according to manufacturer's
protocol. Briefly, 50% AminoLink resin was added into a Pierce Spin
Column at room temperature. Primary antibodies (p-PRAS40; dilution
1:10) were added into the resin in the spin column and incubated on
a rotator at room temperature for 120 min. Subsequently, the total
protein lysates, collected using IP Lysis Buffer, following the
protocol of this kit, of experimental group and the controls
(including the negative control and the blank control) were added
to the resin and incubated with gentle rocking overnight at 4˚C.
The negative control used IgG instead of the p-PRAS40 antibody.
Positive control was used as the whole control protein to
immunoblot directly. The eluent was subjected to 12% SDS-PAGE
followed by western blot analysis using the 14-3-3 antibody
(dilution 1:500).
Statistical analysis
Continuous variables were presented as the mean ±
SD. Each experiment was repeated at least three times. Data were
analyzed using SPSS 17.0 (SPSS, Inc.) and bar graphs were created
using GraphPad Prism (version 5.0; GraphPad Software, Inc.). If the
data distribution was normal, comparisons among ≥3 groups were
calculated with a one-way ANOVA test followed by Tukey's test. If
the data distribution was not normal, comparisons were calculated
using Kruskal-Wallis followed by Dunn's test for non-parametric
analysis. P<0.05 was considered to indicate a statistically
significant difference.
Results
p-PRAS40 levels are elevated following
SE
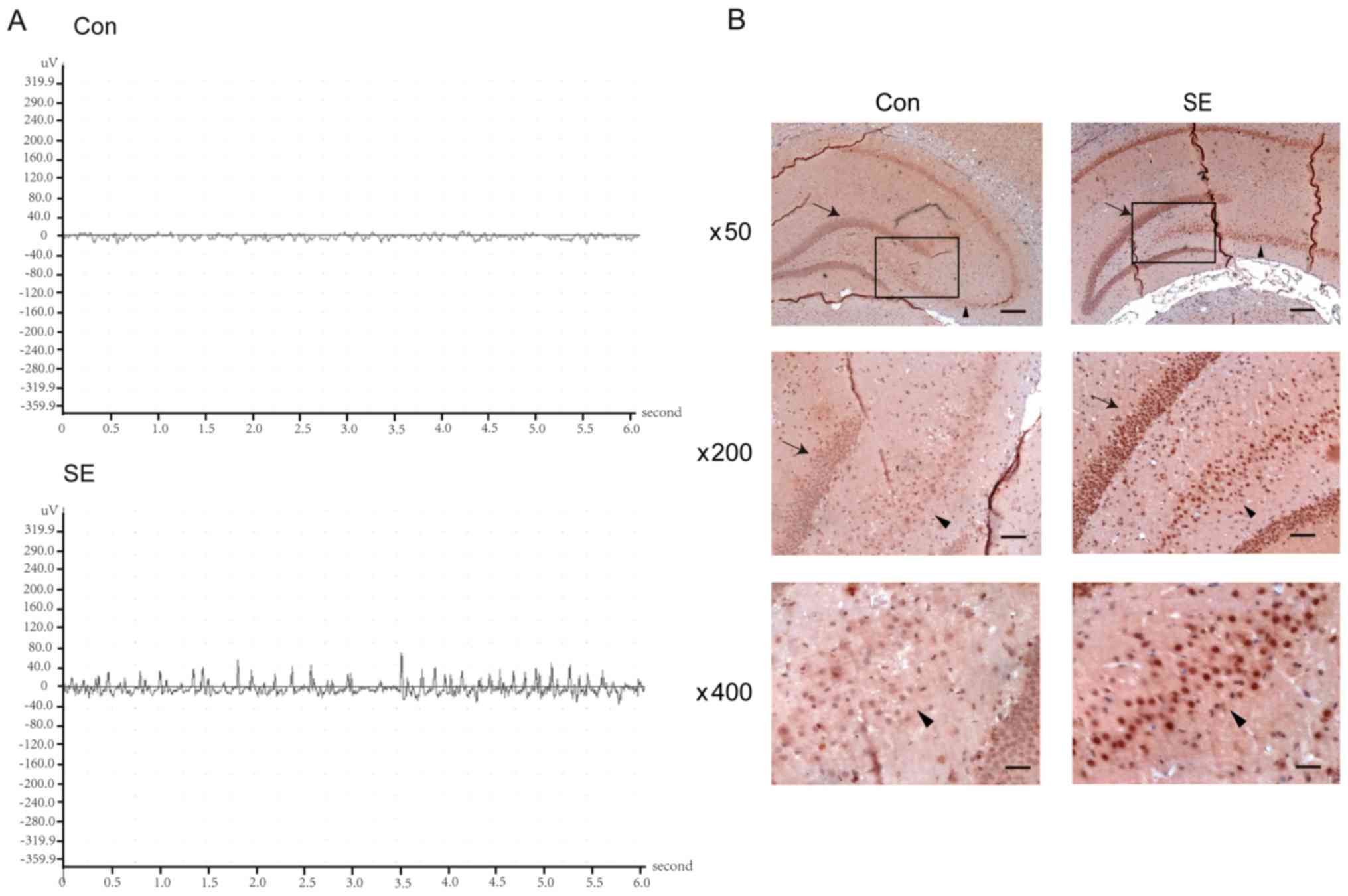
First, the present study established a
pilocarpine-induced rat model to simulate SE. The EEG identified
that the SE group produced high amplitudes (between 100-200 µV)
with superimposed, mildly fast and poly-spike waves in the θ range.
However, the control group was recorded with mildly synchronous and
well-organized waves that were in the normal amplitude range
(between 5-50 µV). In addition, the control showed no focal sharp
or spike wave activities (Fig.
1A).
To investigate whether the expression of p-PRAS40
was elevated in the rat hippocampus following SE, IHC was used to
detect the expression of p-PRAS40. Compared with the control group,
p-PRAS40 positive cells in the dentate gyrus and Cornu Ammonis
(CA)3 regions of the hippocampus were markedly increased in the
SE-3d group (Fig. 1B).
The western blot analysis results suggested that
p-PRAS40 expression was significantly upregulated at the indicated
times following seizures, while the expression of PRAS40 was
slightly increased. Quantification of the ratio of p-PRAS40 and
PRAS40 demonstrated a significant increase in a time-dependent
manner (Fig. 2A and B). These results indicated that p-PRAS40
was significantly elevated over time following acute SE.
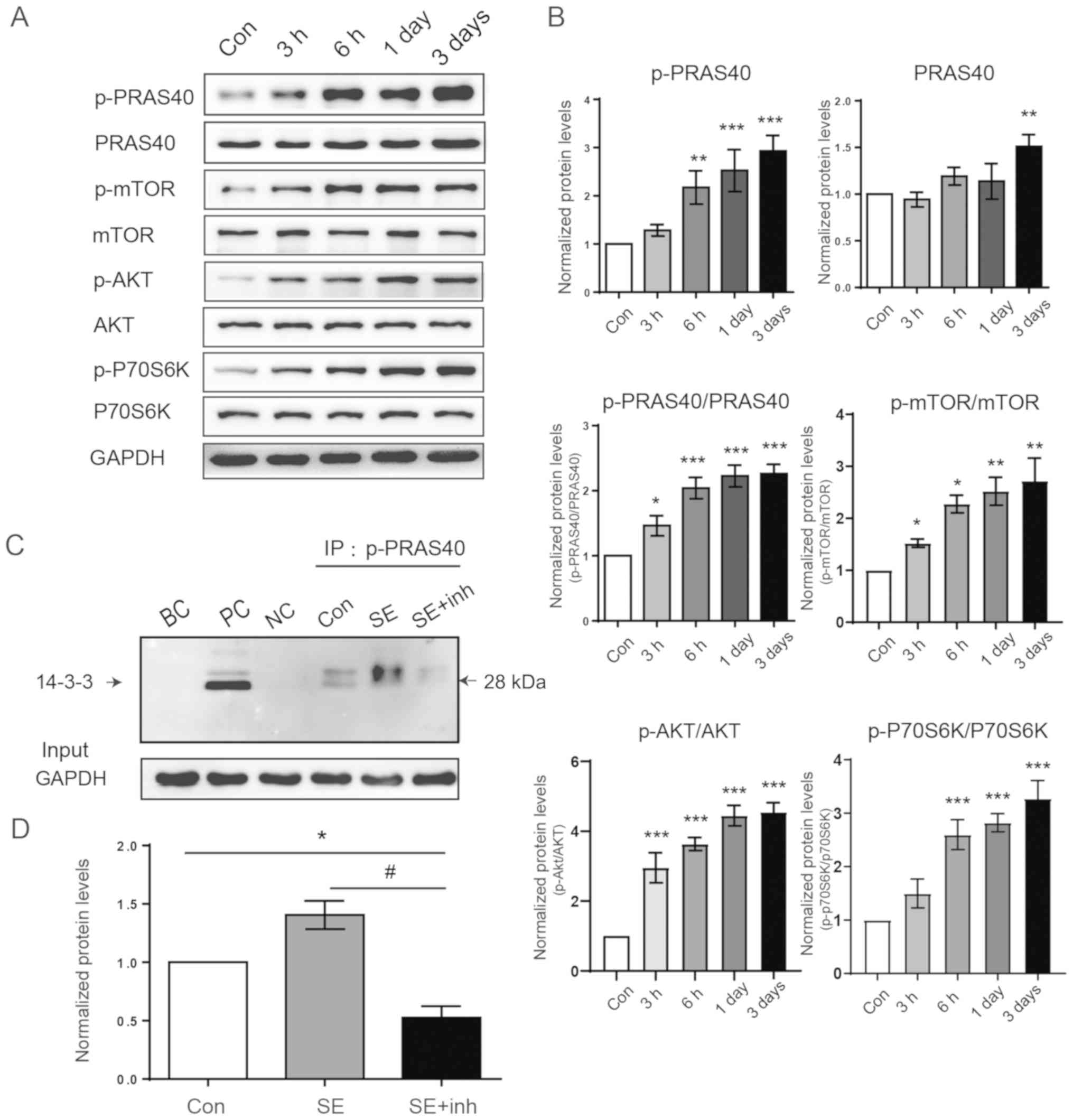 | Figure 2p-PRAS40 combines with 14-3-3 protein
to activate the mTOR signaling pathway. (A) Western blot analysis
of p-PRAS40, PRAS40, p-mTOR, mTOR, p-AKT, AKT, p-P70S6K and P70S6K
expression levels in the rat hippocampus following SE at the
indicated times. (B) Representative proteins of the PI3K/AKT/mTOR
signaling pathway were increased with the upregulation of p-PRAS40
(n=4 per group). (C) Proteins from the control, SE and SE + inh
groups were immunoprecipitated with p-PRAS40 and immunoblotted with
the 14-3-3 antibody. The NC used IgG instead of the p-PRAS40
antibody. PC was used as the whole control protein to immunoblot
directly. (D) Co-IP analysis demonstrated that the combination of
p-PRAS40 with 14-3-3 protein was enhanced following SE and the
inhibition of p-AKT significantly suppressed the combination of
these two proteins. Data were analyzed using ANOVA (n=3).
*P<0.05, **P<0.01 and
***P<0.001 vs. control; #P<0.05 vs. SE
group. NC, negative control; PC, positive control, which is the
whole lysate; BC, Blank control; Co-IP, Co-immunoprecipitation; SE,
status epilepticus; p-, phosphorylated; PRAS40, proline-rich AKT
substrate of 40 kDa; Con, control; inh, inhibitor; P70S6K,
ribosomal protein S6 kinase 1. |
p-PRAS40 activates the PI3K/AKT/mTOR
pathway via combination with the 14-3-3 scaffold protein
Since PRAS40 is downstream of p-AKT, and the
phosphorylation of PRAS40 can lead to the activation of the mTOR
pathway (21), PI3K/AKT/mTOR pathway
activation was detected in the present study. The protein
expression levels of p-mTOR, p-AKT, p-P70S6K and p-PRAS40 in all of
the experimental groups were significantly higher compared with the
control group, and were increased in a time-dependent manner
(Fig. 2A and B).
The combination of p-PRAS40 and 14-3-3 scaffold
protein leads to the activation of the mTOR pathway (21), and thus Co-IP was used to detect this
combination. It was found that p-PRAS40/14-3-3 was markedly
increased 3 days following SE compared with the control group
(Fig. 2C and D). Moreover, LY3023414, the inhibitor of
AKT phosphorylation, significantly reduced the binding of p-PRAS40
and 14-3-3 scaffold protein compares with the SE group (Fig. 2C and D).
Increased expression of p-PRAS40 is
associated with reduced levels of autophagy flux
The mTOR pathway inhibits the initiation of
autophagy and the phosphorylation of PRAS40 regulates mTOR activity
(5,14); therefore, the current study detected
alterations in p-PRAS40 expression, mTOR activity and autophagy
flux following SE. The increase in the ratio of LC3-II to LC3-I and
the decrease in P62 expression levels indicated that the levels of
autophagy were significantly elevated 3 h following SE (Fig. 3A and B). However, with the increase of p-PRAS40
and the activation of the mTOR pathway, the autophagy levels
decreased in a time-dependent manner.
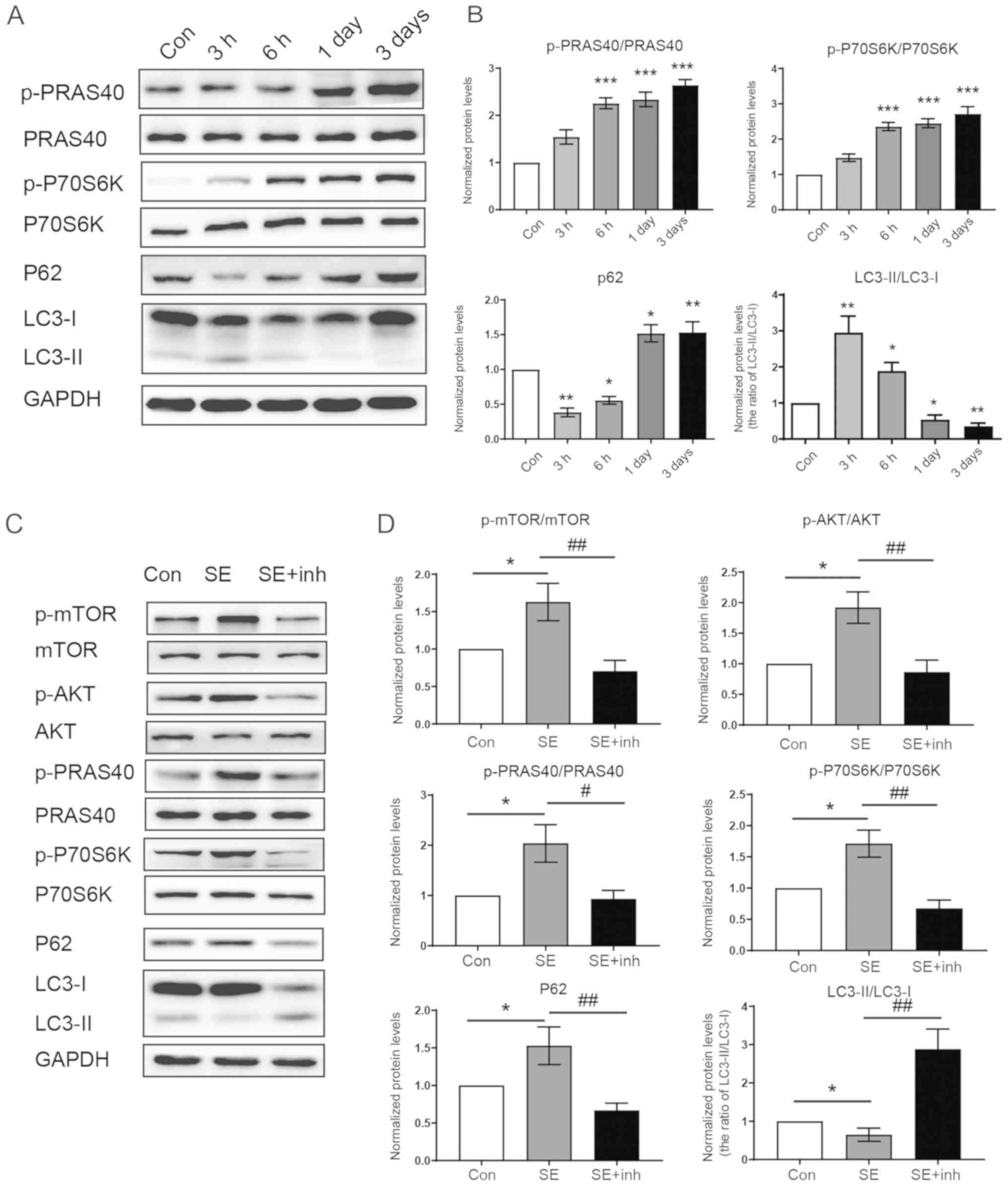 | Figure 3Increased p-PRAS40 expression
activates the mTOR signaling pathway to suppress autophagy. (A)
Protein expression levels of p-PRAS40, PRAS40, p-P70S6K, P70S6K,
P62 and LC3 was detected by western blotting. (B) p-PRAS40 and
p-P70S6K expression levels were increased following SE at the
indicated times. However, the autophagy markers LC3 and P62 were
decreased over time (n=4 per group). (C) Western blot analysis of
the PI3K/AKT/mTOR signaling pathway expression and autophagy levels
in the SE group and the SE + inh group, when the p-AKT pathway was
inhibited using LY3023414. (D) After p-AKT was inhibited by
LY3023414 in the SE + inh group, the autophagy level was increased
with the decreased levels of p-PRAS40 and AKT/mTOR
pathway-associated proteins. Data were analyzed using ANOVA (n=4).
*P<0.05, **P<0.01 and
***P<0.001 vs. control; #P<0.05 and
##P<0.01 vs. SE group. SE, status epilepticus; p-,
phosphorylated; PRAS40, proline-rich AKT substrate of 40 kDa; Con,
control; inh, inhibitor; P70S6K, ribosomal protein S6 kinase 1;
LC3, light chain 3. |
To confirm the p-PRAS40 could regulate the
autophagy, LY3023414 was used to inhibit the PI3K/AKT signaling
pathway and p-PRAS40 in the SE + inh group. As shown in Fig. 3C, after the inhibition of p-PRAS40,
the expression levels of components of the mTOR pathway were
decreased compared the SE groups, including p-mTOR and p-P70S6K.
The rate of conversion of LC3-II to LC3-I was increased, the
expression levels of P62 were decreased, which indicated that the
levels of autophagy in the SE + inh group were significantly higher
compared to the SE groups These results suggested that p-PRAS40 may
regulate the autophagy flux.
Discussion
The mTOR pathway is important in the epileptogenic
mechanism. Moreover, PRAS40 is a subunit of mTORC1 and p-PRAS40 can
regulate mTOR activity (21).
However, to the best of our knowledge, there are few reports
examining the relationship between PRAS40 and SE. The present
results suggested that p-PRAS40 expression was abnormally elevated
following SE. In addition, the IHC staining identified that
p-PRAS40 positive cells increased significantly in the rat
hippocampal dentate gyrus and CA3 region 3 days following SE.
Western blotting results also demonstrated this effect and
indicated an increasing trend over time, which suggested that
p-PRAS40 was increased continuously after SE.
The dentate gyrus is composed of granular cells,
whose axons are covered in mossy fibers that project into the CA3
region (22). Pyramidal cells in the
CA3 region are reported to be more active during epileptic activity
than in other brain areas (22).
Furthermore, mossy-fiber sprouting (MFS) as a result of damage to
pyramidal cells in the CA3 region, the rupture of mossy fibers and
abnormal synaptic connections between lateral growing axons and
granular cells in the dentate gyrus, is the pathological basis of
chronic temporal epilepsy (23). The
present results indicated that p-PRAS40 may participate in the
formation of chronic epileptogenic focus; however, this requires
further investigation. IHC staining demonstrated that the vast
majority of p-PRAS40 existed in neuronal nuclei, which was
consistent with previous reports that p-PRAS40 (Thr246) is mainly
located in nucleus (24-26).
PRAS40 is both the substrate of the PI3K/AKT pathway
and the subunit of mTORC1(5).
Therefore, it was hypothesized that PRAS40 may be the key link
between the PI3K/AKT and mTOR pathways following SE. The western
blotting results suggested that p-PRAS40 expression was elevated
following SE, and the expression of upstream PI3K and p-AKT, as
well as the p- of downstream P70S6K were increased. P70S6K is a
substrate of mTORC1 and p-P70S6K is considered to be a reliable
marker of mTOR activation (27).
Thus, it could be speculated that following SE, activation of the
PI3K/AKT pathway promoted p-PRAS40, which in turn increased the
activity of the mTOR pathway. In addition, the current findings
demonstrated that the increasing expression levels of PI3K, p-AKT
and p-P70S6K were similar to that of p-PRAS40 at different time
points following SE, which could also indicate that PRAS40 was
associated with the PI3K/AKT and mTOR pathways following SE.
p-PRAS40 binds to the 14-3-3 scaffold protein and
then separates from mTORC1(6). In
the present study, Co-IP was used to detect the status of this
combination. It was found that the levels of p-PRAS40 combined with
14-3-3 were higher in the SE group compared with the control group,
but the difference was not statistically significant. Nevertheless,
this combination could be significantly inhibited by LY3023414, an
inhibitor of PI3K, which may partially attenuate the prior defects.
Both PI3K/AKT/PRAS40/mTOR pathway activation and increasing the
expression of p-PRAS40 combined with 14-3-3 were detected following
SE, and therefore it was speculated that the interaction of
p-PRAS40 with 14-3-3 may contribute to the dissociation of p-PRAS40
with mTORC1 and the activation of the mTOR pathway.
Autophagy is a double-edged sword. Under normal
physiological conditions, autophagy can help to promote cell
survival and maintain homeostasis by eliminating damaged cell
organelles and toxic metabolites (28). However, excessive autophagy can
accelerate cell death, which is associated with disease development
(18). Previous studies have
reported insufficient autophagy in chronic neurological diseases,
such as Parkinson's disease (29),
Alzheimer's disease (30) and
Lafora's disease (31). Moreover,
autophagy contributes to the elimination of abnormal protein
aggregates in cells and may provide protection against disease
(13), and there has been a similar
report in chronic epilepsy. Hosseinzadeh et al (32) revealed that cannabidiol could enhance
the induction of the autophagy pathway as a protective mechanism in
temporal epilepsy. Furthermore, Ni et al (33) reported that insufficient autophagy
existed in neonatal rats with chronic epilepsy, which exhibited
consistent trends with rat hippocampal MFS and cognitive deficits.
However, an abnormal increase in autophagy was demonstrated in
acute neurological disorders such as encephalitis (34) and cerebral infarction (35), which may be associated with acute
neurocyte death. Controversy remains regarding how autophagy
changes after SE. For instance, previous studies have suggested
that the autophagy level increase following SE (15,16), but
others have indicated that autophagy after SE has a dynamic course
(17-19).
Since mTOR is one of major pathways regulating autophagy that can
inhibit autophagy initiation, it was hypothesized that PRAS40 may
participate in the regulation of autophagy by influencing mTOR
activity.
LC3 is an important protein involved in
autophagosome formation (36).
Furthermore, LC3 participates in the elongation of bilayer
bio-membranes, cytoplasm and defective protein, as well as
autophagosome formation (37). In
the cytoplasm, LC3 exists as LC3-I. When autophagosomes begin to
form, LC3-I transforms to LC3-II, which binds to the autophagosome
membrane (38). Therefore, the ratio
of LC3-II/LC3-I was used in the present study to represent
autophagy level. P62 was first reported as a polyubiquitin-binding
protein in autophagy (39). It has
been shown that P62 can bind to ubiquitinated protein and produce
ubiquitinated protein aggregates, which serve an essential role in
the degradation of ubiquitinated proteins in autophagosomes
(40). Thus, P62 expression is the
opposite to that of autophagy degradation, that is, the greater the
levels of degradation, the lower the P62 expression (41). Therefore, increasing LC3-II/LC3-I
levels and decreasing levels of P62 occurring concurrently suggest
that the autophagy flux is unobstructed. Thus, LC3-II/LC3-I and P62
were detected in the present study to evaluate the autophagy
flux.
In the current study, p-PRAS40 expression, mTOR
pathway activity and autophagy flux were detected at various time
points following SE. Autophagy levels were significantly elevated
at 3 h post-SE, and gradually decreased over time. In addition,
p-PRAS40 expression was increased and the mTOR pathway was found to
be more active, with a prolonged duration of SE. Thus, it was
speculated that autophagy was abnormally elevated due to various
factors, such as inflammation and oxidative stress, occurring
shortly following SE induction, which then gradually decreased with
increased p-PRAS40 levels and mTOR activation. Since an
experimental group earlier than 3 h post-SE was not used in the
current study, the mechanism underlying the increase in autophagy
was not observed, which was a limitation of the study design.
However, to investigate the aforementioned hypothesis, a LY3023414
pretreated SE group was established at 3 days after SE (highest
expression of p-PRAS40 and lowest autophagy level) and the
PI3K/AKT/PRAS40/mTOR pathway and autophagy flux were detected in
the control, SE-3d and SE-3d + inh groups. The results demonstrated
that there was abnormal activation of the PI3K/AKT/PRAS40/mTOR
pathway and decreased autophagy in the SE-3d group, and that
LY3023414 pretreatment could inhibit the activation of this pathway
and increase the levels of autophagy, which suggested that p-PRAS40
participated in the mTOR-associated downregulation of
autophagy.
Previous studies have reported that the abnormal
elevation of autophagy following SE may be associated with
neurocyte death (15,16). Therefore, it was hypothesized that
p-PRAS40 and mTOR activation may have a protective effect on
neurocyte after SE by inhibiting autophagy. However, abnormal
activation of the mTOR pathway participates in chronic epilepsy
development (42), and thus it was
speculated that long-term high expression of p-PRAS40 and
activation of the mTOR pathway may be associated with epileptic
foci formation, which should be further investigated in future
research.
In conclusion, to the best of our knowledge, the
present study was the first to identify the role of p-PRAS40 and
its associated mechanism contributing to SE pathogenesis.
Currently, the available antiepileptic drugs suppress seizures and
are used as a symptomatic therapy (1). Thus, these drugs are a short-term
medication that can prevent the development of epilepsy. The
present results may represent a novel and promising therapeutic
target with antiepileptogenic effects. Therefore, further studies
should focus on PRAS40 function in other types of epilepsy, such as
temporal lobe epilepsy, as well as investigating treatments that
target p-PRAS40.
Acknowledgements
The authors would like to thank the support from
technician Mrs. Jieqing Jiang at the Department of Forensic
Medicine, Fudan University.
Funding
This study was supported by Shanghai Key Laboratory
of Crime Scene Evidence, Institute of Forensic Science, Shanghai
Public Security Bureau (grant nos. 2015XCWZK01 and 2017XCWZK170),
Funds for Youth in Department of Forensic Medicine, School of Basic
Medical Sciences, Fudan University (grant no. FY2014-01), Sponsored
by the Medical Scientific Research Project of Xuhui District, 2018
(grant no. SHXH201818) and Remote Forensic Consultation Center,
Collaborative Innovation Center of Judicial Civilization, China
University of Political Science and Law (grant no. XUF101010). The
funders had no role in study design, data collection and analysis,
decision to publish or preparation of the manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YS and BL designed the study, and analyzed and
interpreted the data. JL, YF and MZ contributed to and conducted
the experiments and contributed to writing the manuscript. XW, LL,
MH, AX and KZ made substantial contributions to the experiments,
the acquisition of data, interpretation of experiments and revised
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Experimental
Animal Ethics Committee of the Basic Medical College of Fudan
University.
Patient consent for publication
Not applicable.
Competing interest
The authors declare that they have no competing
interests.
References
|
1
|
Betjemann JP and Lowenstein DH: Status
epilepticus in adults. Lancet Neurol. 14:615–624. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Trinka E, Cock H, Hesdorffer D, Rossetti
AO, Scheffer IE, Shinnar S, Shorvon S and Lowenstein DH: A
definition and classification of status epilepticus-report of the
ILAE task force on classification of status epilepticus. Epilepsia.
56:1515–1523. 2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wong M: Mammalian target of rapamycin
(mTOR) inhibition as a potential antiepileptogenic therapy: From
tuberous sclerosis to common acquired epilepsies. Epilepsia.
51:27–36. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kovacina KS, Park GY, Bae SS, Guzzetta AW,
Schaefer E, Birnbaum MJ and Roth RA: Identification of a
proline-rich Akt substrate as a 14-3-3 binding partner. J Biol
Chem. 278:10189–10194. 2003.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Nascimento EB, Snel M, Guigas B, van der
Zon GCM, Kriek J, Maassen JA, Jazet IM, Diamant M and Ouwens DM:
Phosphorylation of PRAS40 on Thr246 by PKB/AKT facilitates
efficient phosphorylation of Ser183 by mTORC1. Cell Signal.
22:961–967. 2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Thedieck K, Polak P, Kim ML, Molle KD,
Cohen A, Jenö P, Arrieumerlou C and Hall MN: PRAS40 and PRR5-like
protein are new mTOR interactors that regulate apoptosis. PLoS One.
2(e1217)2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kazi AA and Lang CH: PRAS40 regulates
protein synthesis and cell cycle in C2C12 myoblasts. Mol Med.
16:359–371. 2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hong-Brown LQ, Brown CR, Kazi AA, Huber
DS, Pruznak AM and Lang CH: Alcohol and PRAS40 knockdown decrease
mTOR activity and protein synthesis via AMPK signaling and changes
in mTORC1 interaction. J Cell Biochem. 109:1172–1184.
2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Lipton JO and Sahin M: The neurology of
mTOR. Neuron. 84:275–291. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
San YZ, Liu Y, Zhang Y, Shi PP and Zhu YL:
Peroxisome proliferator-activated receptor-γ agonist inhibits the
mammalian target of rapamycin signaling pathway and has a
protective effect in a rat model of status epilepticus. Mol Med
Rep. 12:1877–1883. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Brewster AL, Lugo JN, Patil VV, Lee WL,
Qian Y, Vanegas F and Anderson AE: Rapamycin reverses status
epilepticus-induced memory deficits and dendritic damage. PLoS One.
8(e57808)2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wang SJ, Bo QY, Zhao XH, Yang X, Chi ZF
and Liu XW: Resveratrol pre-treatment reduces early inflammatory
responses induced by status epilepticus via mTOR signaling. Brain
Res. 1492:122–129. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Kim KH and Lee MS: Autophagy-a key player
in cellular and body metabolism. Nat Rev Endocrinol. 10:322–337.
2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Munson MJ and Ganley IG: MTOR, PIK3C3, and
autophagy: Signaling the beginning from the end. Autophagy.
11:2375–2376. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Cao L, Xu J, Lin Y, Zhao X, Liu X and Chi
Z: Autophagy is upregulated in rats with status epilepticus and
partly inhibited by vitamin E. Biochem Biophys Res Commun.
379:949–953. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Dong Y, Wang S, Zhang T, Zhao X, Liu X,
Cao L and Chi Z: Ascorbic acid ameliorates seizures and brain
damage in rats through inhibiting autophagy. Brain Res.
1535:115–123. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Benz AP, Niquet J, Wasterlain CG and Rami
A: Status epilepticus in the immature rodent brain alters the
dynamics of autophagy. Curr Neurovasc Res. 11:125–135.
2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Rami A, Benz AP, Niquet J and Langhagen A:
Axonal accumulation of lysosomal-associated membrane protein 1
(LAMP1) accompanying alterations of autophagy dynamics in the rat
hippocampus upon seizure-induced injury. Neurochem Res. 41:53–63.
2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Shacka JJ, Lu J, Xie ZL, Uchiyama Y, Roth
KA and Zhang J: Kainic acid induces early and transient autophagic
stress in mouse hippocampus. Neurosci Lett. 414:57–60.
2007.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Racine RJ: Modification of seizure
activity by electrical stimulation. II. Motor seizure.
Electroencephalogr Clin Neurophysiol. 32:281–294. 1972.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Dibble CC and Cantley LC: Regulation of
mTORC1 by PI3K signaling. Trends Cell Biol. 25:545–555.
2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Sloviter RS: The functional organization
of the hippocampal dentate gyrus and its relevance to the
pathogenesis of temporal lobe epilepsy. Ann Neurol. 35:640–654.
1994.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Sutula TP and Dudek FE: Unmasking
recurrent excitation generated by mossy fiber sprouting in the
epileptic dentate gyrus: An emergent property of a complex system.
Prog Brain Res. 163:541–563. 2007.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Nascimento EB, Fodor M, van der Zon GC,
Jazet IM, Meinders AE, Voshol PJ, Vlasblom R, Baan B, Eckel J,
Maassen JA, et al: Insulin-mediated phosphorylation of the
proline-rich Akt substrate PRAS40 is impaired in insulin target
tissues of high-fat diet-fed rats. Diabetes. 55:3221–3228.
2006.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Saito A, Hayashi T, Okuno S, Nishi T and
Chan PH: Modulation of proline-rich akt substrate survival
signaling pathways by oxidative stress in mouse brains after
transient focal cerebral ischemia. Stroke. 37:513–517.
2006.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Saito A, Narasimhan P, Hayashi T, Okuno S,
Ferrand-Drake M and Chan PH: Neuroprotective role of a proline-rich
Akt substrate in apoptotic neuronal cell death after stroke:
Relationships with nerve growth factor. J Neurosci. 24:1584–1593.
2004.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Feliciano DM, Su T, Lopez J, Platel JC and
Bordey A: Single-cell Tsc1 knockout during corticogenesis generates
tuber-like lesions and reduces seizure threshold in mice. J Clin
Invest. 121:1596–1607. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
28
|
Luo CL, Li BX, Li QQ, Chen XP, Sun YX, Bao
HJ, Dai DK, Shen YW, Xu HF, Ni H, et al: Autophagy is involved in
traumatic brain injury-induced cell death and contributes to
functional outcome deficits in mice. Neuroscience. 184:54–63.
2011.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wang K, Huang J, Xie W, Huang L, Zhong C
and Chen Z: Beclin1 and HMGB1 ameliorate the α-synuclein-mediated
autophagy inhibition in PC12 cells. Diagn Pathol.
11(15)2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Shin JY, Park HJ, Kim HN, Oh SH, Bae JS,
Ha HJ and Lee PH: Mesenchymal stem cells enhance autophagy and
increase β-amyloid clearance in Alzheimer disease models.
Autophagy. 10:32–44. 2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Aguado C, Sarkar S, Korolchuk VI, Criado
O, Vernia S, Boya P, Sanz P, de Córdoba SR, Knecht E and
Rubinsztein DC: Laforin, the most common protein mutated in Lafora
disease, regulates autophagy. Hum Mol Genet. 19:2867–2876.
2010.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hosseinzadeh M, Nikseresht S, Khodagholi
F, Naderi N and Maghsoudi N: Cannabidiol post-treatment alleviates
rat epileptic-related behaviors and activates hippocampal cell
autophagy pathway along with antioxidant defense in chronic phase
of pilocarpine-induced seizure. J Mol Neurosci. 58:432–440.
2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ni H, Zhao DJ and Tian T: Ketogenic diet
change cPLA2/clusterin and autophagy related gene expression and
correlate with cognitive deficits and hippocampal MFs sprouting
following neonatal seizures. Epilepsy Res. 120:13–18.
2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Jin R, Zhu W, Cao S, Chen R, Jin H, Liu Y,
Wang S, Wang W and Xiao G: Japanese encephalitis virus activates
autophagy as a viral immune evasion strategy. PLoS One.
8(e52909)2013.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Li L, Tian J, Long MK, Chen Y, Lu J, Zhou
C and Wang T: Protection against experimental stroke by ganglioside
GM1 is associated with the inhibition of autophagy. PLoS One.
11(e144219)2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Schaaf MB, Keulers TG, Vooijs MA and
Rouschop KM: LC3/GABARAP family proteins: Autophagy-(un)related
functions. FASEB J. 30:3961–3978. 2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Sini P, James D, Chresta C and Guichard S:
Simultaneous inhibition of mTORC1 and mTORC2 by mTOR kinase
inhibitor AZD8055 induces autophagy and cell death in cancer cells.
Autophagy. 6:553–554. 2010.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Lamark T, Svenning S and Johansen T:
Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays
Biochem. 61:609–624. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Nezis IP, Simonsen A, Sagona AP, Finley K,
Gaumer S, Contamine D, Rusten TE, Stenmark H and Brech A: Ref(2)P,
the drosophila melanogaster homologue of mammalian p62, is required
for the formation of protein aggregates in adult brain. J Cell
Biol. 180:1065–1071. 2008.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Bartlett BJ, Isakson P, Lewerenz J,
Sanchez H, Kotzebue RW, Cumming RC, Harris GL, Nezis IP, Schubert
DR, Simonsen A and Finley KD: p62, ref(2)P and ubiquitinated
proteins are conserved markers of neuronal aging, aggregate
formation and progressive autophagic defects. Autophagy. 7:572–583.
2011.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Buckmaster PS, Ingram EA and Wen X:
Inhibition of the mammalian target of rapamycin signaling pathway
suppresses dentate granule cell axon sprouting in a rodent model of
temporal lobe epilepsy. J Neurosci. 29:8259–8269. 2009.PubMed/NCBI View Article : Google Scholar
|