Introduction
Neuroinflammation is a common disease-associated
event, which may affect the progression of multiple
neurodegenerative diseases, including Alzheimer's disease (AD),
Parkinson's disease (PD), traumatic brain injury and stroke
(1,2). Under normal conditions, microglial
cells, the major immune cells of the brain, are the first line of
defense in the innate immune responses and tissue repair of the
central nervous system (CNS), and maintain CNS homeostasis through
their precise activation (3,4).
Sustained over-activation of microglial cells may cause neuronal
death or tissue damage mediated by the excessive production or
release of pro-inflammatory mediators including tumor necrosis
factor-α (TNF-α), nitric oxide (NO), interleukin 6 (IL-6), IL-1β,
inducible NO synthase (iNOS), reactive oxygen species and
cyclooxygenase-2 (COX-2) (5,6).
Therefore, inhibiting the excessive activation of microglia may be
an effective anti-inflammatory approach for attenuating the
progression of multiple neurodegenerative diseases.
Lipopolysaccharide (LPS), as an activator of
inflammation, is a major component of the outer membrane in
gram-negative bacteria, and is commonly used as a pro-inflammatory
agent to generate inflammation models (7). Several signaling pathways are involved
in microglia-associated neuroinflammation. In general, NF-κB is
located in the cytoplasm and binds to the NF-κB inhibitor α
protein. Upon stimulation with LPS, IκBα is phosphorylated rapidly
and degraded, which leads to the release of p50/ transcription
factor p65 (p65) NF-κB heterodimers. NF-κB dimers move into the
nucleus and bind to inflammation-associated genes, resulting in the
transcriptional activation of pro-inflammatory mediators (8). In addition, MAPKs have been
demonstrated to serve key roles in the inflammatory response and
are involved in various cellular processes (9). Research indicates that LPS may induce
the phosphorylation of three major MAPK pathways, p38, JNK and ERK,
which activates the production of pro-inflammatory cytokines
(10). Taken together, the NF-κB
and MAPK signaling pathways are crucial for the modulation of
inflammation in various neurological diseases (11,12).
The P2X7 receptor (P2X7R), a purinergic receptor, is
expressed in the microglia, neurons and astrocytes of the CNS. In
various neurodegenerative processes, P2X7R is overactivated due to
ATP release, resulting in anion imbalance and triggering of cell
death (13,14). The activation of P2X7R is involved
in several signaling pathways, including the NF-κB, MAPK and NFAT
pathways (15). P2X7 activation
leads to microglial activation and facilitates the production of
IL-1β, IL-6 and TNF-α, additionally aggravating cell damage in
neurodegenerative diseases (16).
Recently, Wang et al (17)
demonstrated that brilliant blue G (BBG), a selective and
non-competitive P2X7R antagonist, serves a neuroprotective role by
attenuating microglial activation in an LPS-induced PD model.
Whether the MAPK/NF-κB pathway is involved in the anti-inflammatory
effect of BBG in LPS-induced PD models remains unclear.
Therefore, the aim of the present study was to
investigate whether P2X7 may be regarded as a key upstream factor
that additionally activates the MAPK/NF-κB signaling pathways that
are involved in LPS-induced neuroinflammation.
Materials and methods
Reagents and antibodies
BBG, LPS and MTT were purchased from Sigma-Aldrich;
Merck KGaA. Fetal bovine serum (FBS) was obtained from Gibco;
Thermo Fisher Scientific, Inc., Dulbecco's modified Eagle's medium
(DMEM) was obtained from HyClone; GE Healthcare Life Sciences. The
BCA protein assay kit (cat. no. P0012), penicillin/streptomycin
(cat. no. C0222), protease inhibitor (cat. no. ST505),
phosphorylated protease inhibitor (cat. no. P1096), DAPI (cat. no.
C1002), RIPA lysis buffer (cat. no. P0013B), SB203580 (cat. no.
S1863), SP600125 (cat. no. S1876), PD98059 (cat. no. S1805) and
Nuclear and Cytoplasmic Extraction kit (cat. no. P0027) were
obtained from Beyotime Institute of Biotechnology. BAY 11-7082 was
purchased from Selleck Chemicals (cat. no. S2913). ELISA kits for
TNF-α (cat. no. 70-EK2208-24), IL-6 (cat. no. 70-EK106/2-24) and
IL-1β (cat. no. 70-EK101B-24) were purchased from Hangzhou Multi
Sciences (Lianke) Biotech Co., Ltd. The primers for the reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) were
purchased from Shanghai GeneChem Co., Ltd. The following primary
antibodies were purchased from Cell Signaling Technology, Inc.:
anti-Lamin B (cat. no. 12586), p38 (cat. no. 8690), phosphorylated
(p)-p38 (cat. no. 4511), JNK (cat. no. 9252), p-JNK (cat. no.
4668), ERK (cat. no. 4795), p-ERK (cat. no. 4370), NF-κB p65 (cat.
no. 8242), COX-2 (cat. no. 4852) and iNOS (cat. no. 13120).
Anti-TLR4 was purchased from Abcam (cat. no. 47093). Anti-P2X7R
were purchased from Alomone Labs (cat. no. APR-004). Anti-β-actin
was purchased from ProteinTech Group, Inc. (cat. no. 66009-1-lg).
The anti-mouse IRDye® 680RD-conjugated (cat. no.
C70124-05) and anti-rabbit IRDye® 800CW-conjugated (cat.
no. C11117-05) secondary antibodies were purchased from LI-COR
Biosciences.
Cell culture
BV2 cells were purchased from the China Center for
Type Culture Collection. BV2 cells were cultured in DMEM medium
supplemented with 10% FBS and 1% penicillin/streptomycin, and
incubated at 37˚C in a humidified atmosphere of 5% CO2.
LPS was dissolved in phosphate-buffered saline (PBS) as a stock
solution (1 mg/ml), and stored at -20˚C. BV-2 cells
(5x104) were seeded into 96- or 6-well plates,
respectively, and exposed to LPS (1 µg/ml) in the presence or
absence of 1 µmol/ml BBG for 24 h. The cells were pretreated with
different inhibitors (BAY 11-7082, 10 µM; PD98059, 20 µM; SP203580,
20 µM; and SP600125, 20 µM) for 1 h prior to stimulation with 1
µg/ml LPS for 24 h.
MTT assay
BV-2 cells (5x104) were seeded in 96-well
plates and treatment with LPS (1 µg/ml) in the presence or absence
of 5 µmol/ml BBG for 24 h. Then, cells were cultured in fresh
medium and 5 mg/ml MTT for an additional 4 h at 37˚C. The
supernatant then was removed and 150 µl DMSO was added. The optical
density of the medium was detected at 570 nm using a microplate
reader (BioTek Instruments, Inc.). For relative quantification, the
value of absorbance in each group was normalized to that in the
control group.
Immunocytochemistry
BV-2 cells (5x106) were seeded into
12-well plates overnight, and treated with LPS in the presence or
absence of BBG for 24 h. Following the treatment, the supernatant
was removed via pipette and the cells were fixed with 4%
paraformaldehyde (PFA) for 30 min at 37˚C. Then, cells were
permeabilized with 0.2% Triton X-100 for 20 min at 37˚C, and
blocked with 5% donkey serum (Vicmed) for 30 min at 37˚C. The cells
were incubated with monoclonal rabbit NF-κB p65 (1:800) at 4˚C
overnight and then incubated with a secondary antibody Alexa
Fluor® 594-conjugated donkey anti-rabbit secondary
antibodies (1:500; Invitrogen; Thermo Fisher Scientific, Inc.) for
2 h at 37˚C. Nuclei were then stained with 10% DAPI (Beyotime
Institute of Biotechnology) for 10 min at 37˚C. Fluorescence images
were captured with a fluorescencemicroscope (magnification, x40;
Olympus Corporation).
ELISA assays
BV2 cells were seeded in 24-well plate at a density
of 1x105 cells/well, treated with 1 µmol/ml BBG and
stimulated with LPS for 24 h. Following this treatment step, the
culture supernatants were collected by centrifugation (1,000 x g
for 5 min; 4˚C). The levels of inflammatory cytokines TNF-α, IL-1β
and IL-6 were detected by ELISA according to the manufacturer's
protocol.
Western blot analysis
For whole cell lysates, the cells were lysed in 200
µl RIPA lysis buffer and then 1% protease inhibitor and 1%
phosphorylated protease inhibitor were added. After incubation on
ice for 10 min, the lysates were centrifuged at 12,000 x g for 30
min at 4˚C, and the supernatant was collected. According to the
Nuclear and Cytoplasmic Extraction kit instructions, the
cytoplasmic and nuclear proteins were extracted. The proteins
concentration was quantified using the BCA protein assay kit.
Protein samples (50 µg/lane) were loaded on 10-12% SDS-PAGE and
transferred to nitrocellulose filter membranes. The membranes were
then blocked with 5% skimmed milk for 2 h at room temperature. The
following primary antibodies were incubated with: anti-Lamin B1
(1:1,000), NF-κB p65 (1:1,000), p-p38 (1:1,000), p38 (1:1,000),
p-JNK (1:1,000), JNK (1:1,000), p-ERK (1:1,000), ERK (1:1,000),
COX-2 (1:1,000), iNOS (1:1,000), P2X7R (1:500), TLR4 (1:500) and
β-actin (1:10,000). Following washing with TBST three times, the
membranes were incubated with an anti-mouse IRDye®
680RD-conjugated antibody (1:10,000) or an anti-rabbit
IRDye® 800CW-conjugated antibody (1:10,000). The bands
were determined with Odyssey® Imaging Systems (LI-COR
Biosciences) and protein bands were quantified with ImageJ software
(version 1.5.2; National Institutes of Health).
RT-qPCR
Total RNA from BV2 cells was extracted using
TRIzol® reagent (Thermo Fisher Scientific, Inc.) and
then RNA (1 µg) was reverse transcribed to cDNA using HiScriptQ RT
SuperMix for qPCR (Vazyme Biotech Co., Ltd.). The cDNA (2 µl) was
amplified using a sequence detection system SYBR-Green Master Mix
(Vazyme Biotech Co., Ltd.). RT-qPCR was performed using a
LightCycler® 480 Real-Time PCR system (Roche
Diagnostics) according to the manufacturer's protocol. The cycling
parameters were: 95˚C for 5 min, followed by 40 cycles at 95˚C for
10 sec and 60˚C for 30 sec, 95˚C for 15 sec, 60˚C for 60 sec and
95˚C for 15 sec. RT-qPCR primers were as followed: TNF-α forward,
CAACGGCATGGATCTCAAAG; TNF-α reverse, GTCGTTGCTTGGTTCTCCTTG; IL-6
forward, TTGCCTTCTTGGGACTGATG; IL-6 reverse, GAATTGCCATTGCACAACTCT;
IL-1β forward, GCCCATCCTCTGTGACTCATG; IL-1β reverse,
GTCGTTGCTTGGTTCTCCTTG; GAPDH forward, TGGTGAAGGTCGGTGAAC; and GAPDH
reverse, GCTCCTGGAAGATGGTGATGG. GAPDH was used as the control. The
relative RNA expression of TNF-α, IL-6 and IL-1β was analyzed
according to the 2-ΔΔCq method (18).
Statistical analysis
Each experiment was repeated three times. Data are
expressed as mean ± standard error of the mean. Statistical
significance of data was analyzed using analysis of variance
followed by Fisher's Least Significant Difference tests. GraphPad
Prism 6.0 (GraphPad Software, Inc.) and SPSS 19.0 software (IBM
Corp.) were used to generate graphs and perform statistical
analysis. P<0.05 was considered to indicate a statistically
significant difference.
Results
Effects of BBG on the viability of BV2
cells
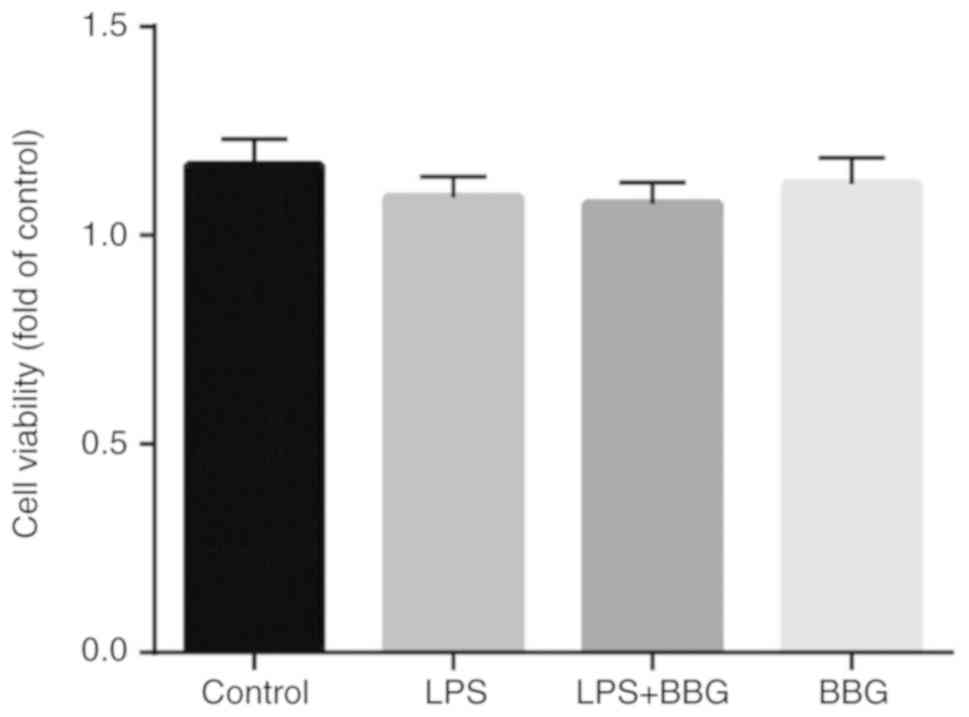
To evaluate the cytotoxic effects of BBG on BV2
cells, cell viability was examined using the MTT assay. The cells
were treated with 1 µM BBG in the presence or absence of LPS (1
µg/ml) for 24 h. As demonstrated in Fig. 1, BBG did not affect cell viability
with or without LPS, suggesting that BBG is not harmful to BV-2
cells.
BBG inhibits LPS-induced inflammation
mediators in BV2 cells
To investigate whether BBG suppresses the production
of pro-inflammatory cytokines in LPS-induced BV2 cells, RT-qPCR was
used. The results revealed that LPS markedly increased the mRNA and
secretion levels of IL-1β, IL-6 and TNF-α. Co-treatment with BBG
significantly decreased the level of these molecules (Fig. 2A-C). Similarly, western blot
analysis revealed that LPS significantly upregulated the expression
of iNOS and COX-2 proteins in BV2 cells, and that this inflammatory
effect was suppressed by BBG (Fig.
2D). Additionally, compared with the control group, the TLR4
protein level was significantly increased in LPS-induced BV2 cells,
while BBG treatment significantly downregulated the protein level
of TLR4. Furthermore, the TLR4 protein level was not significantly
altered following the addition of ATP, a P2X7R agonist (Fig. 2E). BBG markedly inhibited the
protein expression of P2X7R in LPS-induced BV2 cells. By contrast,
no significant change was detected after the addition of ATP
(Fig. 2E).
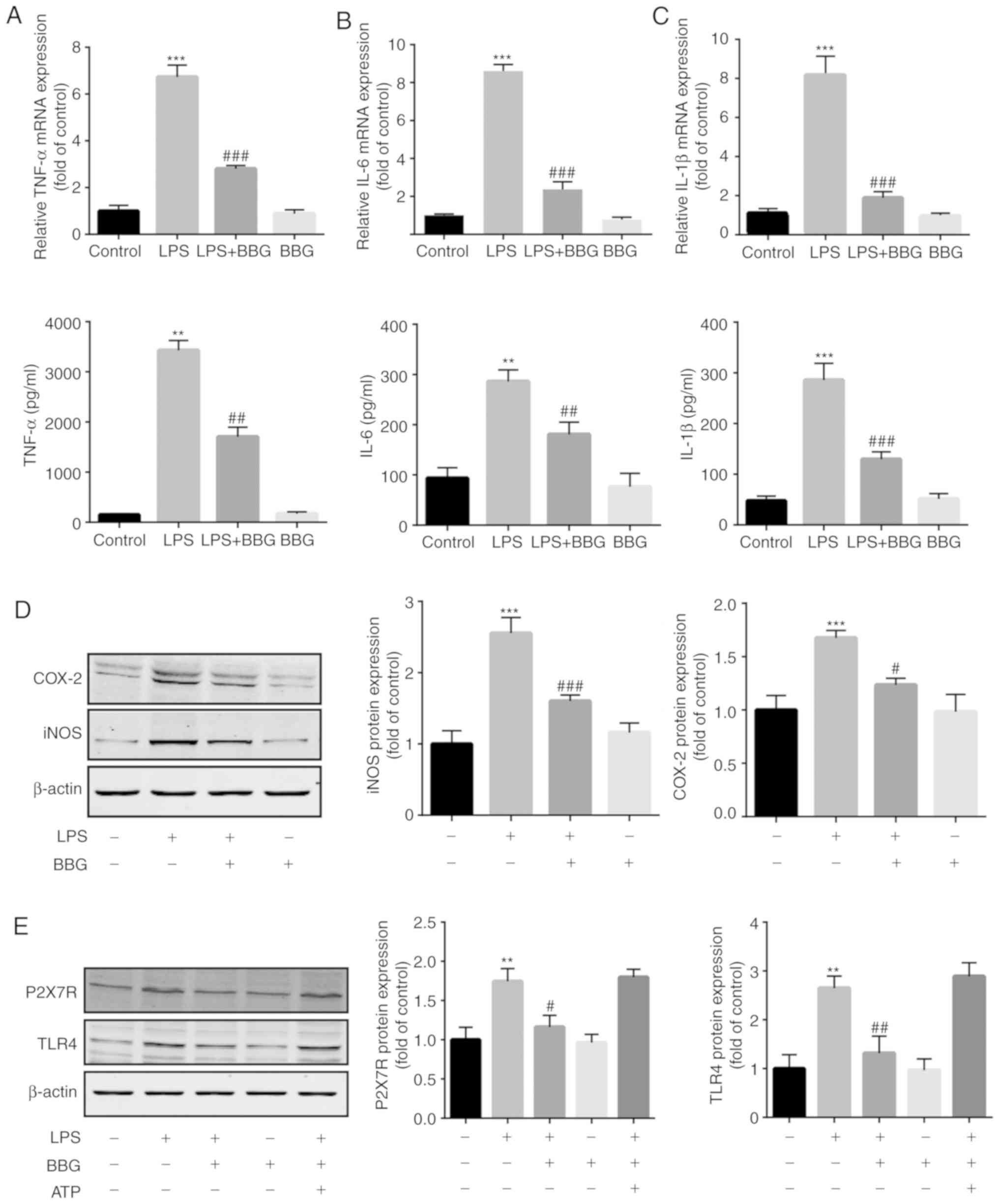 | Figure 2Effect of BBG on pro-inflammatory
cytokines and the expression of iNOS and COX-2 in LPS-stimulated
BV2 cells. BV-2 cells were treated with 1 µM BBG in the presence or
absence of LPS (1 µg/ml) for 24 h. (A-C) The level of (A) TNF-α,
(B) IL-1β and (C) IL-6 was measured by reverse
transcription-quantitative polymerase chain reaction and ELISA. (D)
The protein levels of iNOS and COX-2 were analyzed by western blot
analysis. (E) The protein levels of TLR4 and P2X7R were analyzed by
western blot analysis. Cells were incubated with LPS for 22 h in
the presence or absence of ATP (0.1 mM) for an additional 2 h. Data
are expressed as the mean ± standard error of the mean of three
independent experiments. **P<0.01 and
***P<0.001 vs. control group; #P<0.05,
##P<0.01 and ###P<0.001 vs. LPS-induced
group. iNOS, inducible nitric oxide synthase; COX-2,
cyclooxygenase-2; LPS, lipopolysaccharide; TLR-4, toll-like
receptor-4; BBG, brilliant blue G; TNF-α, tumour necrosis factor-α;
IL, interleukin; P2X7R, P2x purinoceptor 7 receptor. |
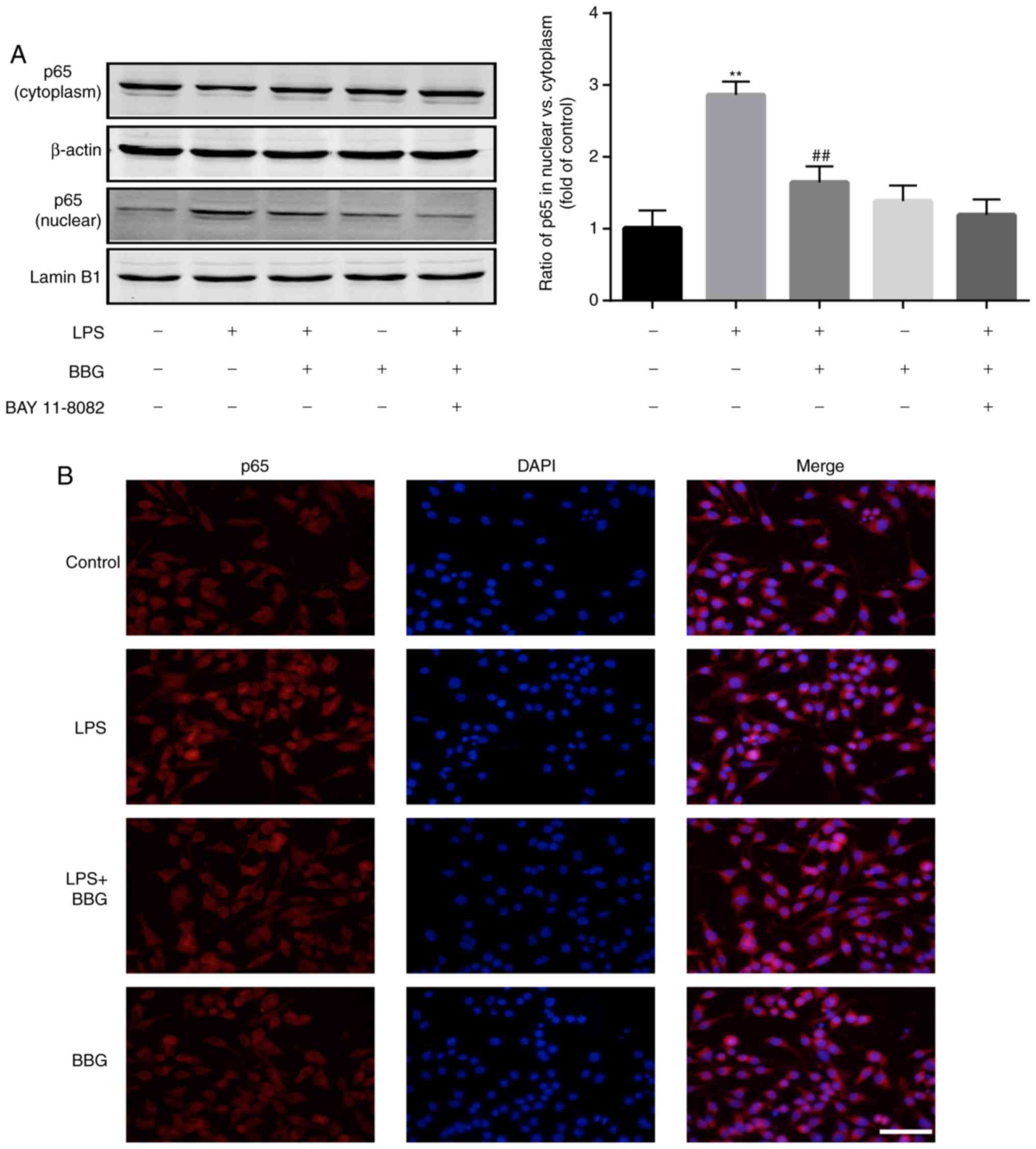
BBG inhibits NF-κB activation in
LPS-induced BV2 cells
As NF-κB is a vital transcription factor
contributing to the expression of iNOS and inflammatory responses,
the present study investigated whether NF-κB was activated or not
under treatment with BBG.
The cytosolic and nuclear proteins were extracted,
and western blot analysis indicated that the protein level of p65
was increased in the nuclear fraction of the LPS-treated group;
when the cells were additionally treated with BBG, the nuclear
translocation was inhibited. However, pretreatment with the NF-κB
inhibitor BAY 11-7082 for 1 h further significantly suppressed
NF-κB p65 nuclear translocation (Fig.
3A). Furthermore, immunofluorescence analysis suggested that
BBG inhibited NF-κB p65 nuclear translocation (Fig. 3B).
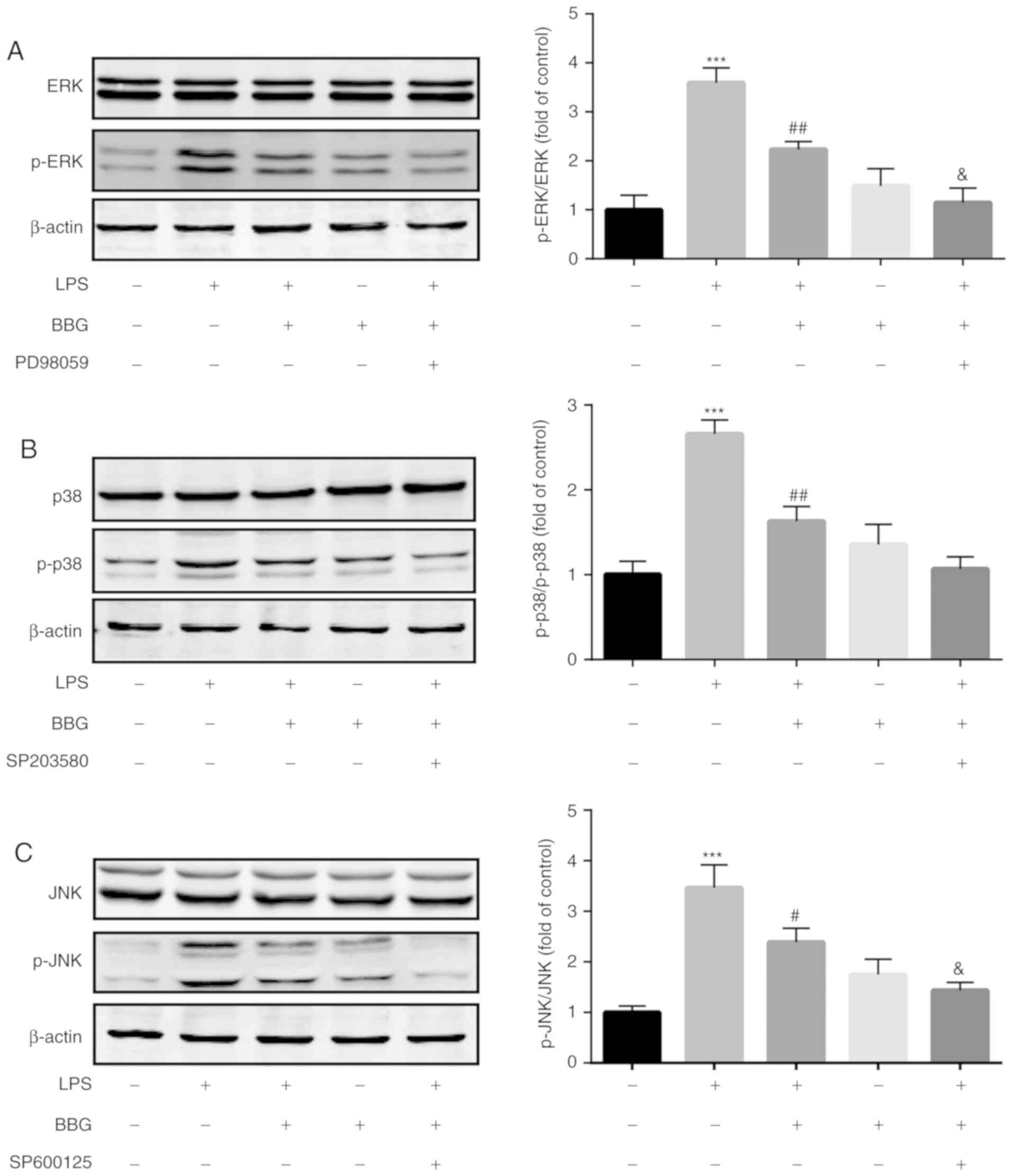
BBG inhibits the phosphorylation of
MAPKs in LPS-induced BV-2 cells
The present study also investigated whether BBG
affects the phosphorylation of MAPKs. As demonstrated in Fig. 4, compared with the control group,
the phosphorylation levels of p38, JNK and ERK1/2 were
significantly upregulated in LPS-treated group. However, the
phosphorylation levels of these proteins were significantly
decreased after the addition of BBG. Besides, co-treatment with
MAPK inhibitors (p38 inhibitor SP203580, JNK inhibitor SP600125 and
ERK inhibitor PD98059) additionally decreased the phosphorylation
levels of p38, JNK and ERK.
Discussion
Neuroinflammation is considered to be an important
contributor to the development and progression of several
neurodegenerative diseases, including PD and AD (19,20).
Over-activated microglial cells trigger neurotoxic effects due to
the increase in inflammatory mediators [NO and prostaglandin E2
(PGE2)] and various toxic cytokines (21). Therefore, an effective way to delay
the progression of various neurodegenerative diseases is through
suppressing the secretion of pro-inflammatory mediators by
over-activated microglial cells (22). The present study identified that
BBG, a selective and non-competitive P2X7R antagonist, notably
lessened the production of inflammatory mediators and the release
of pro-inflammatory cytokines in an LPS-induced inflammation model.
A previous study demonstrated that P2X7R primarily exists in
microglia, astrocytes and neuronal cells in the CNS (23). Concomitantly, P2X7R has been
demonstrated to be involved in inflammatory responses caused by LPS
stimulation, and the activation of P2X7R may directly facilitate
the maturation and release of pro-inflammatory cytokines (24). The present study suggested that
co-treatment with BBG markedly inhibited the mRNA expression of
IL-1β, IL-6 and TNF-α in LPS-stimulated microglial cells. In
addition, it has been demonstrated that an excessive production of
iNOS and COX-2 in microglia may aggravate inflammatory disorders
(25). iNOS and COX-2, as
pro-inflammatory enzymes, may promote the generation of PGE2 and
NO, impair respiratory chain complexes I and II, and produce
multiple deleterious reactive molecules (26). The results suggested that
co-treatment with BBG markedly decreased the production of iNOS and
COX-2. These data demonstrated that BBG has anti-neuroinflammatory
properties by attenuating production of the pro-inflammatory
cytokines by LPS-induced microglial cells.
TLR4 has been demonstrated to be involved in
neuroinflammation, and is upregulated in response to nerve injury.
TLR4 recognizes exogenous ligands such as LPS, leading to the
activation of TLR4 and promoting the production of a variety of
inflammatory cytokines. Previous data suggest that inhibiting or
knocking out TLR4 effectively reverses neuroinflammation or
neuropathic pain (27). Consistent
with a previous study (27), the
present results suggested that BBG treatment decreased the
LPS-induced elevation of the TLR4 protein level. By contrast, no
significance change was observed following ATP treatment.
NF-κB is a dimeric transcription factor that
regulates the expression of multiple genes and serves a critical
role in cellular signaling pathways against immunity, inflammation
and cell death (28). It has been
suggested that P2X7R may activate the NF-κB signaling pathway, and
it has a close association with inflammatory diseases (29). A previous study suggested that P2X7R
regulates the matrix metalloproteinase 13 (MMP-13) and NF-κB
pathways in cartilage tissue and mediates OA-induced pain and
inflammation. Furthermore, NF-κB signaling inhibitors may suppress
the expression of P2X7R and MMP-13, and relieve OA-induced pain and
inflammation (30). Similar to
these data, A438079, a P2X7R antagonist, decreases NF-κB
activation, intensifies the caspase-1 expression in lamina propria
immune cells and suppresses pro-inflammatory cytokine production in
colon tissues, in addition to relieving murine colitis (29). In the present study, the results
suggested that BBG significantly inhibited the nuclear
translocation of NF-κB p65, and this inhibitory effect was
additionally enhanced following pretreatment with the NF-κB
inhibitor BAY 11-7082. These results suggested that the NF-κB
signaling pathway participates in the regulation of the production
of pro-inflammatory mediators by BBG. MAPKs are serine/threonine
kinases, and regulate the expression of genes including p38 MAPK,
ERK and JNK, which are associated with immune and inflammatory
responses and are upregulated in LPS-stimulated macrophages and
microglia (31). It has been
demonstrated that P2X7R contributes to the phosphorylation of p38
MAPK during intracerebral hemorrhage (32). The activation of p38 MAPK further
leads to the production of active caspase-3, and ultimately to cell
death (33). Furthermore, the
inhibition of P2X7R and MAPKs provided significant neuroprotection
in a subarachnoid hemorrhage or intracerebral hemorrhage model
(34). Previous studies have also
demonstrated the marked activation of p38 MAPK and P2X7R in PD
models (35,36). In addition, p38 MAPK signaling
pathway inhibitors or P2X7R antagonists provide a significant
neuroprotection effect against damage to the substantia nigra and
striatum in PD models (35). In the
present study, BBG significantly decreased the levels of
phosphorylated p38, ERK and JNK in LPS-induced BV2 cells, and
co-treatment with MAPK inhibitors (SB203580, SP600125 and PD98059)
further suppressed the levels of these kinases, suggesting that the
anti-inflammatory effect may be attributed to the suppression of
the MAPK signaling pathway.
In conclusion, the results of the present study
demonstrated that BBG significantly inhabited the inflammatory
response in LPS-induced BV-2 cells. The anti-inflammatory mechanism
of BBG may be mediated via the suppression of the activation of the
MAPK/NF-κB signaling pathways. These data indicated that BBG may be
an effective agent for the treatment of neuroinflammation in
neurodegenerative diseases.
Acknowledgements
Not applicable.
Funding
Not applicable.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JX conceived and designed the experiments. WJ, FH
and WWW performed the experiments. WW and FH wrote the manuscript.
WW and WJ conducted the data analysis. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hanamsagar R and Bilbo SD: Sex differences
in neurodevelopmental and neurodegenerative disorders: Focus on
microglial function and neuroinflammation during development. J
Steroid Biochem Mol Biol. 160:127–133. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Heneka MT, Carson MJ, El Khoury J,
Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T,
Vitorica J, Ransohoff RM, et al: Neuroinflammation in Alzheimer's
disease. Lancet Neurol. 14:388–405. 2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Colonna M and Butovsky O: Microglia
function in the central nervous system during health and
neurodegeneration. Annu Rev Immunol. 35:441–468. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Biber K, Moller T, Boddeke E and Prinz M:
Central nervous system myeloid cells as drug targets: current
status and translational challenges. Nat Rev Drug Discov.
15:110–124. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Liu RP, Zou M, Wang JY, Zhu JJ, Lai JM,
Zhou LL, Chen SF, Zhang X and Zhu JH: Paroxetine ameliorates
lipopolysaccharide-induced microglia activation via differential
regulation of MAPK signaling. J Neuroinflammation.
11(47)2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yoshida Y, Yoshimi R, Yoshii H, Kim D, Dey
A, Xiong H, Munasinghe J, Yazawa I, O’Donovan MJ, Maximova OA, et
al: The transcription factor IRF8 activates integrin-mediated TGF-β
signaling and promotes neuroinflammation. Immunity. 40:187–198.
2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wilms H, Sievers J, Rickert U,
Rostami-Yazdi M, Mrowietz U and Lucius R: Dimethylfumarate inhibits
microglial and astrocytic inflammation by suppressing the synthesis
of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model
of brain inflammation. J Neuroinflammation. 7(30)2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Sarkar FH, Li Y, Wang Z and Kong D:
NF-kappaB signaling pathway and its therapeutic implications in
human diseases. Int Rev Immunol. 27:293–319. 2008.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Lai JL, Liu YH, Liu C, Qi MP, Liu RN, Zhu
XF, Zhou QG, Chen YY, Guo AZ and Hu CM: Indirubin inhibits
LPS-induced inflammation via TLR4 abrogation mediated by the NF-κB
and MAPK signaling pathways. Inflammation. 40:1–12. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Chew J, Biswas S, Shreeram S, Humaidi M,
Wong ET, Dhillion MK, Teo H, Hazra A, Fang CC, López-Collazo E, et
al: WIP1 phosphatase is a negative regulator of NF-kappaB
signalling. Nat Cell Biol. 11:659–666. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
11
|
Zhao H, Wang SL, Qian L, Jin JL, Li H, Xu
Y and Zhu XL: Diammonium glycyrrhizinate attenuates
Aβ(1-42)-induced neuroinflammation and regulates MAPK and NF-κB
pathways in vitro and in vivo. CNS Neurosci Ther. 19:117–124.
2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Leyns CEG, Ulrich JD, Finn MB, Stewart FR,
Koscal LJ, Serrano JR, Robinson GO, Anderson E, Colonna M and
Holtzman DM: TREM2 deficiency attenuates neuroinflammation and
protects against neurodegeneration in a mouse model of tauopathy.
Proc Natl Acad Sci USA. 114:11524–11529. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Volonte C, Apolloni S, Skaper SD and
Burnstock G: P2X7 receptors: channels, pores and more. CNS Neurol
Disord Drug Targets. 11:705–721. 2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Chen S, Ma Q, Krafft PR, Chen Y, Tang J,
Zhang J and Zhang JH: P2X7 receptor antagonism inhibits p38
mitogen-activated protein kinase activation and ameliorates
neuronal apoptosis after subarachnoid hemorrhage in rats. Crit Care
Med. 41:e466–e474. 2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Skaper SD, Debetto P and Giusti P: The
P2X7 purinergic receptor: from physiology to neurological
disorders. FASEB J. 24:337–345. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhao H, Pan P, Yang Y, Ge H, Chen W, Qu J,
Shi J, Cui G, Liu X, Feng H and Chen Y: Endogenous hydrogen
sulphide attenuates NLRP3 inflammasome-mediated neuroinflammation
by suppressing the P2X7 receptor after intracerebral haemorrhage in
rats. J Neuroinflammation. 14(163)2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Wang XH, Xie X, Luo XG, Shang H and He ZY:
Inhibiting purinergic P2X7 receptors with the antagonist brilliant
blue G is neuroprotective in an intranigral lipopolysaccharide
animal model of Parkinson's disease. Mol Med Rep. 15:768–776.
2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Rahimifard M, Maqbool F, Moeini-Nodeh S,
Niaz K, Abdollahi M, Braidy N, Nabavi SM and Nabavi SF: Targeting
the TLR4 signaling pathway by polyphenols: A novel therapeutic
strategy for neuroinflammation. Ageing Res Rev. 36:11–19.
2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Baima ET, Guzova JA, Mathialagan S, Nagiec
EE, Hardy MM, Song LR, Bonar SL, Weinberg RA, Selness SR, Woodard
SS, et al: Novel insights into the cellular mechanisms of the
anti-inflammatory effects of NF-kappaB essential modulator binding
domain peptides. J Biol Chem. 285:13498–13506. 2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Le W, Rowe D, Xie W, Ortiz I, He Y and
Appel SH: Microglial activation and dopaminergic cell injury: An in
vitro model relevant to Parkinson's disease. J Neurosci.
21:8447–8455. 2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Glass CK, Saijo K, Winner B, Marchetto MC
and Gage FH: Mechanisms underlying inflammation in
neurodegeneration. Cell. 140:918–934. 2010.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Yu Q, Guo Z, Liu X, Ouyang Q, He C,
Burnstock G, Yuan H and Xiang Z: Block of P2X7 receptors could
partly reverse the delayed neuronal death in area CA1 of the
hippocampus after transient global cerebral ischemia. Purinergic
Signal. 9:663–675. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Petes C, Wynick C, Guzzo C, Mehta D, Logan
S, Banfield BW, Basta S, Cooper A and Gee K: IL-27 enhances
LPS-induced IL-1β in human monocytes and murine macrophages. J
Leukoc Biol. 102:83–94. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Cesario A, Rocca B and Rutella S: The
interplay between indoleamine 2,3-dioxygenase 1 (IDO1) and
cyclooxygenase (COX)-2 in chronic inflammation and cancer. Curr Med
Chem. 18:2263–2271. 2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Cai B, Seong KJ, Bae SW, Chun C, Kim WJ
and Jung JY: A synthetic diosgenin primary amine derivative
attenuates LPS-stimulated inflammation via inhibition of NF-kappaB
and JNK MAPK signaling in microglial BV2 cells. Int
Immunopharmacol. 61:204–214. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Stokes JA, Cheung J, Eddinger K, Corr M
and Yaksh TL: Toll-like receptor signaling adapter proteins govern
spread of neuropathic pain and recovery following nerve injury in
male mice. J Neuroinflammation. 10(148)2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Mattson MP, Culmsee C, Yu Z and Camandola
S: Roles of nuclear factor kappaB in neuronal survival and
plasticity. J Neurochem. 74:443–456. 2000.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wan P, Liu X, Xiong Y, Ren Y, Chen J, Lu
N, Guo Y and Bai A: Extracellular ATP mediates inflammatory
responses in colitis via P2 x7 receptor signaling. Sci Rep.
6(19108)2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Hu H, Yang B, Li Y, Zhang S and Li Z:
Blocking of the P2X7 receptor inhibits the activation of the MMP-13
and NF-kappaB pathways in the cartilage tissue of rats with
osteoarthritis. Int J Mol Med. 38:1922–1932. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kim EK and Choi EJ: Pathological roles of
MAPK signaling pathways in human diseases. Biochim Biophys Acta.
1802:396–405. 2010.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Chu K, Yin B, Wang J, Peng G, Liang H, Xu
Z, Du Y, Fang M, Xia Q and Luo B: Inhibition of P2X7 receptor
ameliorates transient global cerebral ischemia/reperfusion injury
via modulating inflammatory responses in the rat hippocampus. J
Neuroinflammation. 9(69)2012.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Papp L, Vizi ES and Sperlagh B: P2X7
receptor mediated phosphorylation of p38MAP kinase in the
hippocampus. Biochem Biophys Res Commun. 355:568–574.
2007.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wen Z, Mei B, Li H, Dou Y, Tian X, Shen M
and Chen G: P2X7 participates in intracerebral hemorrhage-induced
secondary brain injury in rats via MAPKs signaling pathways.
Neurochem Res. 42:2372–2383. 2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Kumar S, Mishra A and Krishnamurthy S:
Purinergic antagonism prevents mitochondrial dysfunction and
behavioral deficits associated with dopaminergic toxicity induced
by 6-OHDA in rats. Neurochem Res. 42:3414–3430. 2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wu F, Wang Z, Gu JH, Ge JB, Liang ZQ and
Qin ZH: p38(MAPK)/p53-Mediated Bax induction contributes to neurons
degeneration in rotenone-induced cellular and rat models of
Parkinson's disease. Neurochem Int. 63:133–140. 2013.PubMed/NCBI View Article : Google Scholar
|