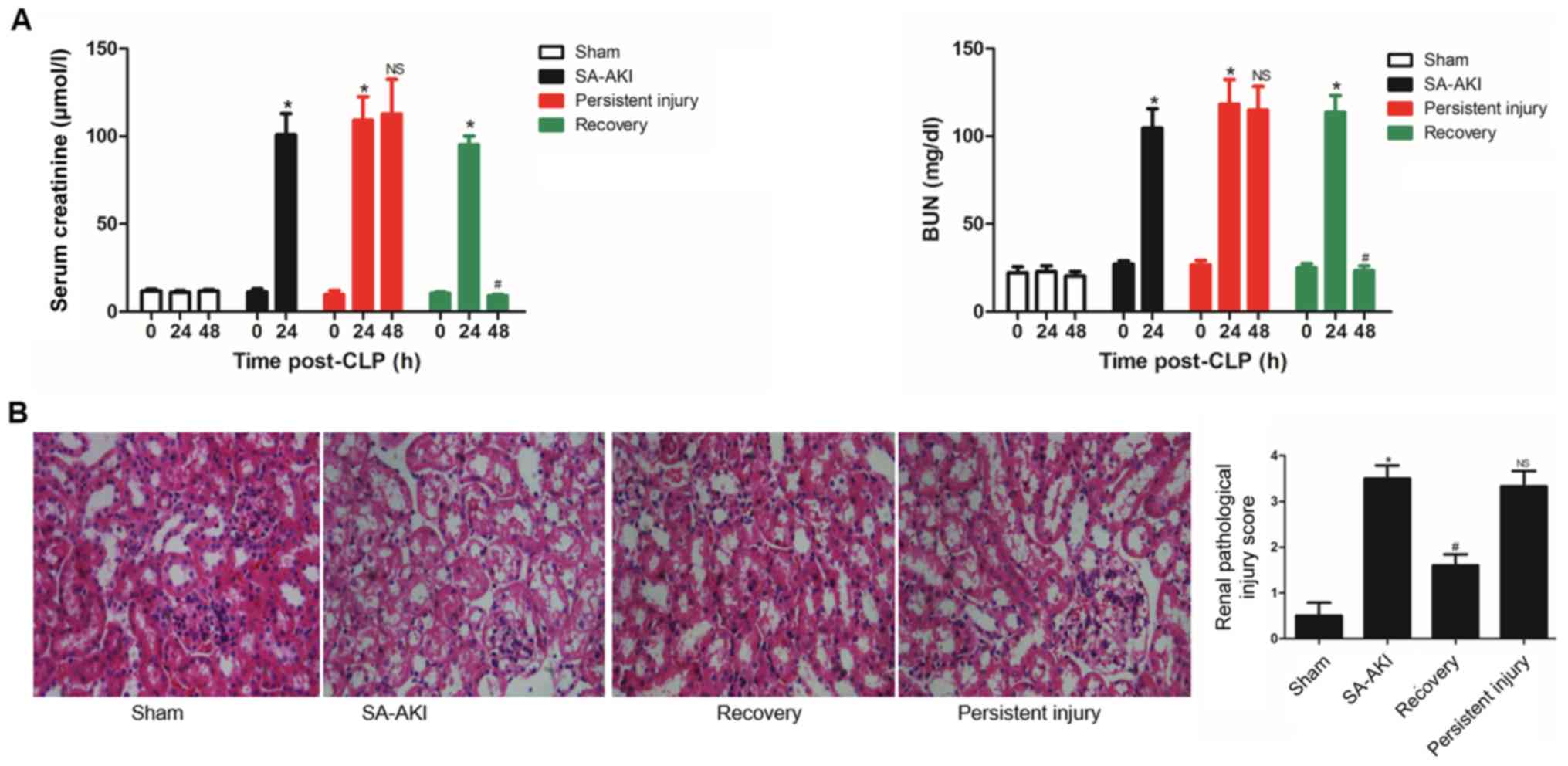
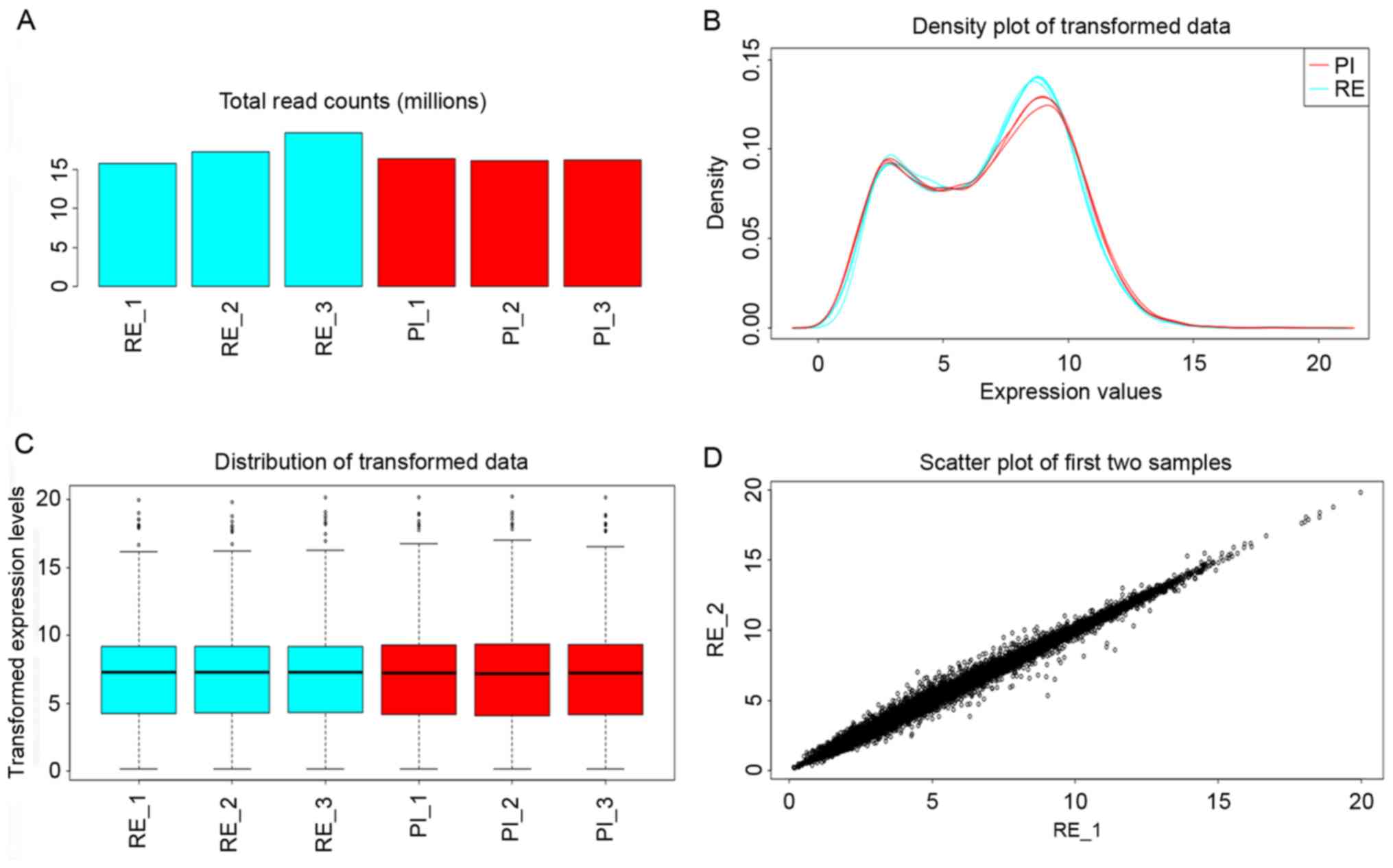
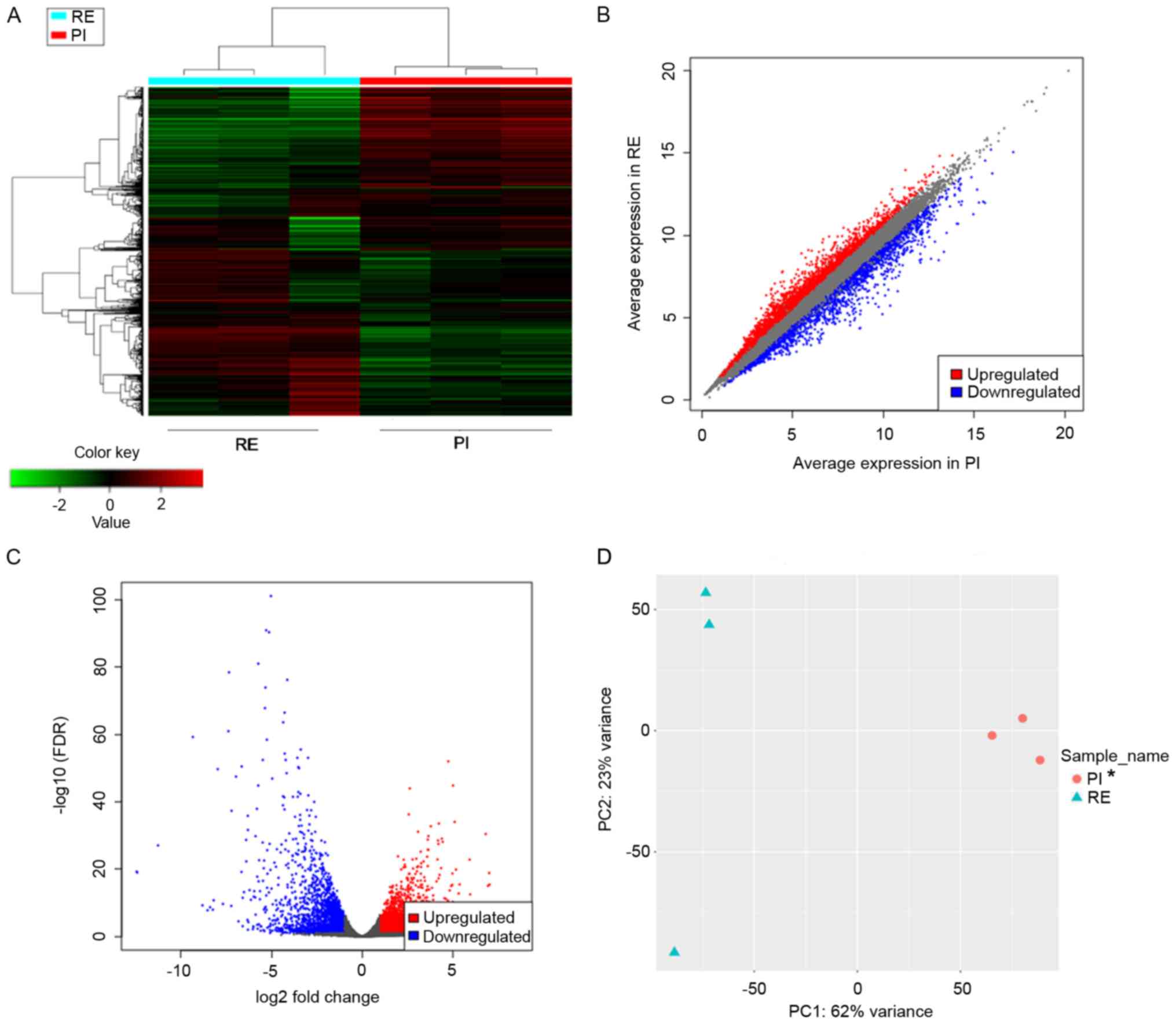
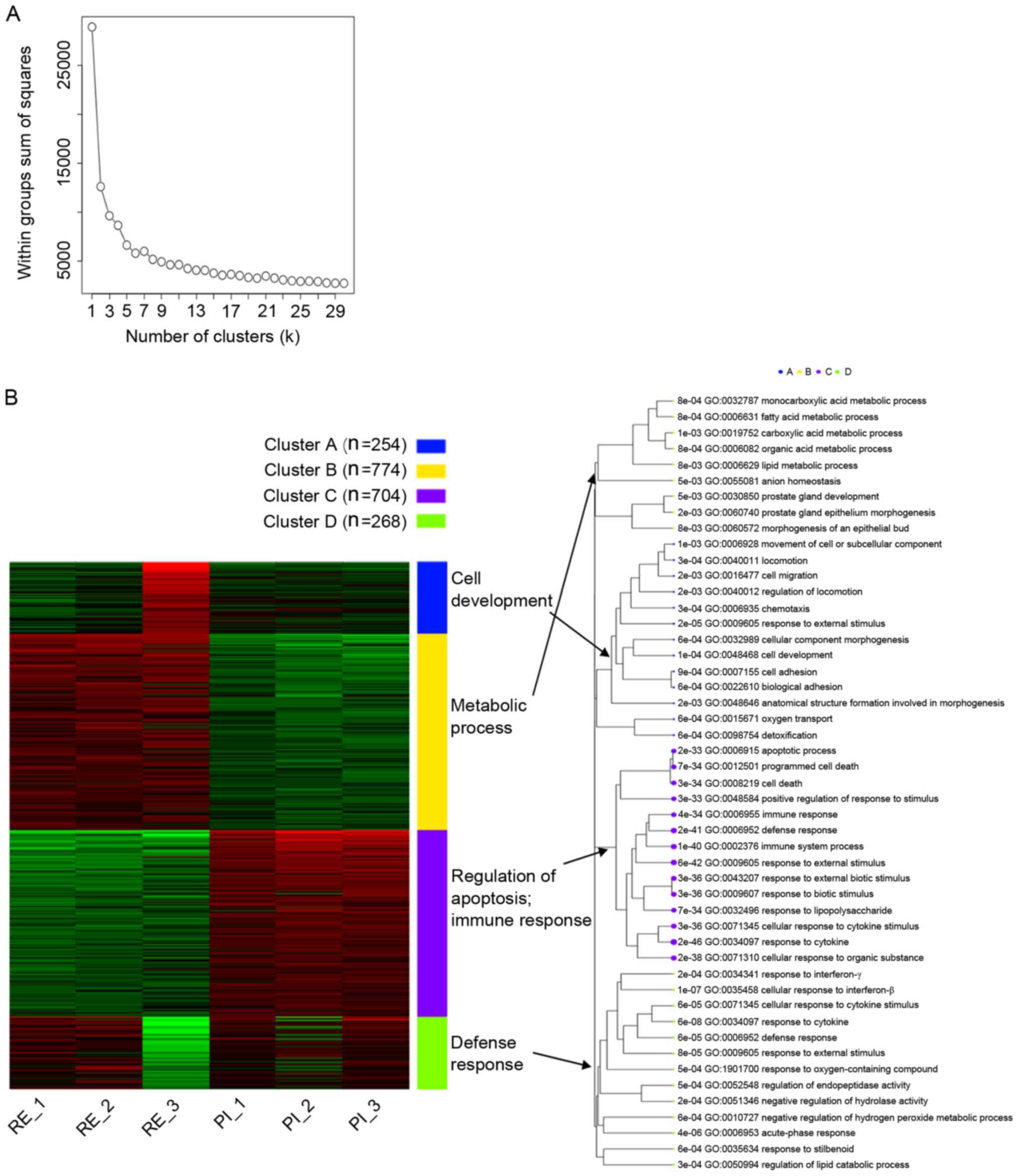
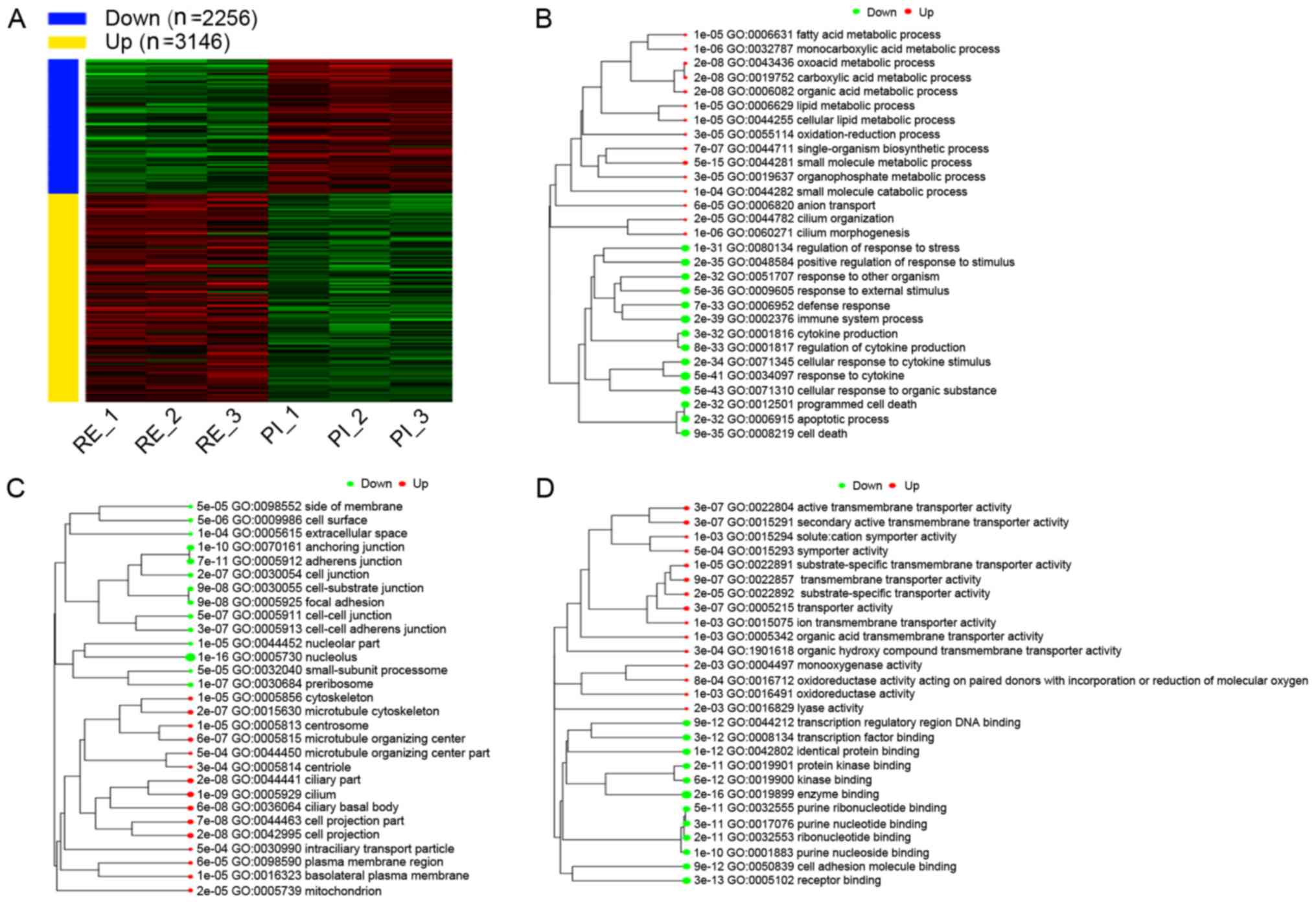
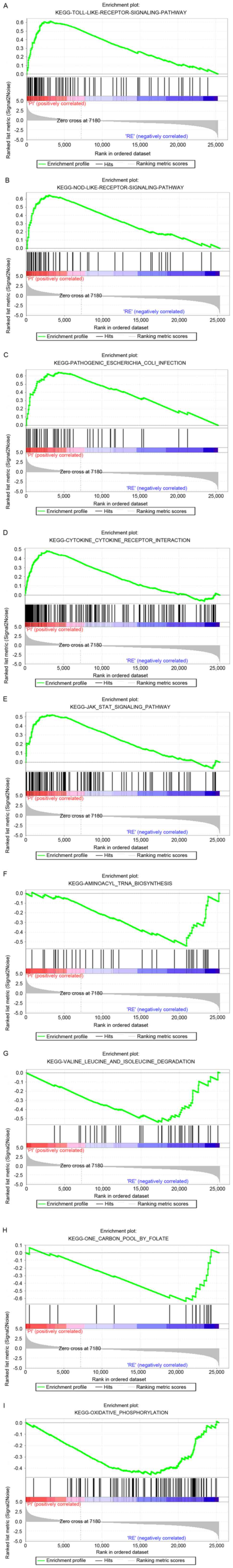
|
1
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (Sepsis-3). JAMA.
315:801–810. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kellum JA and Lameire N: KDIGO AKI
Guideline Work Group: Diagnosis, evaluation, and management of
acute kidney injury: A KDIGO summary (Part 1). Crit Care.
17(204)2013.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Hoste EA, Lameire NH, Vanholder RC, Benoit
DD, Decruyenaere JM and Colardyn FA: Acute renal failure in
patients with sepsis in a surgical ICU: Predictive factors,
incidence, comorbidity, and outcome. J Am Soc Nephrol.
14:1022–1030. 2003.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Uchino S, Kellum JA, Bellomo R, Doig GS,
Morimatsu H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, et al:
Acute renal failure in critically ill patients: A multinational,
multicenter study. JAMA. 294:813–818. 2005.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Morrell ED, Kellum JA, Pastor-Soler NM and
Hallows KR: Septic acute kidney injury: Molecular mechanisms and
the importance of stratification and targeting therapy. Crit Care.
18(501)2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Bagshaw SM, Uchino S, Bellomo R, Morimatsu
H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, Gibney N, et al:
Septic acute kidney injury in critically ill patients: Clinical
characteristics and outcomes. Clin J Am Soc Nephrol. 2:431–439.
2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Bouchard J, Acharya A, Cerda J,
Maccariello ER, Madarasu RC, Tolwani AJ, Liang X, Fu P, Liu ZH and
Mehta RL: A prospective international multicenter study of AKI in
the intensive care unit. Clin J Am Soc Nephrol. 10:1324–1331.
2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Poston JT and Koyner JL: Sepsis associated
acute kidney injury. BMJ. 364(k4891)2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Tran M, Tam D, Bardia A, Bhasin M, Rowe
GC, Kher A, Zsengeller ZK, Akhavan-Sharif MR, Khankin EV,
Saintgeniez M, et al: PGC-1α promotes recovery after acute kidney
injury during systemic inflammation in mice. J Clin Invest.
121:4003–4014. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Stark R, Grzelak M and Hadfield J: RNA
sequencing: The teenage years. Nat Rev Genet. 20:631–656.
2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ge SX, Son EW and Yao R: iDEP: An
integrated web application for differential expression and pathway
analysis of RNA-Seq data. BMC Bioinformatics.
19(534)2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Goldstein B, Giroir B and Randolph A:
International Consensus Conference on Pediatric Sepsis:
International pediatric sepsis consensus conference: Definitions
for sepsis and organ dysfunction in pediatrics. Pediatr Crit Care
Med. 6:2–8. 2005.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wu A, Hinds CJ and Thiemermann C:
High-density lipoproteins in sepsis and septic shock: Metabolism,
actions, and therapeutic applications. Shock. 21:210–221.
2004.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Murch O, Collin M, Hinds CJ and
Thiemermann C: Lipoproteins in inflammation and sepsis. I. Basic
science. Intensive Care Med. 33:13–24. 2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Vyroubal P, Chiarla C, Giovannini I,
Hyspler R, Ticha A, Hrnciarikova D and Zadak Z: Hypocholesterolemia
in clinically serious conditions-review. Biomed Pap Med Fac Univ
Palacky Olomouc Czech Repub. 152:181–189. 2008.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Tsai MH, Peng YS, Chen YC, Lien JM, Tian
YC, Fang JT, Weng HH, Chen PC, Yang CW and Wu CS: Low serum
concentration of apolipoprotein A-I is an indicator of poor
prognosis in cirrhotic patients with severe sepsis. J Hepatol.
50:906–915. 2009.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chenaud C, Merlani PG, Roux-Lombard P,
Burger D, Harbarth S, Luyasu S, Graf JD, Dayer JM and Ricou B: Low
apolipoprotein A-I level at intensive care unit admission and
systemic inflammatory response syndrome exacerbation. Crit Care
Med. 32:632–637. 2004.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Barlage S, Gnewuch C, Liebisch G, Wolf Z,
Audebert FX, Glück T, Fröhlich D, Krämer BK, Rothe G and Schmitz G:
Changes in HDL-associated apolipoproteins relate to mortality in
human sepsis and correlate to monocyte and platelet activation.
Intensive Care Med. 35:1877–1885. 2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Bonville DA, Parker TS, Levine DM, Gordon
BR, Hydo LJ, Eachempati SR and Barie PS: The relationships of
hypocholesterolemia to cytokine concentrations and mortality in
critically ill patients with systemic inflammatory response
syndrome. Surg Infect (Larchmt.). 5:39–49. 2004.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Svahn SL, Ulleryd MA, Grahnemo L, Ståhlman
M, Borén J, Nilsson S, Jansson JO and Johansson ME: Dietary omega-3
fatty acids increase survival and decrease bacterial load in mice
subjected to staphylococcus aureus-induced sepsis. Infect Immun.
84:1205–1213. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Spite M, Norling LV, Summers L, Yang R,
Cooper D, Petasis NA, Flower RJ, Perretti M and Serhan CN: Resolvin
D2 is a potent regulator of leukocytes and controls microbial
sepsis. Nature. 461:1287–1291. 2009.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chen F, Fan XH, Wu YP, Zhu JL, Wang F, Bo
LL, Li JB, Bao R and Deng XM: Resolvin D1 improves survival in
experimental sepsis through reducing bacterial load and preventing
excessive activation of inflammatory response. Eur J Clin Microbiol
Infect Dis. 33:457–464. 2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhuo Y, Zhang S, Li C, Yang L, Gao H and
Wang X: Resolvin D1 promotes SIRT1 expression to counteract the
activation of STAT3 and NF-κB in mice with septic-associated lung
injury. Inflammation. 41:1762–1771. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kerr JF, Wyllie AH and Currie AR:
Apoptosis: A basic biological phenomenon with wide-ranging
implications in tissue kinetics. Br J Cancer. 26:239–257.
1972.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ayala A, Perl M, Venet F, Lomas-Neira J,
Swan R and Chung CS: Apoptosis in sepsis: Mechanisms, clinical
impact and potential therapeutic targets. Curr Pharm Des.
14:1853–1859. 2008.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Harjai M, Bogra J, Kohli M and Pant AB: Is
suppression of apoptosis a new therapeutic target in sepsis?
Anaesth Intensive Care. 41:175–183. 2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Green DR, Galluzzi L and Kroemer G:
Mitochondria and the autophagy-inflammation-cell death axis in
organismal aging. Science. 333:1109–1112. 2011.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Galley HF: Oxidative stress and
mitochondrial dysfunction in sepsis. Br J Anaesth. 107:57–64.
2011.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Rocha M, Herance R, Rovira S,
Hernandez-Mijares A and Victor VM: Mitochondrial dysfunction and
antioxidant therapy in sepsis. Infect Disord Drug Targets.
12:161–178. 2012.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Siskind LJ, Kolesnick RN and Colombini M:
Ceramide channels increase the permeability of the mitochondrial
outer membrane to small proteins. J Biol Chem. 277:26796–26803.
2002.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Garcia-Ruiz C, Colell A, Mari M, Morales A
and Fernandez-Checa JC: Direct effect of ceramide on the
mitochondrial electron transport chain leads to generation of
reactive oxygen species. Role of mitochondrial glutathione. J Biol
Chem. 272:11369–11377. 1997.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Sureshbabu A, Patino E, Ma KC, Laursen K,
Finkelsztein EJ, Akchurin O, Muthukumar T, Ryter SW, Gudas L, Choi
AMK and Choi ME: RIPK3 promotes sepsis-induced acute kidney injury
via mitochondrial dysfunction. JCI Insight.
3(e98411)2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Rodgers LS and Fanning AS: Regulation of
epithelial permeability by the actin cytoskeleton. Cytoskeleton
(Hoboken). 68:653–660. 2011.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Jacobson JR and Garcia JG: Novel therapies
for microvascular permeability in sepsis. Curr Drug Targets.
8:509–514. 2007.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Gustot T: Multiple organ failure in
sepsis: Prognosis and role of systemic inflammatory response. Curr
Opin Crit Care. 17:153–159. 2011.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Ma J, Chen C, Barth AS, Cheadle C, Guan X
and Gao L: Lysosome and cytoskeleton pathways are robustly enriched
in the blood of septic patients: A meta-analysis of transcriptomic
data. Mediators Inflamm. 2015(984825)2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
de Montmollin E and Annane D: Year in
review 2010: Critical care-multiple organ dysfunction and sepsis.
Crit Care. 15(236)2011.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Goldenberg NM, Steinberg BE, Slutsky AS
and Lee WL: Broken barriers: A new take on sepsis pathogenesis. Sci
Transl Med. 3(88ps25)2011.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Chousterman BG, Swirski FK and Weber GF:
Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol.
39:517–528. 2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Vertii A, Ivshina M, Zimmerman W, Hehnly
H, Kant S and Doxsey S: The centrosome undergoes Plk1-independent
interphase maturation during inflammation and mediates cytokine
release. Dev Cell. 37:377–386. 2016.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Sperandeo MP, Andria G and Sebastio G:
Lysinuric protein intolerance: Update and extended mutation
analysis of the SLC7A7 gene. Hum Mutat. 29:14–21. 2008.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Lesina M, Wörmann SM, Morton J,
Diakopoulos KN, Korneeva O, Wimmer M, Einwächter H, Sperveslage J,
Demir IE, Kehl T, et al: RelA regulates CXCL1/CXCR2-dependent
oncogene-induced senescence in murine Kras-driven pancreatic
carcinogenesis. J Clin Invest. 126:2919–2932. 2016.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Chen Y, Guo L, Lang H, Hu X, Jing S, Luo
M, Xu G and Zhou Z: Effect of a stellate ganglion block on acute
lung injury in septic rats. Inflammation. 41:1601–1609.
2018.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Zhu T, Wang DX, Zhang W, Liao XQ, Guan X,
Bo H, Sun JY, Huang NW, He J, Zhang YK, et al: Andrographolide
protects against LPS-induced acute lung injury by inactivation of
NF-κB. PLoS One. 8(e56407)2013.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Ni S, Miao K, Zhou X, Xu N, Li C, Zhu R,
Sun R and Wang Y: The involvement of follistatin-like protein 1 in
osteoarthritis by elevating NF-κB-mediated inflammatory cytokines
and enhancing fibroblast like synoviocyte proliferation. Arthritis
Res Ther. 17(91)2015.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Lim CP and Cao X: Structure, function, and
regulation of STAT proteins. Mol Biosyst. 2:536–550.
2006.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Lu R, Zhang YG and Sun J: STAT3 activation
in infection and infection-associated cancer. Mol Cell Endocrinol.
451:80–87. 2017.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Ghilardi N, Ziegler S, Wiestner A, Stoffel
R, Heim MH and Skoda RC: Defective STAT signaling by the leptin
receptor in diabetic mice. Proc Natl Acad Sci USA. 93:6231–6235.
1996.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Devi SJ, Madhav MS, Kumar GR, Goel AK,
Umakanth B, Jahnavi B and Viraktamath BC: Identification of abiotic
stress miRNA transcription factor binding motifs (TFBMs) in rice.
Gene. 531:15–22. 2013.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Zamanighomi M, Lin Z, Wang Y, Jiang R and
Wong WH: Predicting transcription factor binding motifs from
DNA-binding domains, chromatin accessibility and gene expression
data. Nucleic Acids Res. 45:5666–5677. 2017.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Eckert D, Buhl S, Weber S, Jäger R and
Schorle H: The AP-2 family of transcription factors. Genome Biol.
6(246)2005.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Xu X, Liu Z, Huang H, Zheng K, Hu X, Zhang
Z and Qiu M: AP-2α and AP-2β regulate dorsal interneuron
specification in the spinal cord. Neuroscience. 340:232–242.
2017.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Suyama K, Kabuyama Y, Suzuki S, Kawasaki
Y, Suzuki J, Suzuki H and Homma Y: Induction of transcription
factor AP-2 by cytokines and prostaglandins in cultured mesangial
cells. Am J Nephrol. 21:307–314. 2001.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Mi X, Tang W, Chen X, Liu F and Tang X:
Mitofusin 2 attenuates the histone acetylation at collagen IV
promoter in diabetic nephropathy. J Mol Endocrinol. 57:233–249.
2016.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Hobert O: Gene regulation by transcription
factors and microRNAs. Science. 319:1785–1786. 2008.PubMed/NCBI View Article : Google Scholar
|
|
57
|
He L and Hannon GJ: MicroRNAs: Small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531.
2004.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Dieter C, Assmann TS, Costa AR, Canani LH,
de Souza BM, Bauer AC and Crispim D: MiR-30e-5p and MiR-15a-5p
expressions in plasma and urine of type 1 diabetic patients with
diabetic kidney disease. Front Genet. 10(563)2019.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Sun SQ, Zhang T, Ding D, Zhang WF, Wang
XL, Sun Z, Hu LH, Qin SY, Shen LH and He B: Circulating
MicroRNA-188, -30a, and -30e as early biomarkers for
contrast-induced acute kidney injury. J Am Heart Assoc.
5(e004138)2016.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Galicia JC, Naqvi AR, Ko CC, Nares S and
Khan AA: MiRNA-181a regulates Toll-like receptor agonist-induced
inflammatory response in human fibroblasts. Genes Immun.
15:333–337. 2014.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Yuan J, Ji H, Xiao F, Lin Z, Zhao X, Wang
Z, Zhao J and Lu J: MicroRNA-340 inhibits the proliferation and
invasion of hepatocellular carcinoma cells by targeting JAK1.
Biochem Biophys Res Commun. 483:578–584. 2017.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Li Y, Fan X, He X, Sun H, Zou Z, Yuan H,
Xu H, Wang C and Shi X: MicroRNA-466l inhibits antiviral innate
immune response by targeting interferon-alpha. Cell Mol Immunol.
9:497–502. 2012.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Ma F, Liu X, Li D, Wang P, Li N, Lu L and
Cao X: MicroRNA-466l upregulates IL-10 expression in TLR-triggered
macrophages by antagonizing RNA-binding protein
tristetraprolin-mediated IL-10 mRNA degradation. J Immunol.
184:6053–6059. 2010.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Rout AK, Udgata SR, Dehury B, Pradhan SP,
Swain HS, Behera BK and Das BK: Structural bioinformatics insights
into the CARD-CARD interaction mediated by the mitochondrial
antiviral-signaling protein of black carp. J Cell Biochem.
120:12534–12543. 2019.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Creagh EM and O'Neill LA: TLRs, NLRs and
RLRs: A trinity of pathogen sensors that co-operate in innate
immunity. Trends Immunol. 27:352–357. 2006.PubMed/NCBI View Article : Google Scholar
|