Introduction
Breast cancer (BC) is one of the most malignant and
common types of cancer among women worldwide and the sixth leading
cause of cancer-associated mortality among Chinese women (1). China had 12.2% of global cases and
9.6% of cancer-related deaths of BC in 2012 (1,2). Solid
tumors can occur in the lactiferous ducts, lobules of the mammary
glands and in interstitial tissues of patients with BC (3). Worldwide, cancer of the lactiferous
ducts and lobules accounted for 90% of all BC in 2018 (Korea Breast
Cancer Society; www.kbcs.or.kr). On the molecular
level, BC can be divided into four subtypes: Hormone
receptor-positive BC, human epidermal growth factor receptor 2
(HER2)-positive BC, triple-negative BC (TNBC) and basal-like BC
(4). Among these types of cancer,
the luminal estrogen receptor (ER)-positive, HER2-negative subtype
accounted for ~70% of patients with BC in China in 2012 (1,2).
Endocrine therapies, including aromatase inhibitors,
selective ER downregulators, gonadotropin-releasing hormone and
selective ER modulators, are effective therapeutic strategies
targeting ER and HER2 in the clinical treatment of patients with BC
(5). Although endocrine therapy is
successful in clinical practice, patients can develop resistance to
these therapies. Several important molecular signaling pathways
have been identified to contribute to resistance, including
estrogen-independent activation of the ER and cell-cycle regulation
by cyclin D-CDK4/6, epigenetic pathways, heat shock protein 90 and
an immunogenic pathway including cytotoxic T-lymphocyte associated
protein 4 and programmed cell death 1 ligand 1/2- programmed cell
death 1(6). Notably, the PI3K/Akt/
mTOR pathway is the most common oncogenic pathway with a crucial
role in growth, survival, proliferation and differentiation of
cancer cells (7,8). The PTEN signaling pathway serves as an
upstream regulator of mTOR, an antagonist of PI3K/Akt signaling and
a modulator of numerous cellular processes (9-11).
The Akt, tuberous sclerosis complex 2 and live kinase B1 signaling
pathways have been reported to inhibit mTOR activation in cancer
cells (12-15).
Therefore, BC could be controlled by inhibiting the mTOR
pathway.
Ubiquitin-specific peptidases (USPs) regulate cell
proliferation and apoptosis by ubiquitination and deubiquitination
(16,17). USPs determine the accumulation
levels of proteins through post-translational modifications in
cells by controlling the conjugation and removal of ubiquitin
(18). Additionally, the
dysfunction of USPs leads to oncogenic progression in cells
(19,20). USP10 regulates signaling factors
associated with cell proliferation, apoptosis and cancer
metabolism. Several studies have reported that UPS10 regulates the
PTEN signaling pathway to inhibit cancer cell growth and invasion,
and c-Myc transcription to suppress cancer formation and to affect
cellular sensitivity to DNA damage (21-23).
A recent study reported that USP10 expression was decreased in
hepatocellular carcinoma, leading to poor prognosis (24). Consequently, it may be hypothesized
that the regulation of USP10 expression inhibits the mTOR signaling
pathway and the treatment and prognosis of patients.
Inflammation, a biological response to adverse
physical or chemical stimuli, serves a role in cancer development
and metastasis via the release of proinflammatory cytokines,
including TNF-α, IL-6 and IL-1β, to activate the key transcription
factor NF-κB (25). Plasminogen
activator inhibitor (PAI-1) is a member of the serine protease
inhibitor protein family (26).
PAI-1 knockout mice exhibited low levels of inflammation compared
with wild-type mice (27),
indicating that this protein may serve a role in the inflammatory
response. Furthermore, a previous study indicated that the PAI-1
level was closely associated with BC metastasis (28). Previous studies supported the
hypothesis that chronic inflammation promoted cancer development
(29,30). Certain evidence has indicated that
inflammatory factors such as cyclooxygenase-2 (COX2) and
lipoxygenase (LOX) were upregulated in BC (31-33)
and COX2 in ER-negative BC and TNBC was associated with poor
prognosis (34). COX and LOX
metabolic products serve a role in BC (35). In addition, apoptosis is important
for cancer cell therapy. Previous studies reported that numerous
signaling pathways and molecules regulated BC cell apoptosis,
including osteocyte signaling (36), miRNAs (37,38)
and TNF-related apoptosis-inducing ligand (TRAIL) signaling
(39), indicating that the control
of inflammation and apoptosis was important for BC treatment.
Clostridium difficile toxin B was reported to inhibit the
inflammatory response by suppressing the COX2 level and to activate
apoptosis in BC (40). In addition,
ursolic acid inhibits BC development by activating apoptosis and
suppressing the inflammatory response (41), indicating that inflammation is
negatively associated with BC cell growth (42). However, NF-κB, a key inflammatory
signaling transcription factor, activates anti- and pro-apoptotic
genes (42), indicating that the
regulation of inflammation and apoptosis is complex.
Panax ginseng has been used as a medicinal plant in
China for thousands of years (43).
The ginseng extract is composed of ginsenoside (the ginseng
saponin), acanthosides, senticosides, triterpene saponins,
flavonoid, vitamins, minerals and polysaccharides (44,45).
Ginsenosides are the main ingredients reported to inhibit tumor
metastasis in cells (46).
Additionally, ginseng-derived polysaccharides were previously
believed to exhibit antitumor effects and were isolated as the
antitumor fraction for the first time in 1994(47). Ginseng polysaccharide (GPS) has been
demonstrated to exhibit low toxicity (47). Furthermore, GPS stimulates
nonspecific immune cells and activates natural killer cells and
macrophages to protect the host against foreign antigens and tumor
growth (48-50).
Furthermore, GPS activates macrophages and inhibits tumor
angiogenesis and metastasis (51-53).
Therefore, the current study was performed to investigate the
function of GPS in BC cell proliferation. The inflammatory response
and apoptosis were analyzed. Additionally, the regulatory effect of
signaling pathways associated with the inflammatory response on
GPS-mediated inhibition of BC cell proliferation was assessed. The
findings indicated a novel mechanism by which GPS may inhibit BC
cell proliferation.
Materials and methods
Cell culture and transfection
assays
The human breast adenocarcinoma cell line MDA-MB-231
obtained from the American Type Culture Collection was cultured in
DMEM (Gibco; Thermo Fisher Scientific, Inc.) with glutamine
(Sigma-Aldrich; Merck KGaA) and supplemented with 10% FBS and 100
µg/ml penicillin and streptomycin (Gibco; Thermo Fisher Scientific,
Inc.) at 37˚C. Ikaros family zing finger protein 1 (IKZF1) or p65
cDNAs were synthesized by Sangon Biotech Co., Ltd. and pcDNA3.1 (+)
(Invitrogen; Thermo Fisher Scientific, Inc.) was used for
constructing overexpression (OX) vectors. Subsequently, 2 µg of
pcDNA3.1 (+) empty vector, IKZF1 OX and p65 OX plasmids were
transfected (seeding density, 1x106 cells) on day 0
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) and Opti-MEM I Reduced Serum Medium (Gibco;
Thermo Fisher Scientific, Inc.), according to the manufacturer's
protocol. On day 1, 24 h post-transfection, the cells were confluent
and the IKZF1 or p65 OX solutions were replaced with DMEM with
glutamine supplemented with 10% FBS and 100 µg/ml penicillin and
streptomycin. These transfected MDA-MB-231 cells were used for the
subsequent experiments.
Western blotting
MDA-MB-231 cells treated with 100 µM or 200 µM GPS
dissolved in ddH2O (Xi'an Virgin Biotechnology Co.,
Ltd.) for 24 h at 37˚C or transfected with IKZF1 or p65 OX plasmids
for 24 h following GPS treatment were harvested in an ice-cold
lysis solution (7M urea, 2M thiourea, 2% CHAPS, 40 mM Tris base, 40
mM dithiothreitol and 1% protease inhibitor) to obtain whole-cell
extracts. Cells in the control group were treated with equal
amounts of ddH2O. The protein concentration was measured
by using BCA protein assay kit (Cell Signaling Technology, Inc.).
Total proteins from each sample (20 µg/lane) were separated using
SDS-PAGE (10% gel) and transferred onto Immobilon-P transfer
membranes (Merck KGaA). The membranes were incubated in 1X TBS
containing 5% skim milk and 0.05% Tween-20 for 1-2 h at room
temperature and subsequently incubated with primary antibodies at
4˚C overnight. The following primary antibodies were used:
Anti-vimentin (cat. no. ab193555; 1:1,000; Abcam), anti-E-cadherin
(cat. no. ab194982; 1:1,000; Abcam), anti-p53 (cat. no. ab32389;
1:2,000; Abcam), anti-cleaved (c)-Caspase-3 (cat. no. ab2302;
1:2,000; Abcam), anti-Caspase-3 (cat. no. ab13847; 1:2,000; Abcam),
anti-PAI-1 (cat. no. ab66705; 1:2,000; Abcam), anti-TNF-α (cat. no.
ab1793; 1:2,000; Abcam), anti-phosphorylated p-JNK (Thr183/Tyr185;
cat. no. 4668; 1:1,000; Cell Signaling Technology, Inc.), anti-JNK1
+ JNK2 + JNK3 (cat. no. ab179461; 1:1,000; Abcam), anti-p-Akt
(Ser473; cat. no. 4060; 1:2,000; Cell Signaling Technology, Inc.),
anti-Akt (cat. no. 4691; 1:1,000; Cell Signaling Technology, Inc.),
IKZF1 (cat. no. H-100; 1:2,000; Santa Cruz Biotechnology, Inc.),
IκBα (cat. no. ab32518; 1:2,000, Abcam), anti-p-IκBα (cat. no.
sc-8404; 1:500; Santa Cruz Biotechnology, Inc.), anti-NF-κB p50
(cat. no. ab109752; 1:2,000; Abcam), anti-NF-κB p65 (cat. no.
ab16502; 1:2,000; Abcam) and anti-GAPDH (cat. no. ab8245; 1:2,000;
Abcam). The membranes were washed twice with 1X PBS and incubated
with anti-mouse or anti-rabbit horseradish peroxidase-conjugated
secondary antibodies (cat. nos. 7074 and 7076; 1:2,000, Cell
Signaling Technology) for the corresponding primary antibodies
synthesized from mouse or rabbit secondary antibodies for 1 h at
room temperature. Reaction products were visualized using an ECL
Western Blotting Detection system (GE Healthcare). Quantification
of relative band densities was performed by scanning densitometry
using ImageJ software (version 2; National Institute of
Health).
Cell proliferation analysis
MDA-MB-231 cells were plated in a volume of 150 µl
at a density of 2x103 cells/well into 96-well plates to
determine the cell proliferation rate. Cell proliferation ability
was analyzed following treatment with 50 or 100 µM GPS dissolved in
ddH2O (Xi'an Virgin Biotechnology Co., Ltd.) for 24, 48
and 72 h at 37˚C. Control cells were treated with equal volumes of
ddH2O. Cell proliferation in the IKZF1 OX and p65
OX-transfected cells was analyzed using a Cell Counting Kit-8
(Dojindo Molecular Technologies, Inc.), following a previously
published method (54).
Chromatin immunoprecipitation (ChIP)
assay
The ChIP assay was performed on MDA-MB-231 cells
using a chromatin immunoprecipitation assay kit (cat. no. 17-295;
EMD Millipore), according to the manufacturer's protocol. An
anti-NF-κB p65 antibody (~1 µg; cat. no. ab16502; 1:2,000; Abcam)
was used for immunoprecipitation and no-antibody IP was used as the
negative control. Following immunoprecipitation, a wash buffer
[0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl (pH 8.1) and
150 mM NaCl] was used to wash the precipitates. The DNA
immunoprecipitated using the antibodies was compared with the DNA
precipitated without the addition of antibodies using quantitative
PCR (qPCR). A SYBR-Green Master Mix (Bio-Rad Laboratories, Inc.)
was used to perform the qPCR on an Illumina Eco 3.0 (Illumina,
Inc.). The following thermocycling conditions were used: An initial
denaturation at 95˚C for 3 min; 40 cycles of denaturation for 30
sec at 95˚C, annealing for 30 sec at 58˚C and extension at 72˚C for
30 sec; followed by a final extension at 72˚C for 5 min. The Ct
value of each ChIP DNA fraction was normalized to the input DNA
fraction Ct value for the same qPCR Assay to account for chromatin
sample preparation differences using the 2-ΔΔCq method
(50) and GAPDH was used as the
negative control. The 1.5 kb of IKZF1 (NP_001207694.1) promoter
sequences were downloaded from the NCBI database (https://www.ncbi.nlm.nih.gov/) and the three pairs of
primers were designed. The primers used for ChIP-PCR were as
follows: 1 forward, 5'-TCCTGAGTTGCTTCCCACT-3' and reverse,
5'-GGTGTGTCCCAGTAACAT-3'; 2 forward, 5'-GGGCAGAAGGAAAAGTGTCA-3' and
reverse, 5'-CCAAAGGAATGTGAGCTCGT-3'; 3 forward
5'-GACCACCCCTCACATTCAAC-3' and reverse, 5'-TGGCAGTTGAGAATCAGTGG-3';
and GAPDH forward, 5'-GACCTGCCGTCTAGAAAAAC-3' and reverse,
5'-CTGTAGCCAAATTCGTTGTC-3'.
Electrophoretic mobility shift assay
(EMSA)
p65 open reading frame sequences were synthesized
and subcloned into T-easy vectors (Promega Corporation) and moved
to XhoI and EcoRI restriction enzyme sites of pET28a
(+) expression vectors to produce p65 recombinant proteins. The
resulting pET28a-p65 plasmids were used for the transformation of
Escherichia coli (E. coli) strain BL21 DE3 (Tiangen
Biotech, Co., Ltd.). Recombinant His-p65 proteins were harvested
following 4 h of 0.5 mM isopropyl β-D-1-thiogalactopyranoside
(Sigma-Aldrich; Merck KGaA) treatment at 30˚C by centrifugation
12,000 rpm at 4˚C for 30 min. E. coli cells expressing
His-p65 were suspended in 1x PBS solution (Tiangen Biotech, Co.,
Ltd.) and lysed by sonication (130-150 V; amplitude 40 µM) at 4˚C
for 5 min. Crude extracts were purified by adding 1 ml of Mag-Beads
His-Tag (Sangon Biotech, Co., Ltd.). The beads and crude extract
were incubated overnight in a rotating instrument (BaoDragon, Inc.)
at 4˚C and the beads washed with 1x PBS solution 5 times at 4˚C.
Proteins were eluted by adding 100 mM imidazole (Sigma-Aldrich;
Merck KGaA) and protein concentrations were measured using a BCA
kit (cat. no. BCA1; Merck KGaA), according to the manufacturer's
protocol. Protein were further dialyzed by removing imidazole in 1x
PBS solution using dialysis tubing (Sangon Biotech, Co., Ltd.). The
recombinant protein was dissolved in 1x PBS (Tiangen Biotech, Co.,
Ltd.). For EMSA, a standard binding reaction was performed in a
total volume of 20 µl by incubating 1 µg of purified protein with
40,000 cpm of a 32P-labeled DNA probe (PCR fragments
amplified in the aforementioned ChIP assay) and 1 µg of poly dI-dC,
which blocked the nonspecific binding of the protein to probe DNA
in the reaction buffer [25 mM HEPES-KOH (pH 7.5), 100 mM KCl, 0.1
mM EDTA, 10% (v/v) glycerol and 1mM DTT] at room temperature for 30
min. The binding reaction products were resolved on an 8%
polyacrylamide gel run in 0.5X TBE buffer and bands were detected
using and x-ray film (Sangon Biotech, Co., Ltd.) by detecting the
32P radiation signal.
Statistical analysis
Statistical analysis was performed with a Prism
software package (version no. 5.0; GraphPad Software, Inc.). Data
are presented as mean ± standard error. Comparisons between two
groups and the determination of statistical significance was
performed using the Student's t-test. Comparisons between more than
two groups were performed using one-way ANOVA, followed by
Bonferroni's multiple comparisons test. P<0.05 was considered to
indicate a statistically significant difference.
Results
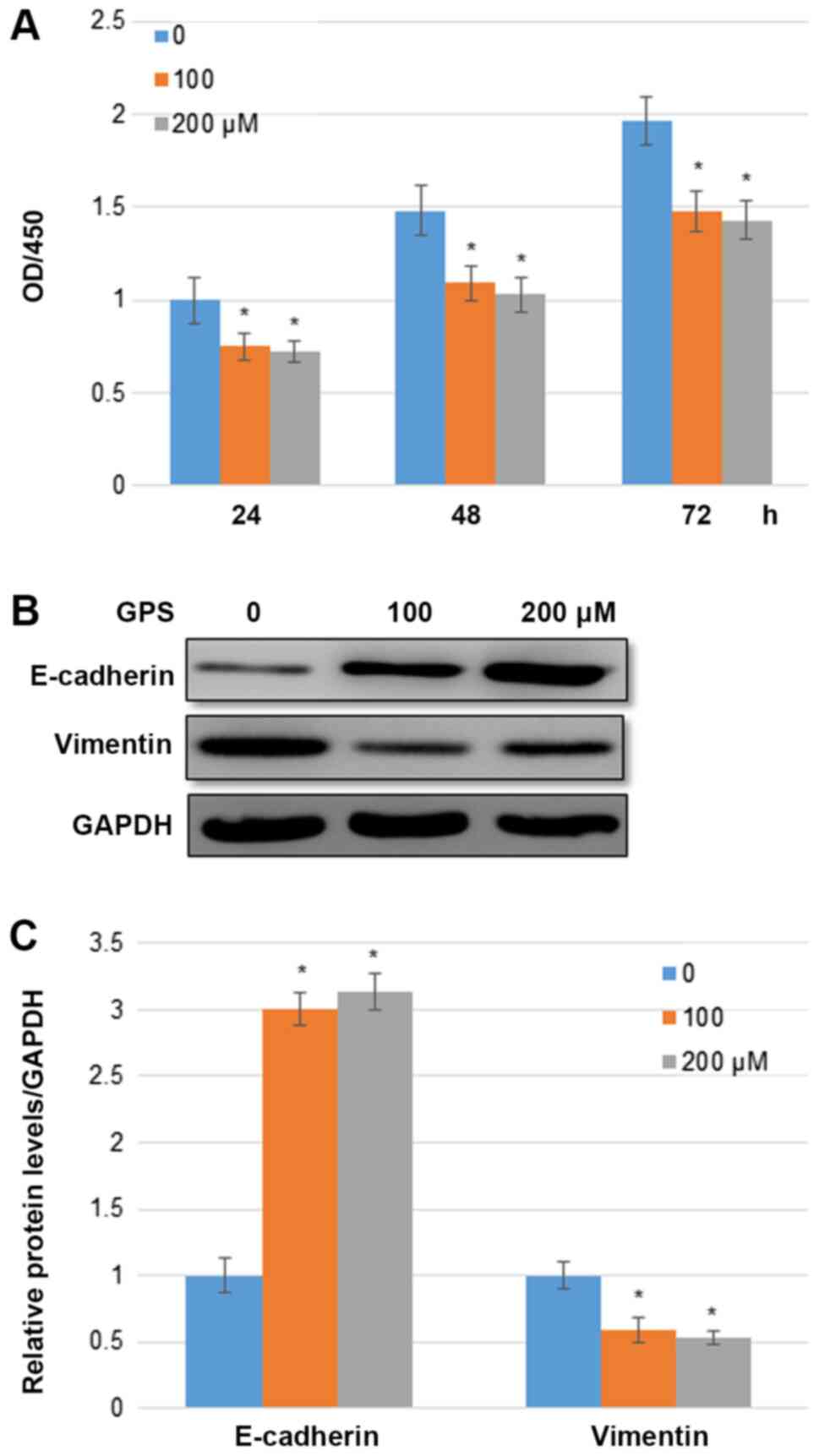
GPS treatment inhibits the
proliferation of MDA-MB-231 cells
A BC cell line MDA-MB-231 was used in the current
study to analyze the role of GPS in BC cells. Compared with the
control group, 100 and 200 µM GPS significantly inhibited
MDA-MB-231 cell proliferation following 24, 48 and 72 h of
treatment. The changes in the cell proliferation rate following
treatment with 100 and 200 µM GPS had fold changes in OD values of
0.75 and 0.72, respectively, after 24 h; 0.699 and 0.682,
respectively, after 48 h; and 0.668 and 0.661, respectively, after
72 h compared with the control group (Fig. 1A). Treatment with 100 µM GPS
significantly enhanced the E-cadherin level; however, the vimentin
levels was reduced compared with the control group. The percentage
changes in the E-cadherin level following treatment with 100 and
200 µM GPS were ~2.993 and 3.113, respectively. The percentage
changes in the vimentin level following treatment with 100 and 200
µM GPS were ~0.562 and 0.529, respectively, compared with the
control group (Fig. 1B and C).
GPS treatment activates the
inflammatory response and apoptosis in MDA-MB-231 cells
As discussed above, GPS treatment inhibited
MDA-MB-231 cell proliferation. Therefore, the present study
subsequently examined the inflammatory and apoptotic response in
MDA-MB-231 cells after 100 µM GPS treatment. Apoptotic markers,
including the c-Caspase-3/Caspase-3 ratio and p53 expression level,
and inflammatory response markers, including PAI-1 and TNF-α, were
detected in cells with and without GPS treatment. The western
blotting results demonstrated that the GPS treatment enhanced the
protein levels of p53, c-Caspase-3/Caspase-3, TNF-α and PAI-1 after
stimulation for 24 h stimulation, while the level of Caspase-3
remained unchanged. The fold-changes in c-Caspase-3/Caspase-3, p53,
PAI-1 and TNF-α after GPS treatment was 2.928, 2.194, 1.889 and
2.643, respectively, compared with the control group (Fig. 2). These results indicated that GPS
treatment activated the inflammatory response and apoptosis in
MDA-MB-231 cells.
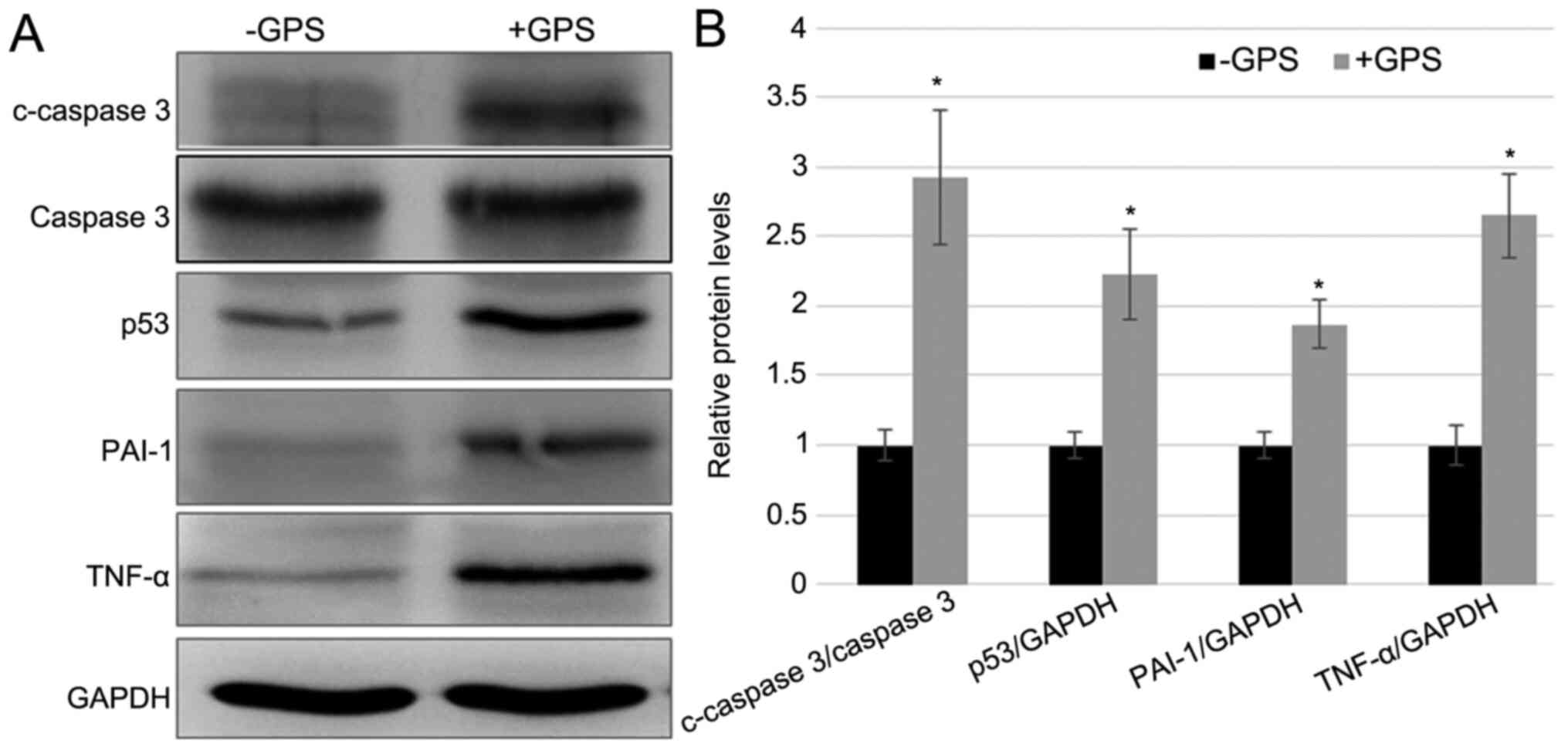 | Figure 2GPS treatment induced the expression
of inflammatory and apoptosis markers in MDA-MB-231 cells. (A)
Western blotting was performed to analyze the levels of apoptotic
markers, including the c-Caspase-3/Caspase-3 ratio and p53, and
inflammatory response markers, including PAI-1 and TNF-α. GAPDH was
used as the loading control. (B) The relative band densities shown
were calculated using the ratios of c-Caspase-3/Caspase-3,
p53/GAPDH, PAI-1/GAPDH and TNF-α/GAPDH levels. Data are presented
as mean ± standard error. Experiments were performed in triplicate.
*P<0.05 vs. the -GPS group. GPS, ginseng
polysaccharide; PAI-1, plasminogen activator inhibitor 1; TNF-α,
tumor necrosis factor-α; c, cleaved; p, phosphorylated; +GPS, cells
treated with GPS; -GPS, cells untreated with GPS. |
GPS treatment activates Akt, JNK and
IκBα
To explore whether GPS treatment influenced the
phosphorylation of Akt and JNK, western blotting was performed to
evaluate the levels of total (t)-Akt and t-JNK, and p-AKT and
p-JNK. The results indicated that GPS treatment enhanced p-AKT and
p-JNK levels without affecting the t-AKT and t-JNK levels (Fig. 3A and B). Since, as demonstrated above, GPS
treatment activated the inflammatory response by the induction of
PAI-1 and TNF-α, the phosphorylation of IκBα, an NF-κB signaling
regulator, was examined. Western blotting results demonstrated that
GPS activated the phosphorylation of IκB-α without affecting the
t-IκB-α level. The fold changes in p-Akt/t-Akt, p-JNK/t-JNK and
p-IκB-α/ t-IκB-α ratios after the GPS treatment were 1.758, 1.638
and 1.953, respectively, compared with the control group (Fig. 3A and B). These results indicated that GPS
supplementation activated the phosphorylation of AKT, JNK and
IκB-α.
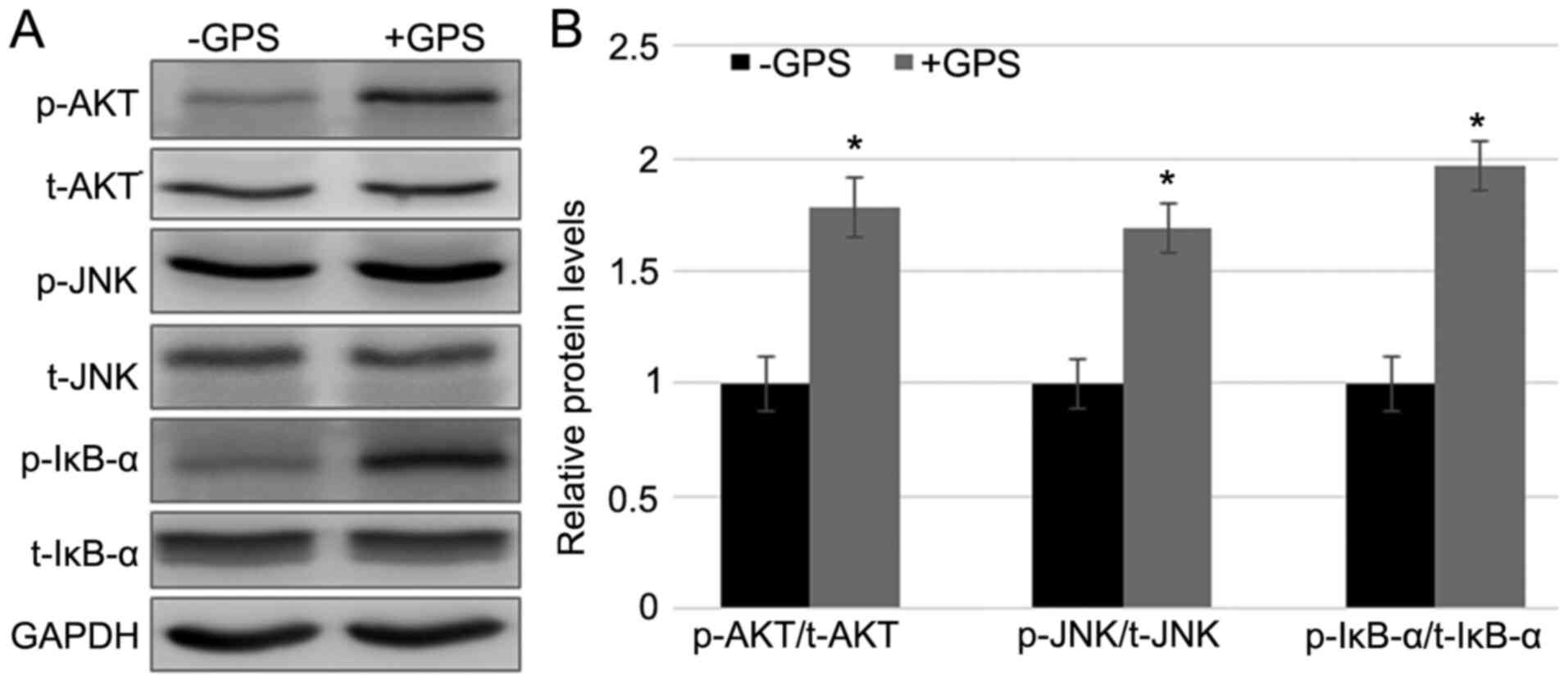 | Figure 3GPS treatment activates Akt, JNK and
IκB-α in MDA-MB-231 cells. (A) Western blotting was performed to
analyze the t- and p-Akt, JNK and IκB-α levels. GAPDH was used as
the loading control. (B) Relative band densities were calculated.
Data are presented as mean ± standard error. Experiments were
performed in triplicate. *P<0.05 vs. the -GPS group.
GPS, ginseng polysaccharide; Akt, protein kinase B; JNK, c-Jun
N-terminal kinase; IκB-α, inhibitor κ B-α; t, total; p,
phosphorylated; +GPS, cells treated with GPS; -GPS, cells untreated
with GPS. |
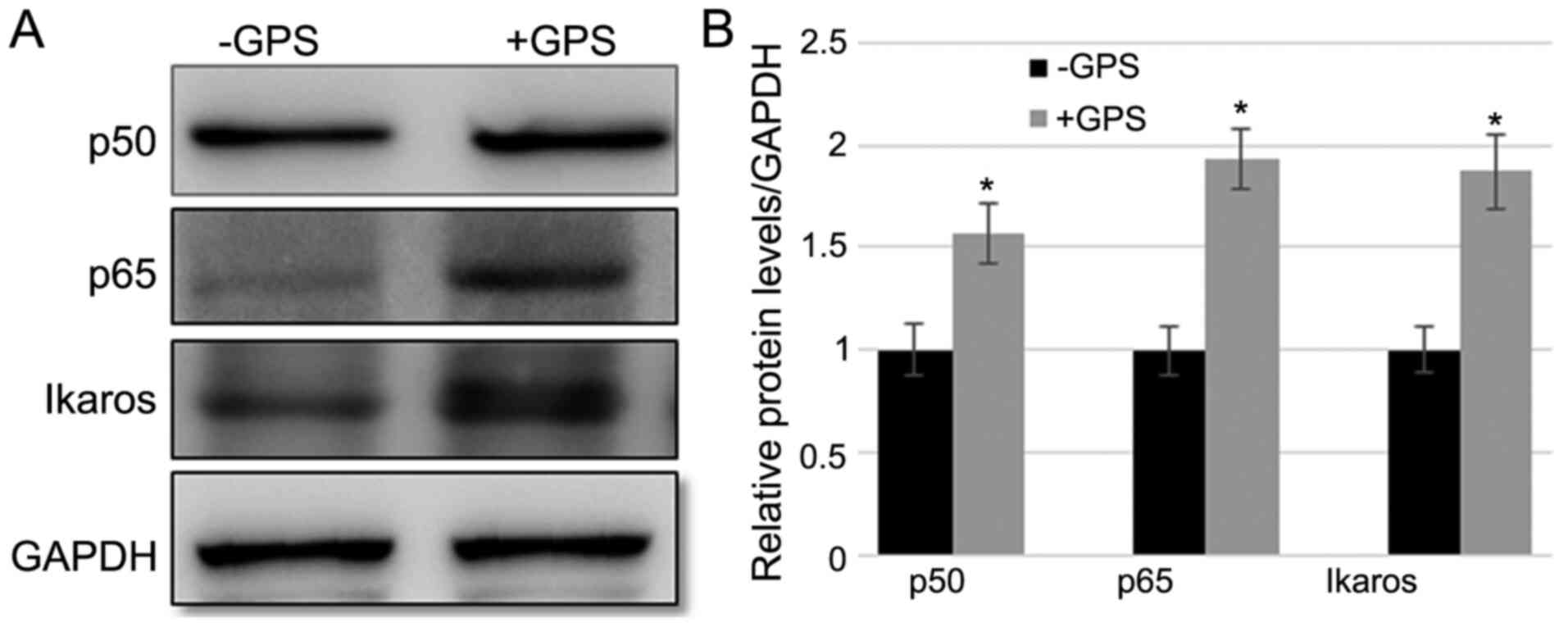
GPS treatment leads to the
accumulation of p65, p50 and IKZF1 in MDA-MB-231 cells
As demonstrated above, GPS treatment increased the
levels of p-IκB-α, a key NF-κB signaling regulator. Subsequently,
the changes in the p65 level following GPS treatment were analyzed.
Western blotting indicated that GPS treatment led to the
accumulation of p65 and p50 proteins in MDA-MB-231 cells, compared
with the control group (Fig. 4A and
B). Furthermore, the level of IKZF1
was determined. The data indicated that GPS treatment enhanced the
IKZF1 expression level compared with the control group (Fig. 4A and B). The fold changes in p50, p65 and IKZF1
after GPS treatment were 1.544, 1.909 and 1.859, respectively,
compared with the control group.
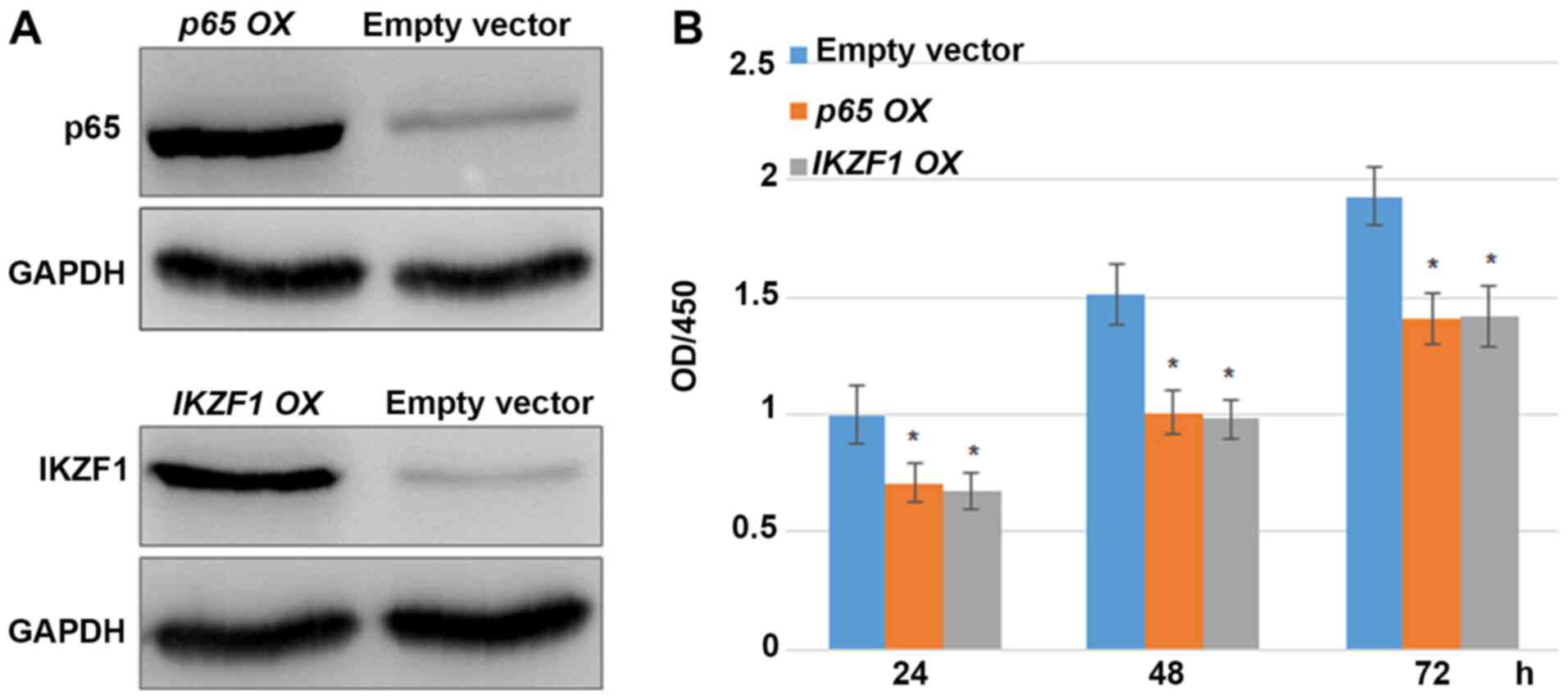
p65 or IKZF1 overexpression inhibits
the proliferation of MDA-MB-231 cells
p65 and IKZF1 were overexpressed in MDA-MB-231 cells
to analyze whether p65 and IKZF1 had an influence on MDA-MB-231
cell proliferation. Western blotting results indicated that p65 and
IKZF1 levels were markedly higher in the OX groups compared with
the empty vector control group (Fig.
5A). Furthermore, MDA-MB-231 cell proliferation was examined in
p65- or IKZF1-OX groups. CCK-8 assay results indicated that the
overexpression of p65 or IKZF1 inhibited cell proliferation
compared with the empty vector control (Fig. 5B). The fold changes in the cell
proliferation rate following the overexpression of p65 and IKZF1
were 0.712 and 0.671, respectively, after 24 h; 0.669 and 0.649,
respectively, after 48 h; and 0.731 and 0.736, respectively, after
72 h compared with the control group.
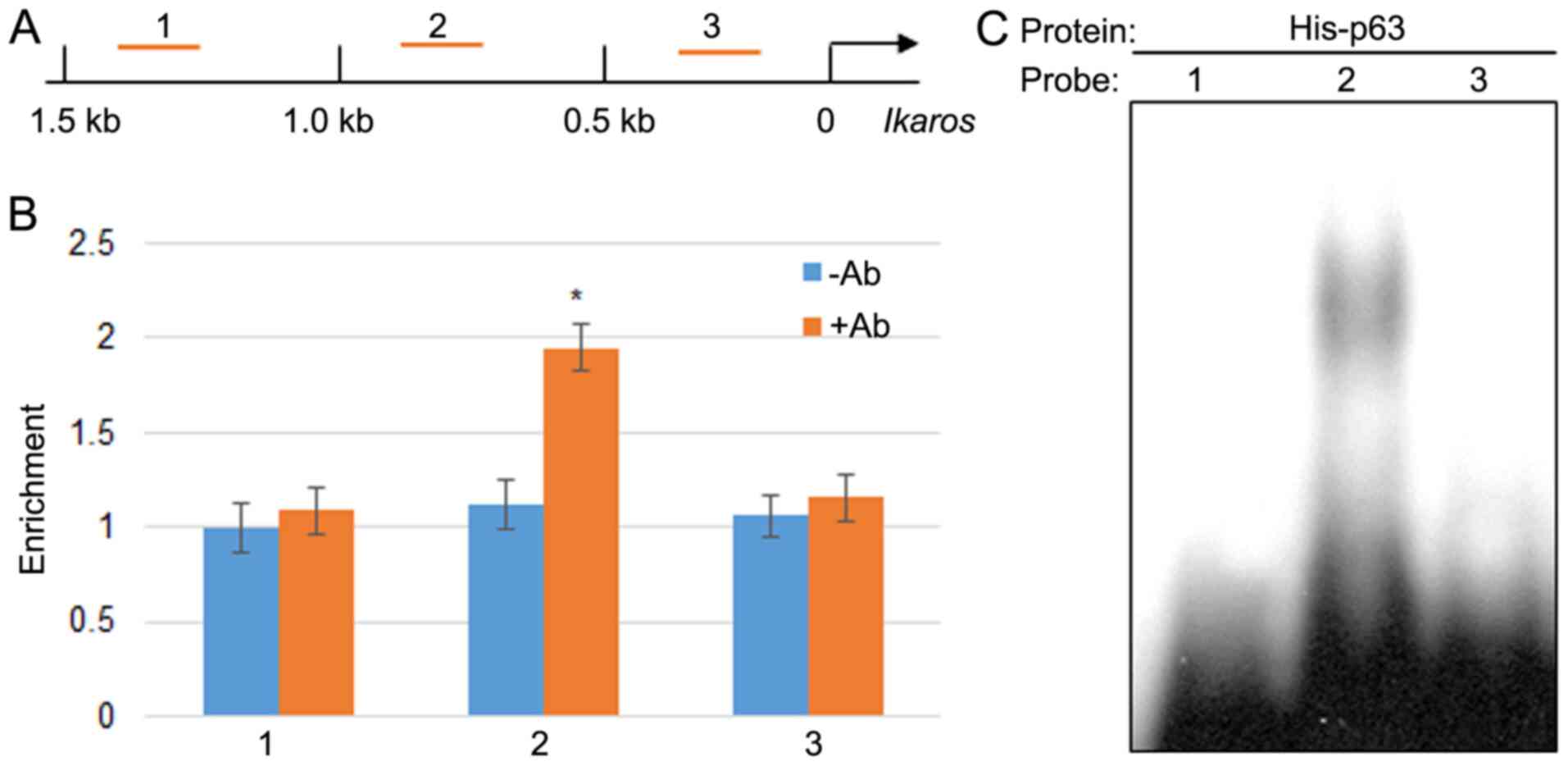
p65 directly binds to the promoter of
IKZF1
As aforementioned, the current results indicated
that GPS promoted the expression of p65 and IKZF1, and inhibited
MDA-MB-231 cell proliferation. Therefore, the possibility of a
direct interaction between p65 and IKZF1 was explored. ChIP assay
using the p65 antibody was performed to determine whether p65 bound
to 1.5 kb of the IKZF1 promoter, which is 1.5 kb upstream from
start codon ATG (Fig. 6A). ChIP-PCR
was performed by scanning 1.5 kb of the promoter using three pairs
of primer with a 0.5 kb distance for each set primer. The results
indicated that p65 may bind to region 2; however, it did not bind
region 1 or 3 within the IKZF1 promoter (Fig. 6B). The fold changes in the
percentage of DNA content by the p65 antibody was 1.734 compared
with the control group without antibody treatment. Further, EMSA
was performed to test the direct binding of p65 to the IKZF1
promoter region. The EMSA results indicated that the p65
recombinant protein bound to region 2 but not regions 1 and 3
(Fig. 6C).
Discussion
In 2012, BC was one of the most common gynecological
malignancies in China (2).
Chemotherapy serves an important role in the treatment of BC;
however, the side effects of chemotherapeutic drugs reduce the
quality of patients' life (1).
Among them, ginsenoside Rh2 and GPS have been reported to exhibit
anticancer effects (46,47). The application of the Chinese
medicine GPS for BC therapy may be a novel treatment approach;
however, the molecular mechanism underlying the inhibition of BC
cells by GPS remains largely unknown.
In the current study, the BC cell line MDA-MB-231
and GPS were used to examine the mechanism underlying the
inhibition of MDA-MB-231 cell proliferation by GPS. GPS is known as
an anticancer molecule (47);
however, it's role in the inhibition of BC cell proliferation
remains unclear. Proliferation is an important step in cancer cell
metastasis in human tissues and is directly associated with disease
severity (47). GPS treatment
induced E-cadherin; however, vimentin levels were suppressed. These
results indicated that GPS inhibited MDA-MB-231 cell viability. The
inflammation and apoptosis status was evaluated by detecting the
expression levels of marker proteins PAI-1 and TNF-α levels to
determine the effect of GPS on the inflammatory response. Western
blotting results indicated that GPS treatment activated
inflammation in MDA-MB-231 cells. These results were further
confirmed by examining the IκB-α phosphorylation level and the
accumulation of two NF-κB components, p65 and p50. The results
suggested that GPS may activate IκB-α to increase the expression of
p65 in the nucleus and, subsequently, activate the expression of
PAI-1 and TNF-α. GPS exhibited inhibitory activity against the p38
MAP kinase pathway, NF-κB and proinflammatory cytokines in
vitro (50). In addition,
ginsenoside Rg3 treatment reduced the COX2 level in mouse skin and
human pro-myelocytic leukemia (HL-60) cells (55), implying a regulatory effect of
ginseng extracts on inflammation in cancer cells. NF-κB is known to
be the upstream regulator of COX2 and Linoleate 9S-lipoxygenase 5
(5LOX), indicating that GPS treatment may downregulate COX2 and
5LOX expression. Previous studies have demonstrated that chronic
inflammation may lead to the cancerous condition (29,30).
Together, these previous studies indicate that GPS may suppress
COX2 and 5LOX to reduce inflammation and BC risk. However, the
results of the current study reported that GPS induced the levels
of inflammation marker proteins at early time points, indicating
that GPS or other ginseng extract-mediated reduction of
inflammation may occur in the later stages of cancer. Further
studies are required to clarify this hypothesis. Furthermore, the
activity of stress-responsive kinases Akt and JNK was examined by
detecting the levels of t- and p-Akt and JNK. JNK functions
downstream of Akt, belongs to the mitogen-activated protein kinase
family and is responsive to cytokines, ultraviolet irradiation,
heat and osmotic stresses (56).
The results of the current study revealed that GPS activated Akt
and JNK, indicating that GPS treatment led to stress in MDA-MB-231
cells. Together, the evidence revealed that GPS activated the
inflammatory response by activating the NF-κB signaling in BC. In
addition, apoptotic markers c-Caspase-3 and p53 were induced by GPS
treatment. Furthermore, GPS treatment inhibited the proliferation
of MDA-MB-231 cells, suggesting that activation of inflammation and
apoptosis by GPS may be associated with the inhibition of
proliferation of BC cells.
IKZF1, a negative regulator of hepatocellular
carcinoma proliferation (57), was
induced by GPS treatment in the current study. In mammalian cells,
NF-κB1 (p50 or its precursor p105), NF-κB2 (p52 or its precursor
p100), Rel (c-Rel), RelA (p65) and RelB are the five members of the
NF-κB/Rel family. The NF-κB/Rel family member contains 300 amino
acids in the N-terminal region termed Rel homolog domain (58). Among them, the p65-p50 heterodimer
is the most abundant active form of NF-κB in numerous cell types
(58). Additionally, p65 and p50
were induced by GPS treatment. Overexpression of p65 or IKZF1 was
revealed to significantly inhibit MDA-MB-231 cell proliferation.
IKZF1 is a transcriptional repressor while p65 is a transcriptional
activator (58); therefore, the
regulatory effect of p65 on the IKZF1 promoter was investigated.
ChIP and EMSA data confirmed that p65 could bind to the IKZF1
promoter, indicating that the GPS-mediated induction of IKZF1 may
be via p65. These data demonstrated that GPS, an anticancer
molecule, inhibited breast cell proliferation possibly by
activating the NF-κB signaling to induce inflammation. GPS
treatment also activated the apoptotic response, which was analyzed
by detecting the c-Caspase-3/Caspase-3 ratio and p53, suggesting
that GPS may activate both apoptosis and inflammation to inhibit
MDA-MB-231 BC cell proliferation. Numerous studies have indicated
that apoptosis was important for the control of BC cell growth
(36-39).
The role of GPS in the activation of apoptosis in BC cells requires
further investigation.
Apoptosis is one of the pathways of cell death;
however, cancer cells survival is promoted by the induction of an
apoptosis resistance mechanism (59). A previous study reported that
aldehyde dehydrogenase family 1 member A3 (ALDH1A3) and sex
determining region Y box 2 (Sox-2) regulated the mechanism of
apoptosis resistance in BC cells (60); however, GPS-mediated regulation of
ALDH1A3 and Sox-2 has not been reported. TNF-α is a mediator of
inflammation and is a member of a family of >20 related proteins
including lymphotoxin-a, CD30 ligand, CD40 ligand, Fas ligand and
TRAIL (61). TRAIL is known to
induce apoptosis in several cell line models; however,
TRAIL-resistant tumors have also been reported and represent a
challenge in cancer therapy (62,63).
Furthermore, microRNA-519a-3p was reported to regulate apoptosis
resistance in a TRAIL-dependent or independent manner in BC
(37); however, the mechanism of
TNF-α-mediated apoptosis resistance requires further investigation
in the context of BC therapy.
Inflammatory factors are upregulated in BC (31-33).
The results of the present study contradicted those of previous
reports, since GPS treatment induced the expression of inflammatory
markers. Considering the present study used only one BC cell line,
MDA-MB-231, which is derived from TNBC, it is important to note
that the effect of GPS on this cell line may not be applicable to
all subtypes of BC. Therefore, further experiments should be
conducted using different BC cell types to verify these results. In
the current study, GPS promoted the expression of pro-inflammatory
and pro-apoptotic markers, and inhibited the proliferation of BC
cells. The results provided a novel molecular mechanism of
GPS-mediated BC cell inhibition and may be used to further explore
the mechanism of BC cell proliferation.
Acknowledgements
Not applicable.
Funding
The current study was supported by the Wenzhou City
Public Welfare Technology Project (grant no. Y20180511).
Availability of data and materials
The datasets used and/or analyzed in the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
HZ and RH designed the experiments. HZ, YY, XZ, TZ
and JX performed the experiments. HZ, YY, BZ, TZ, JX and RH
analyzed data. HZ and RH wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
National Cancer Institute: Cancer topics:
Breast cancer, 2014. Accessed 5 Jan 2015. 2015:1-1.
|
|
2
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Cancer incidence and mortality worldwide:
IARC Cancer Base no. 10. GLOBOCAN 2008. Lyon: International Agency
for Research on Cancer, 2010.
|
|
3
|
Han Z, Wei B, Zheng Y, Yin Y, Li K and Li
S: Breast cancer multi-classification from histopathological images
with structured deep learning model. Sci Rep.
7(4172)2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Prat A, Pineda E, Adamo B, Galván P,
Fernández A, Gaba L, Díez M, Viladot M, Arance A and Muñoz M:
Clinical implications of the intrinsic molecular subtypes of breast
cancer. Breast. 24 (Suppl 2):S26–S35. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sørlie T, Perou CM, Tibshirani R, Aas T,
Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey
SS, et al: Gene expression patterns of breast carcinomas
distinguish tumor subclasses with clinical implications. Proc Natl
Acad Sci USA. 98:10869–10874. 2001.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Wilson S and Chia SK: Treatment algorithms
for hormone receptor positive advanced breast cancer: Applying the
results from recent clinical trials into daily practice-insights,
limitations, and moving forward. Am Soc Clin Oncol Educ Book.
33(e20)2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, 379 metabolism, and disease. Cell. 168:960–976.
2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Dickler MN, Tolaney SM, Rugo HS, Cortés J,
Diéras V, Patt D, Wildiers H, Frenzel M, Koustenis A and Baselga J:
MONARCH 1: Results from phase II study of abemaciclib, a CDK4 and
CDK6 inhibitor, as monotherpay, in patients with HR+/HER2- breast
cancer, after chemotherapy for advanced disease. J Clin Oncol.
34(510)2016.
|
|
9
|
Di Cristofano A, Pesce B, Cordon-Cardo C
and Pandolfi PP: PTEN is essential for embryonic development and
tumour suppression. Nature Genetics. 19:348–355. 1998.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Garcia-Cao I, Song MS, Hobbs RM, Laurent
G, Giorgi C, de Boer VC, Anastasiou D, Ito K, Sasaki AT, Rameh L,
et al: Pandolfi systemic elevation of PTEN induces a
tumor-suppressive metabolic state. Cell. 149:49–62. 2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Song MS, Salmena L and Pandolfi PP: The
functions and regulation of the PTEN tumour suppressor. Nat Rev Mol
Cell Biol. 13:283–296. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Ma X and Blenis J: Molecular mechanisms of
mTOR-mediated translational control. Nat Rev Mol Cell Biol.
10:307–318. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Huang J and Manning BD: A complex
interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc
Trans. 37:217–222. 2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Inoki K, Li Y, Zhu T, Wu J and Guan KL:
TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR
signaling. Nat Cell Biol. 4:648–657. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
15
|
Shaw RJ, Bardeesy N, Manning BD, Lopez L,
Kosmatka M, DePinho RA and Cantley LC: The LKB1 tumor suppressor
negatively regulates mTOR signaling. Cancer Cell. 6:91–99.
2004.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wilkinson KD and Hochstrasser M:
Deubiquitinating enzymes. In: Ubiquitin and Biology of the Cell.
Peters JM, Finley D and Harris JR (eds). Plenum Press, New York,
NY, pp99-120, 1998.
|
|
17
|
Everett RD, Meredith M, Orr A, Cross A,
Kathoria M and Parkinson J: A novel ubiquitin-specific protease is
dynamically associated with the PML nuclear domain and binds to a
herpesvirus regulatory protein. EMBO J. 16:566–577. 1997.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Amerik AY and Hochstrasser M: Mechanism
and function of deubiquitinating enzymes. Biochim Biophys Acta.
1695:189–207. 2004.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhang J, Zhang P, Wei Y, Piao HL, Wang W,
Maddika S, Wang M, Chen D, Sun Y, Hung MC, et al: Deubiquitylation
and stabilization of PTEN by USP13. Nat Cell Biol. 15:1486–1494.
2013.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Hussain S, Zhang Y and Galardy PJ: DUBs
and cancer: The role of deubiquitinating enzymes as oncogenes,
non-oncogenes and tumor suppressors. Cell Cycle. 8:1688–1697.
2009.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhang M, Hu C, Tong D, Xiang S, Williams
K, Bai W, Li GM, Bepler G and Zhang X: Ubiquitin-specific peptidase
10 (USP10) deubiquitinates and stabilizes MutS Homolog 2 (MSH2) to
regulate cellular sensitivity to DNA damage. J Biol Chem.
291:10783–10791. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Lin Z, Yang H, Tan C, Li J, Liu Z, Quan Q,
Kong S, Ye J, Gao B and Fang D: USP10 antagonizes c-Myc
transcriptional activation through SIRT6 stabilization to suppress
tumor formation. Cell Rep. 5:1639–1649. 2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Sun J, Li T, Zhao Y, Huang L, Sun H, Wu H
and Jiang X: USP10 inhibits lung cancer cell growth and invasion by
upregulating PTEN. Mol Cell Biochem. 41:1–7. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Lu C, Ning Z, Wang A, Chen D, Liu X, Xia
T, Tekcham DS, Wang W, Li T, Liu X, et al: USP10 suppresses tumor
progression by inhibiting mTOR activation in hepatocellular
carcinoma. Cancer Lett. 436:139–148. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Chung HY, Cesari M, Anton S, Marzetti E,
Giovannini S, Seo AY, Carter C, Yu BP and Leeuwenburgh C: Molecular
inflammation: Underpinnings of aging and age-related diseases.
Ageing Res Rev. 8:18–30. 2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Douglas EV, Rahul R, Sadiya SK, Mesut E
and Asish KG: Plasminogen activator inhibitor-1 is a marker and a
mediator of senescence. Arterioscler Thromb Vasc Biol.
37:1446–1452. 2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Shin SG, Koh SH, Woo CH and Lim JH: PAI-1
inhibits development of chronic otitis media and tympanosclerosis
in a mouse model of otitis media. Acta Otolaryngol. 134:1231–1238.
2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wei X, Li S, He J, Du H, Liu Y, Yu W, Hu
H, Han L, Wang C, Li H, et al: Tumor-secreted PAI-1 promotes breast
cancer metastasis via the induction of adipocyte-derived collagen
remodeling. Cell Commun Signal. 17(58)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Bhatelia K, Singh K and Singh R: TLRs:
Linking inflammation and breast cancer. Cell Signal. 26:2350–2357.
2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Jiang X and Shapiro DJ: The immune system
and inflammation in breast cancer. Mol Cell Endocrinol.
382:673–682. 2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Harris RE, Casto BC and Harris ZM:
Cyclooxygenase-2 and the inflammogenesis of breast cancer. World J
Clin Oncol. 5:677–692. 2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Erler JT, Bennewith KL, Nicolau M,
Dornhöfer N, Kong C, Le QT, Chi JT, Jeffrey SS and Giaccia AJ:
Lysyl oxidase is essential for hypoxia-induced metastasis. Nature.
440:1222–1226. 2006.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Kirschmann DA, Seftor EA, Fong SFT, Nieva
DR, Sullivan CM, Edwards EM, Sommer P, Csiszar K and Hendrix MJ: A
molecular role for lysyl oxidase in breast cancer invasion. Cancer
Res. 62:4478–4483. 2002.PubMed/NCBI
|
|
34
|
Basudhar D, Glynn SA, Greer M,
Somasundaram V, No JH, Scheiblin DA, Garrido P, Heinz WF, Ryan AE,
Weiss JM, et al: Coexpression of NOS2 and COX2 accelerates tumor
growth and reduces survival in estrogen receptor-negative breast
cancer. Proc Natl Acad Sci USA. 114:13030–13035. 2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Noguchi M, Rose DP, Earashi M and Miyazaki
I: The role of fatty acids and eicosanoid inhibitors in breast
carcinoma. Oncology. 52:265–271. 1995.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Ma YV, Lam C, Dalmia S, Gao P, Young J,
Middleton K, Liu C, Xu H and You L: Mechanical regulation of breast
cancer migration and apoptosis via direct and indirect osteocyte
signaling. J Cell Biochem. 119:5665–5675. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Breunig C, Pahl J, Küblbeck M, Miller M,
Antonelli D, Erdem N, Wirth C, Will R, Bott A, Cerwenka A and
Wiemann S: MicroRNA-519a-3p mediates apoptosis resistance in breast
cancer cells and their escape from recognition by natural killer
cells. Cell Death Dis. 8(e2973)2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Yu B, Gao W, Zhou H, Miao X, Chang Y, Wang
L, Xu M and Ni G: Propofol induces apoptosis of breast cancer cells
by downregulation of miR-24 signal pathway. Cancer Biomark.
21:513–519. 2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Yin N, Yi L, Khalid S, Ozbey U,
Sabitaliyevich UY and Farooqi AA: TRAIL mediated signaling in
breast cancer: Awakening guardian angel to induce apoptosis and
overcome drug resistance. Adv Exp Med Biol. 1152:243–252.
2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Zhang Y, Li Y, Li H, Chen W and Liu W:
Clostridium difficile toxin B recombinant protein inhibits tumor
growth and induces apoptosis through inhibiting Bcl-2 expression,
triggering inflammatory responses and activating C-erbB-2 and Cox-2
expression in breast cancer mouse model. Biomed Pharmacother.
101:391–398. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Luo J, Hu YL and Wang H: Ursolic acid
inhibits breast cancer growth by inhibiting proliferation, inducing
autophagy and apoptosis, and suppressing inflammatory responses via
the PI3K/AKT and NF-κB signaling pathways in vitro. Exp Ther Med.
14:3623–3631. 2017.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Burstein E and Ducket CS: Dying for
NF-kappaB? Control of cell death by transcriptional regulation of
the apoptotic machinery. Curr Opin Cell Biol. 15:732–737.
2003.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Helms S: Cancer prevention and
therapeutics: Panax ginseng. Altern Med Rev. 9:259–274.
2004.PubMed/NCBI
|
|
44
|
Davydov M and Krikorian AD:
Eleutherococcus senticosus (Rupr & Maxim.) Maxim. (Araliaceae)
as an adaptogen: A closer look. J Ethnopharmacol. 72:345–393.
2000.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Lee S, Shin DS, Oh KB and Skin KH:
Antibacterial compounds from leaves of Acanthopanax senticosus.
Arch Pharm Res. 26:40–42. 2003.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Hasegawa H, Suzuki R, Nagaoka T, Tezuka Y,
Kadota S and Saiki I: Prevention of growth and metastasis of murine
melanoma through enhanced natural-killercytotoxicity by fatty acid
conjugate of protopanaxatriol. Biol Pharm Bull. 25:861–866.
2002.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Kiyohara H, Hirano M, Wen XG, Matsumoto T,
Sun XB and Yamada H: Characterization of an antiulcer pectic
polysaccharide from leaves of Panaxginseng C.A. Meyer. Carbohydr
Res. 263:89–101. 1994.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Schepetkin IA and Quinn MT: Botanical
polysaccharides: Macrophage immunomodulation and therapeutic
potential. Int Immunopharmacol. 6:317–333. 2006.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Yoon TJ, Yoo YC, Kang TB, Baek YJ, Huh CS,
Song SK, Lee KH, Azuma I and Kim JB: Prophylactic effect of Korean
mistletoe (Viscum album coloratum) extract on tumor metastasis is
mediated by enhancement of NK cell activity. Int J Immunopharmacol.
20:163–172. 1998.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Shin MS, Lee H, Hong HD and Shin KS:
Characterization of immunostimulatory pectic polysaccharide
isolated from the leaves of Diospyros kaki Tumb (Persimmon). J
Funct Foods. 26:319–329. 2016.
|
|
52
|
Park JY, Shin MS, Kim SN, Kim HY, Kim KH,
Shin KS and Kang KS: Polysaccharides from Korean Citrus hallabong
peels inhibit angiogenesis andbreast cancer cell migration. Int J
Biol Macromol. 85:522–529. 2016.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Lee EH, Park HR, Shin MS, Cho SY, Choi HJ
and Shin KS: Antitumor metastasis activity of pectic polysaccharide
purified from the peels of Korean Citrus Hallabong. Carbohydr
Polym. 111:72–79. 2014.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Xuan YH, Huang BB, Tian HS, Chi LS, Duan
YM, Wang X, Zhu ZX, Cai WH, Zhu YT, Wei TM, et al: High-glucose
inhibits human fibroblast cell migration in wound healing via
repression of bFGF-regulating JNK phosphorylation. PLoS One.
9(e108182)2014.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Keum YS, Han SS, Chun KS, Park KK, Park
JH, Lee SK and Surh YJ: Inhibitory effects of the ginsenoside Rg3
on phorbol ester-induced cyclooxygenase-2 expression, NF-kappaB
activation and tumor promotion. Mutat Res. 523-524:75–85.
2003.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Ip YT and Davis RJ: Signal transduction by
the c-Jun N-terminal kinase (JNK)-from inflammation to development.
Curr Opin Cell Biol. 10:205–219. 1998.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Liu YY, Ge C, Tian H, Jiang JY, Zhao FY,
Li H, Chen TY, Yao M and Li JJ: The transcription factor Ikaros
inhibits cell proliferation by downregulating ANXA4 expression in
hepatocellular carcinoma. Am J Cancer Res. 7:1285–1297.
2017.PubMed/NCBI
|
|
58
|
Baeuerle PA and Baltimore D: NF-kappa B:
Ten years after. Cell. 87:13–20. 1996.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Ahn JY, Choi IS, Shim JY, Yun EK, Yun YS,
Jeong G and Song JY: The immunomodulator ginsan induces resistance
to experimental sepsis by inhibiting Toll-like receptor-mediated
inflammatory signals. Eur J Immunol. 36:37–45. 2006.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Kashii-Magaribuchi K, Takeuchi R, Haisa Y,
Sakamoto A, Itoh A, Izawa Y, Isa M, Fukuzawa M, Murakami M and
Takahashi R: Induced expression of cancer stem cell markers ALDH1A3
and Sox-2 in hierarchical reconstitution of apoptosis-resistant
human breast cancer cells. Acta Histochem Cytochem. 49:149–158.
2016.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Wallach D, Varfolomeev EE, Malinin NL,
Goltsev YV, Kovalenko AV and Boldin M: Tumor necrosis factor
receptor and Fas signaling mechanisms. Ann Rev Immunol. 17:331–367.
1999.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Ehrhardt H, Fulda S, Schmid I, Hiscott J,
Debatin KM and Jeremias I: TRAIL induced survival and proliferation
in cancer cells resistant towards TRAIL-induced apoptosis mediated
by NF-kappaB. Oncogene. 22:3842–3852. 2003.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Trauzold A, Siegmund D, Schniewind B,
Sipos B, Egberts J, Zorenkov D, Emme D, Röder C, Kalthoff H and
Wajant H: TRAIL promotes metastasis of human pancreatic ductal
adenocarcinoma. Oncogene. 25:7434–7439. 2006.PubMed/NCBI View Article : Google Scholar
|