Introduction
Mowat-Wilson syndrome [MWS; Phenotype Mendelian
Inheritance in Man (MIM) no. 235730] was first described in 1998 as
a rare condition characterized by dysmorphic facial features,
delayed motor development, intellectual disability and multisystem
involvement (~1 among 50,000-70,000 live births) (1). The typical facial features of MWS
include hypertelorism, horizontal wedge-shaped eyebrows with medial
flaring, deep-set eyes, a broad nasal bridge, a prominent nasal
tip, uplifted earlobes with a central depression, a prominent,
pointed chin and prognathism (2).
Affected individuals display moderate to severe intellectual
disability, and >70% of individuals with MWS develop epilepsy
(3-5).
Multisystem structural anomalies include Hirschsprung disease
(HSCR), genitourinary anomalies (hypospadias and renal tract
anomalies), congenital heart defects and eye and brain
abnormalities, including agenesis of the corpus callosum,
ventriculomegaly and hippocampal and white matter abnormalities
(6-8).
In addition, individuals with MWS may develop microcephaly and a
short stature as well as musculoskeletal anomalies (5,9,10). Due
to this broad phenotypic spectrum and the varying degrees of
disease severity, clinical diagnosis of MWS is often challenging
but is facilitated by an analysis of the underlying genetic
cause.
MWS has been demonstrated to be associated with
de novo heterozygous loss-of-function alleles of zinc finger
E box-binding homeobox2 (ZEB2), which is a gene that was
previously named ZFHX1B or SMAD interacting protein 1
(SIP1; Gene/Locus MIM no. 605802) and is located on
chromosome 2q22.3 (11,12). ZEB2 spans ~70 kb, comprises
10 exons and 9 introns, and encodes ZEB2, also known as SIP1 or
SMAD interacting protein 1, which has been indicated to be
associated with regulating the TGF-β/BMP/SMAD signaling cascade
(12,13). Exon 1 is non-coding, exon 2 contains
the initiation codon and exon 10 encodes the homeodomain and the
stop codon (3). ZEB2 is
expressed in the majority of human tissues and is critical for the
development and migration of neural-crest cells (14), heart septation and midline
development during early embryogenesis (15).
The ZEB2 protein contains a number of functional
domains, including a nucleosome remodeling and
deacetylase-interaction motif, one zinc-finger (ZF) cluster in the
amino terminal region (N-ZF), a SMAD binding domain, a homeodomain,
a C-terminal binding protein interacting domain, and one ZF cluster
in the carboxyl terminal region (C-ZF) (16).
ZEB2 acts as a transcriptional repressor, and
one of its characterized targets is the E-cadherin promoter, with
the ZEB2 C-ZF being necessary for proper DNA-binding and
transactivation activities (17).
Given the complex domain structure of the protein, it is likely
that besides N- and C-ZF, other domains may also serve important
roles in target gene regulation and consequently embryonic
development.
Up to date, >220 pathogenic variants of
ZEB2 have been associated with MWS, including point
mutations, deletions, duplications and large chromosomal
rearrangements according to the Human Gene Mutation Database (HGMD)
(18). Previous studies have
demonstrated that patients with ZEB2 deletions and
truncations exhibit similar phenotypic severities, but those with
larger deletions have more severe phenotypes compared with those
with smaller deletions (19,20).
It is still unclear, however, to what degree the variable MWS
phenotypes are influenced directly by the nature of the
corresponding ZEB2 mutation. The analysis of
genotype/phenotype associations in MWS is therefore required. For
the majority of children with MWS, the facial features are not
obvious during the early postnatal period but start to develop with
increasing age (4). Therefore,
early on, the identification of a ZEB2 mutation may be the
only way to confirm a suspicion of MWS and to distinguish MWS from
other genetic disorders such as Goldberg-Shprintzen megacolon
syndrome (Phenotype MIM no. 609460), with which MWS shares some
clinical features but which is caused by variants of a different
gene.
The current study described the identification of
two previously described and two novel de novo mutations in
4 unrelated children and associate their specific mutations with
their distinct phenotypes.
Materials and methods
Patients
The current study assessed two cohorts of patients
from the Shenzhen Children's Hospital (Shenzhen, China) between
October 2016 and December 2017. The inclusion criteria were: i)
Diagnosis of epilepsy based on International League Against
Epilepsy criteria (ILAE2017) (21);
ii) patients presented with developmental delays or intellectual
disabilities; and iii) age <18 years. The exclusion criterion
was that individuals with acquired brain injuries, including head
trauma, brain tumors, central nervous system infections, immune
encephalitis and cerebrovascular pathological changes. Cohort A
included 333 patients (sex, 197 males and 136 females) who
presented with epilepsy and whose ages ranged from 1 month to 17
years (median age, 4.4 years). Of these, 209 also exhibited
developmental delays or intellectual disabilities. Cohort B
included 197 patients (sex, 112 males and 85 females) who presented
with developmental delays or intellectual disabilities but not
epilepsy (age range, 3 months to 16 years; median age, 5.6 years).
DNA samples from the patients and their parents were collected. The
four patients in whom ZEB2 mutations were identified
underwent a general medical examination, which included physical
examination, doppler ultrasound of the heart and urinary system,
abdominal X-ray, electrocardiogram, gas chromatography-mass
spectrometry (GC/MS) (22), which
has been widely used in metabolomics analyses of biofluid samples,
tandem mass spectrometry (MS/MS) (23), which is the fundamental platform
technology for proteomics, and as a major technology for peptide
sequencing. Both GC/MS and MS/MS were used in the screening of
metabolic diseases in the present study. In addition, a detailed
clinical assessment of their epilepsy that included
electroencephalogram (24) and MRI
brain studies (25). Their
developmental and general medical history and medical records were
reviewed. Written informed consent was obtained from all parents or
legal guardians of the patients. The current study was approved by
the Ethics Committee of the Shenzhen Children's Hospital (reference
no. 2017014).
Whole genome sequencing, variant
identification and validation
Genomic DNA was isolated from probands' peripheral
leukocyte with the QIAamp DNA Blood Mini Kit (Qiagen GmbH),
according to the manufacturer's protocol. Leukocyte DNA was also
obtained from the parents of the four probands. Sample collection
from the adult participants was performed in Shenzhen Children's
Hospital (Shenzhen, China) at the same time with the probands
between December 12th, 2016 and May 5th, 2017. Totally, eight
parents (sex, four males and four females) participated in the
study (age range, 30-40 years; median age, 34 years). Whole genome
sequencing was conducted using the BGISEQ-500 platform (cat. no.
1000005478; BGI Group) performing paired-end, 100 bp (PE100)
sequencing as described previously (26). Deep sequencing data were aligned to
the reference GRCh37 build (hg19), and variants were called using
the Edico Dragen analysis pipeline (27). The Edico Genome's Dragen Bio-IT
Platform is based on the Dragen Bio-IT Processor, a bioinformatics
application-specific integrated reference-based mapping, aligning,
sorting, de-duplication and variant calling system (28). Variants were annotated using bcfanno
(v1.4; https://github.com/shiquan/bcfanno) in Frequency data
[The Exome Aggregation Consortium (29), the Genome Aggregation Database
(GnomAD; v2.1.1) (30), and
1000Genomes (G1000) (31)] and
gene-disease data [ClinVar (32),
Clinical genomic database (33),
Online MIM (OMIM) (34) and HGMD
(35)]. The predictive programs
SIFT (v 5.2.2) (36), Polyphen2
(v2.2.2) (37), MutationTaster
(NCBI 37/Ensembl 69) (38) and
PROVEAN (v1.1.5) (39) were used to
access the pathogenic potential of the variants. Human Phenotype
Ontology (HPO) items were used to prioritize candidate genes using
Phenolyzer (40), which
incorporates a list of gene-disease databases, compiled from
several data sources, including OMIM, Orphanet (41), ClinVar and Genome Wide Association
Studies Catalog (42). Rare
variants [<1% minor allele frequency in G1000, ExAC-East Asian
population (EAS), and GnomAD-exome-EAS] in HPO candidate genes were
classified into five categories (pathogenic, likely pathogenic,
uncertain significance, likely benign, and benign) according to
American College of Medical Genetics and Genomics (ACMG) Guidelines
(43). Sanger sequencing was
performed to validate the variants and for parental segregation
testing.
Primers for the amplification of the four
ZEB2 variants using PCR were designed using Primer Premier
6.0 (http://www.premierbiosoft.com;
Table I) and synthesized by BGI
Tech Solutions Co., Ltd., who also performed the Sanger sequencing.
The source of DNA was from peripheral blood leukocyte. The DNA
polymerase was supplied by NEBNext® High-Fidelity 2X PCR
Master Mix (cat. no. M0541; New England Biolabs, Inc.).
Thermocycling conditions were: Initial denaturation at 98˚C for 30
sec, followed by 35 cycles of 98˚C for 10 sec; the annealing
temperature was 66˚C for primer pair of P1, P3 and P4 and 63˚C for
primer pair of P2 for 30 sec. Extension temperature was 72˚C for 30
sec per kb. Final extension temperature was 72˚C for 5 min.
 | Table IPrimers for the amplification of four
ZEB2 variants by PCR. |
Table I
Primers for the amplification of four
ZEB2 variants by PCR.
| Patient | Variant | Primer (5'-3') |
|---|
| P1 | c.290G>A
(p.Trp97X) | Forward:
GATGTAACTGCCGCAATGTGA |
| | | Reverse:
GGGGTGGCTGATGTTTCTCA |
| P2 | c.1067_1068insAGACG
(p.Val357Aspfs*15) | Forward:
AGTGCCACTAAACCCGTGTG |
| | | Reverse:
TGTCCTCCCAGGGCAGATAA |
| P3 |
c.2761C>T(p.Arg921X) | Forward:
AGCCCACTGATGGTTTTA |
| | | Reverse:
GAGCCTCTGAACTTGACTTT |
| P4 |
c.3214C>T(p.Gln1072X) | Forward:
GCCTTCTTTCTCGTGCTCCT |
| | | Reverse:
CCATCGATTAGACCGGGGTG |
Multiple sequence alignments were generated for
homologous ZEB2 sequences to evaluate conservation using T-Coffee
(44). Alignments for ZEB2 were
generated using the following sequences: Homo sapiens, NP_055610.1;
Mus musculus, NP_056568.2; Rattus norvegicus, NP_001028873.1;
Gallus gallus, NP_001305395.1; Pan troglodytes, XP_001158120.1
isoform X1; Canis lupus familiaris, XP_005632021.2; Bos taurus,
NP_001069660.2; Danio rerio, NP_001232895.1; Equus caballus,
XP_005601530.2; and Sus scrofa and XP_020932163.1.
Results
Demographics and genotypic
features
WGS was performed in 530 patients with epilepsy,
developmental delays and intellectual disabilities alone or in
combination, as aforementioned. Among these, 4 (~0.75 %) unrelated
patients, ranging in age from 2 years and 4 months to 6 years and 5
months, who harbored heterozygous de novo pathogenic
variants of ZEB2 were identified.
Each of the four distinct mutations introduced
premature stop codons, either directly as a consequence of a codon
change to a stop codon, or indirectly as a consequence of a frame
shift. In patients 1, 2 and 3, the stop codons were located outside
the known protein domains (NM_014795.3:c.290G>A/p.Trp97X;
NM_014795.3: c.1067_1068insAGACG/p.Val357Aspfs*15; and
NM_014795.3:c.2761C>T/p.Arg 921X, respectively) while in patient
4, the stop codon was located in the C-ZF domain
(NM_014795.3:c.3214C>T/p.Gln1072X; Fig. 1). A comparison of the patients'
mutations with the sequences of their corresponding parents by
Sanger sequencing indicated that the mutations occurred de
novo (data not shown). Of the four mutations, the Arg921X
mutation of patient 3 had previously been reported in a number of
patients (2,4,45).
Furthermore, the Trp97X mutation of patient 1 had been reported in
one Chinese population (46), while
the other mutations were, to the best of our knowledge, novel. The
electropherograms of the four mutations in ZEB2 along with
the amino acid conservation in the regions of the mutations in
several vertebrates are presented in Fig. 2. None of these mutations were found
in G1000 and ExAC-EAS databases. Each of the mutations produced a
stop codon at a location that was 100% conserved at the amino acid
level across a number of mammalian and avian species. Bioinformatic
tools (Polyphen2, SIFT, MutationTaster and PROVEAN) were used to
predict that if the truncated proteins were indeed produced, they
would be altered in both structure and function compared with the
corresponding portions of the wild type protein. According to the
rules of ACMG, all of the four mutations aforementioned were
considered pathogenic.
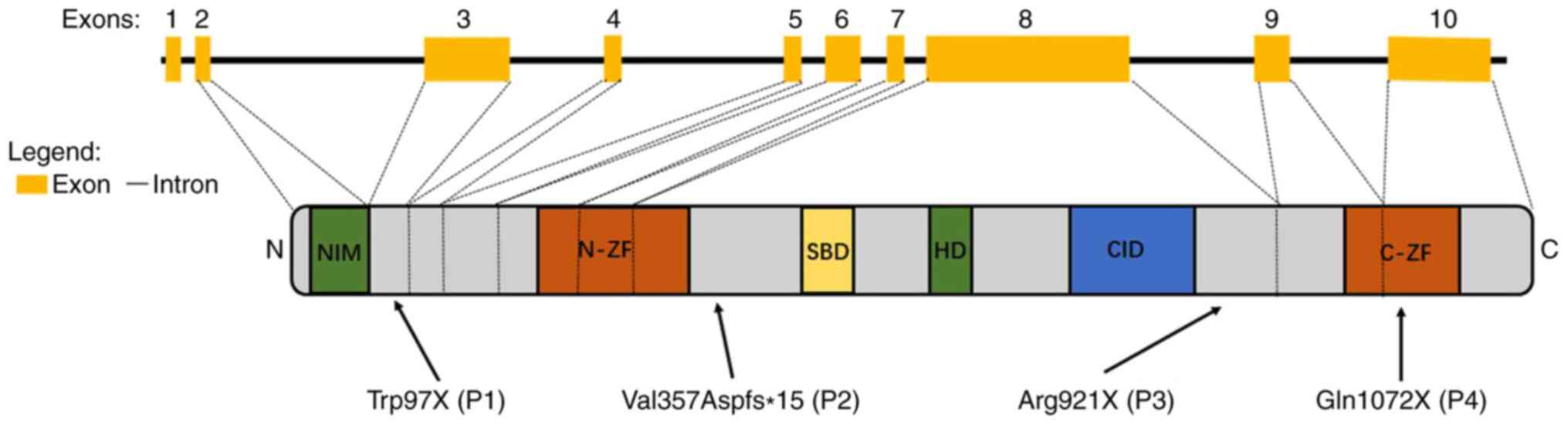 | Figure 1Schematic representation of the
ZEB2 exons and protein structure. The figure represents the
functional domains of ZEB2 and the corresponding exon
distributions: NIM, N-ZF, SBD, HD, CID and C-ZF. Arrows indicate
the localization of the mutations in patients, with the numbers
P1-4 representing patients 1, 2, 3 and 4, respectively.
ZEB2, zinc finger E box-binding homeobox2; NIM, nucleosome
remodeling and deacetylase-interaction motif; N-ZF, zinc-finger
cluster in the amino terminal region; SBD, SMAD binding domain; HD,
homeodomain; CID, C-terminal binding protein interacting domain;
C-ZF, zinc-finger cluster in the carboxyl terminal region. |
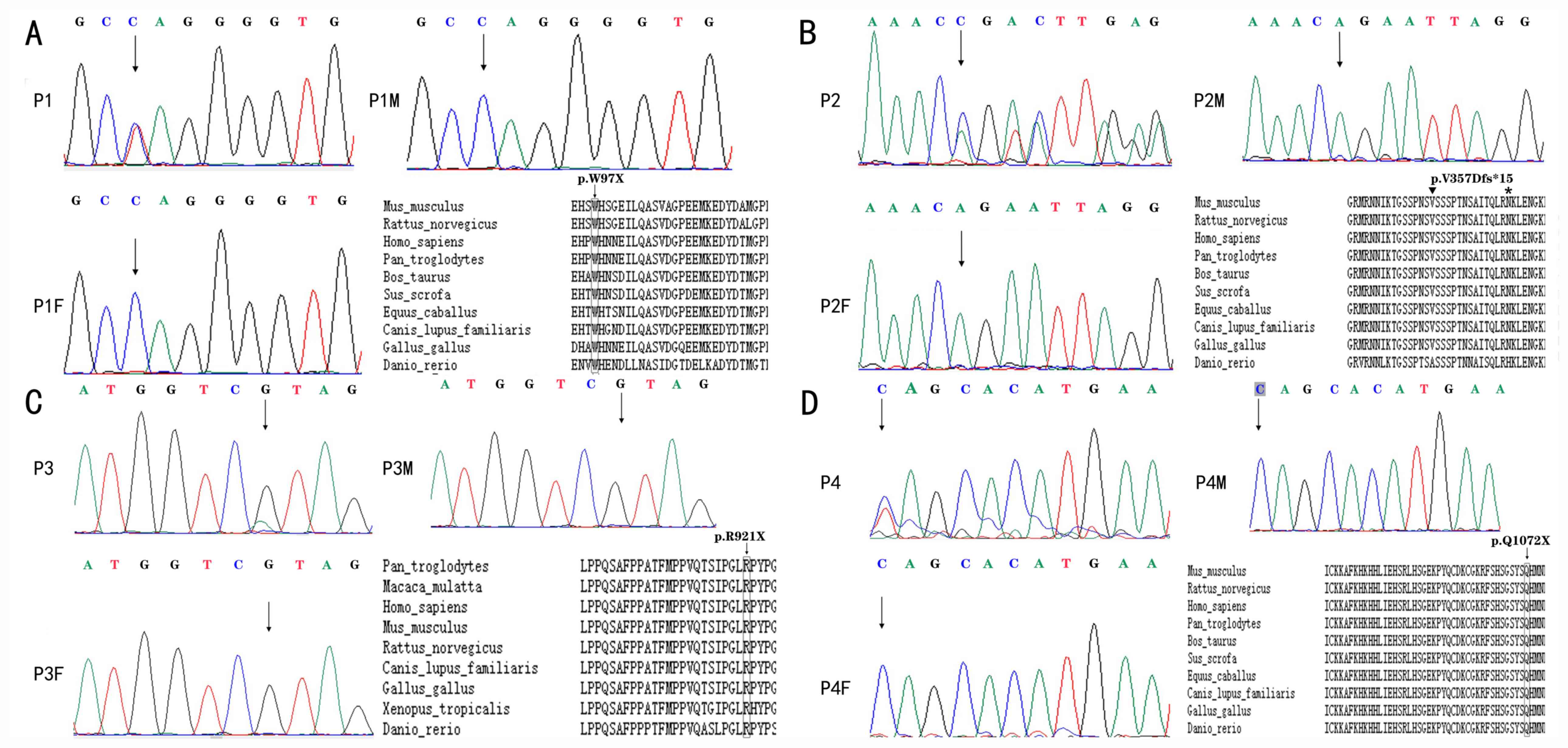 | Figure 2Electropherograms of patients with
Mowat-Wilson syndrome with ZEB2 pathogenic variants.
Heterozygous truncating mutations (nonsense or frameshift
mutations) resulting in premature termination of the
ZEB2-encoded protein are shown. (A) Patient 1, exon 3,
c.290G>A (p.Trp97X). (B) Patient 2, exon 8, c.1067_1068insAGACG
(p. Val357Aspfs*15). Inverted triangle on top of the V, where the
insertion is located; * at the N, where the stop is. (C) Patient 3,
exon 8, c.2761C>T (p. Arg921X). (D) Patient 4, exon 10,
c.3214C>T (p. Gln1072X). Each figure (A-D) also indicates the
conservation among vertebrates of the sequence regions where the
respective premature stop codons are located. Rectangles indicated
the location of the nonsense mutation in the location. Triangle and
* were used to indicate the location of frameshift mutation,
insertion and the stop codon. The ZEB2 accession numbers are as
follows: Homo sapiens, NP_055610.1; Mus musculus, NP_056568.2;
Rattus norvegicus, NP_001028873.1; Gallus gallus, NP_001305395.1;
Pan troglodytes, XP_001158120.1 isoform X1; Canis lupus familiaris,
XP_005632021.2; Bos taurus, NP_001069660.2; Danio rerio,
NP_001232895.1; Equus caballus, XP_005601530.2; and Sus scrofa,
XP_020932163.1. |
Spectrum of phenotypic features
The four patients demonstrated a wide spectrum of
phenotypic features (Table II).
Patient 1 exhibited classical features of MWS, including typical
facial features, delayed motor development, intellectual
disability, epilepsy, hypotonia, microcephaly, frontal cortical
atrophy, HSCR and hypospadias. Patient 2 presented with distinctive
facial characteristics, delayed motor development, intellectual
disability, hypotonia, microcephaly, ventriculomegaly and
constipation. Patient 3 had typical facial features, delayed motor
development, intellectual disability, epilepsy, hypotonia,
microcephaly, myelin dysplasia and constipation. Patient 4
presented with relatively milder symptoms that, although included
distinctive facial features, delayed motor development,
intellectual disability and epilepsy, lacked the more severe
symptoms aforementioned for the other three patients.
| Table IIClinical features and ZEB2
mutations in patients with MWS. |
Table II
Clinical features and ZEB2
mutations in patients with MWS.
| Patient
characteristic | P1 | P2 | P3 | P4 |
|---|
| Age | 3 y | 2 y 4 m | 3 y 2 m | 6 y 5 m |
| Sex | M | F | M | M |
| Exon affected by
the mutation | 3 | 8 | 8 | 10 |
| Mutation | c.290G>A |
c.1067_1068insAGACG | c.2761C>T | c.3214C>T |
| Novel | No | Yes | No | Yes |
| AA | p. Trp97X | p.
Val357Aspfs*15 | p. Arg921X | p. Gln1072X |
| Inheritance | De novo | De novo | De novo | De novo |
| CFA | HT, BNB, PNT, UECD,
PC | HT, BNB, PNT, UECD,
PC | HT, DSE, BNB, PNT,
PC | HT, BNB, PNT, UECD,
PC |
| WA | -a | -a | 3 y | 2 y |
| ID | ++ | ++ | ++ | + |
| Speech | - | - | - | FW |
| Epilepsy
(medication received) | + | - | + (VPA) | + (VPA, LEV) |
| Disposition | Happy | Happy | Happy | Happy |
| Hypotonia | + | + | + | - |
| MC | + | + | + | - |
| BA | FCA | ETV | MD | - |
| HSCR | + | - | - | - |
| CO | - | + | + | - |
| Other organ
malformations | Hypospadias | - | - | - |
At the time of diagnosis, all 4 patients exhibited
some signs of distinctive facial features of MWS, including a broad
nasal bridge, a prominent and pointed chin, a prominent nasal tip,
and uplifted earlobes with a central depression. As an example, the
characteristic facial appearance of patient 2 is presented in
Fig. S1. Patients 1 and 2 are
still unable to walk, patient 3 started to walk at ~3 years, but
the less severely affected patient 4 was able to walk at ~2 years.
The date at which these observations are still valid for patient 1
and 2 was July 30th, 2020; for patient 3 and 4 this was June 25th,
2020. Similar to the differential delay in the ability to walk, the
intellectual disabilities were severe in patients 1-3 but moderate
in patient 4. Among the four patients, only patient 4 could speak
at age 3 years and this patient was able to point at objects kept
out of reach. At 6 years, patient 4 could speak short sentences
with two to five words, and language comprehension was only
moderately impaired. Patient 1 presented with epilepsy at the age
of 2, but the seizures stopped after half a year even without the
use of antiepileptic drugs. The epilepsy of patient 3 and 4 was
easily controlled with one or two types of antiepileptic drugs,
where patient 3 was treated with valproate (20 mg/kg/d) and patient
4 was treated with valproate (25 mg/kg/d) and levetiracetam (30
mg/kg/d). Patients 1, 2 and 3 displayed hypotonia and microcephaly.
MRI indicated frontal cortical atrophy in patient 1,
ventriculomegaly in patient 2 and mild myelin dysplasia in patient
3. In patient 4, however, the MRI was normal. None of the patients
had congenital heart disease, but patient 1 exhibited signs of HSCR
and displayed hypospadias while the others exhibited no signs of
multisystem involvement. The disease severity was based on a
comprehensive evaluation of the phenotypes, including multisystem
involvements, differential delay in the ability to walk and speak,
and MRI findings. In general, patient 1 presented with the most
severe symptoms and patient 4 with the mildest. Patient 3 had no
signs of HSCR, not affected by hypospadias and demonstrated less
severe MRI findings than either patient 1 or patient 2. Patient 2
demonstrated no signs of multisystem involvements and epilepsy and
hence was less severely affected than patient 1.
Discussion
The wide phenotypic spectrum and varying degrees of
disease severity render a clinical diagnosis of MWS difficult,
especially when the typical facial features are not clearly present
at an early age. Therefore, none of the 4 patients in the current
study were identified to carry truncating ZEB2 mutations and
had not been clearly diagnosed as exhibiting MWS despite displaying
signs of the disorder. In fact, it was only after identification of
mutations in ZEB2 that a diagnosis was made.
The detailed clinical assessment showed considerable
patient-to-patient differences in disease manifestations, which
ranged from mild in patient 4 to severe in patient 1. While patient
4 presented with mild delayed motor development, intellectual
disability and epilepsy, but no multisystem involvements, patient 1
exhibited severely delayed motor development, severe intellectual
disability, epilepsy, hypotonia, microcephaly, frontal cortical
atrophy, HSCR and hypospadias. The disease severity in patients 2
and 3 was milder compared with that in patient 1 but more severe
compared with that in patient 4. The molecular genetic analysis
revealed that patient 4 had a truncating mutation in the ultimate
exon 10, which encodes a highly conserved cluster of zinc fingers.
This mutation resembles one described by Ivanovski et al
(47) (c.3031delA, p. S1011Afs*64),
which was associated with mild to moderate intellectual disability,
no seizures and absence of HSCR (47).
As premature stop codons in the ultimate exon do not
lead to nonsense-mediated decay of mRNA and may not alter mRNA
stability or translatability (48),
it can be assumed that the truncated protein accumulated in this
patient. This protein retains a number of protein- and
DNA-interacting domains but lacks part of the domain that is
associated with target gene interaction. Notwithstanding the fact
that MWS-associated ZEB2 alleles seemingly represent
loss-of-function alleles or haploinsufficiency, it is still
theoretically possible that a truncated protein lacking a DNA
interaction domain may act in a dominant-negative fashion. Such
dominant-negative action, however, may be partial and not
devastating for various reasons, for instance due to the fact the
truncated protein is less stable or the retained wild type mRNA and
protein are upregulated (49,50).
Further elucidation of this would require a deeper molecular
analysis of expression and activity of the mutant ZEB2.
In contrast to patient 4, patients 1, 2 and 3 all
exhibited coding region truncations originating in internal exons
(exons 3 and 8) where premature stop codons usually lead to
nonsense-mediated decay of the corresponding mRNAs and hence a
severe reduction or total absence of polypeptides that otherwise
may be derived from them (47).
This would mean that the corresponding alleles are all equally
nonfunctional null-alleles, leaving their carriers with ZEB2
protein derived from just one wild type copy of the gene. The
corresponding symptoms would be consistent with an earlier report
on gene deletions that had established haploid insufficiency of
ZEB2 in MWS (51). Previous
studies have revealed no obvious genotype-phenotype associations in
patients with MWS except in cases where large deletions are
associated with more severe phenotypes (20,52). A
recent study indicated that milder clinical presentations can be
identified with variant ZEB2 proteins that are predicted to
preserve some functionality (47).
Likewise, the present study revealed that the severity of the
corresponding disease phenotypes would seem to depend on where
exactly the truncation is localized, being most severe in patient 1
carrying the allele with the truncation localized closest to the
amino terminus and being mildest in patient 4 with the truncation
closer to the carboxyl terminus. It is conceivable that the
expression level of the wild type allele may be influenced by the
nature of the mutant allele, thereby potentially enabling partial
compensation of the loss of protein from the mutant allele
(53). It is equally conceivable,
however, that alternative splicing events of the mutant mRNA may
lead to altered mRNAs that can, or cannot, be properly translated
depending on the allele. In addition, disease severities may also
be influenced by varying genetic backgrounds of the different
patients. Each of these mechanisms, alone or in combination, may
then influence the phenotypes associated with a given allele
(54).
Alternatively, it is possible that the mutant mRNAs
of patients 1, 2, and 3, as suggested for the mRNA of patient 4,
are not subject to nonsense-mediated decay and are translated,
leading to accumulation of the truncated proteins. If so, it would
appear that the smaller the resulting polypeptide is, the more
severe the phenotype. This may be consistent with the earlier
notion that while ZEB2 deletions lead to severe disease,
intragenic mutations that are predicted to preserve some ZEB2
protein functionality lead to milder clinical manifestations
(47), supporting the notion that
not all alleles are necessarily complete loss-of-function alleles.
Larger proteins, such as those only truncated in exon 8 (as in
patients 2 and 3), would likely preserve more functionality and may
lead to less severe phenotypes than those truncated in exon 3 (as
in patient 1). None of the domains, however, in which the proteins
of patients 1, 2 and 3 are truncated, have been recognized to carry
specific functions despite their general sequence conservation. To
explain the allele-specific disease manifestations, other molecular
or cell biological properties of the corresponding polypeptides
should be investigated, such as differences in specific half-lives
or nuclear/cytoplasmic distributions.
In conclusion, the current study described 4
patients with de novo ZEB2 mutations, two novel and two
recurrent, that were associated with features of MWS. Indeed, it
was only through a molecular genetic analysis that a clear
diagnosis became possible in these patients due to the rarity and
phenotypic variability of MWS. The analysis revealed the
possibility of an association between the nature of the truncating
mutation and the phenotype. The results suggested that not all
ZEB2 alleles found in MWS are complete loss-of-function
alleles. In the current study, the number of patients assessed was
small and additional experiments, including experiments in animal
models, may be necessary to identify the mechanisms by which each
allele is responsible for its distinct associated set of
symptoms.
Supplementary Material
Figure S1. Clinical features of
patient 2 at age 2 years and 4 months. Hypertelorism, broad nasal
bridge, pointed chin, prominent nasal tip and uplifted earlobes
with a central depression are signs of facial abnormalities
presenting in patients with Mowat-Wilson syndrome.
Acknowledgements
Not applicable.
Funding
The current study was supported by the Science and
Technology Innovation Commission of Shenzhen (grant no.
KJYY20151116165726645) and Sanming Project of Medicine in Shenzhen
(grant no. SZSM201812005).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL and JG designed the study. FW designed the study
and administered the project. DZ completed the recruitment of the
patients, collection of the clinical data, analysis of the data,
and draft of the manuscript. LW, JD and HX performed data analysis
and interpretation. TZ, ZY and QD performed data analysis and
provided technical assistance. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The current study was approved by the Ethics
Committee of the Shenzhen Children's Hospital (Shenzhen, China;
reference no. 2017014). All procedures performed in studies
involving human participants were in accordance with the ethical
standards of the institutional and/or national research committee
and with the 1964 Declaration of Helsinki and its later amendments.
Written informed consent was obtained from all parents or legal
guardians of the patients.
Patient consent for publication
Written patient consent for publication was obtained
from patients' legal guardians.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Evans E, Einfeld S, Mowat D, Taffe J,
Tonge B and Wilson M: The behavioral phenotype of Mowat-Wilson
syndrome. Am J Med Genet A. 158A:358–366. 2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Zweier C, Thiel CT, Dufke A, Crow YJ,
Meinecke P, Suri M, Ala-Mello S, Beemer F, Bernasconi S, Bianchi P,
et al: Clinical and mutational spectrum of Mowat-Wilson syndrome.
Eur J Med Genet. 48:97–111. 2005.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Garavelli L and Mainardi PC: Mowat-Wilson
syndrome. Orphanet J Rare Dis. 2(42)2007.PubMed/NCBI
|
|
4
|
Garavelli L, Zollino M, Mainardi PC,
Gurrieri F, Rivieri F, Soli F, Verri R, Albertini E, Favaron E,
Zignani M, et al: Mowat-Wilson syndrome: Facial phenotype changing
with age: Study of 19 Italian patients and review of the
literature. Am J Med Genet A. 149A:417–426. 2009.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Cordelli DM, Garavelli L, Savasta S,
Guerra A, Pellicciari A, Giordano L, Bonetti S, Cecconi I,
Wischmeijer A, Seri M, et al: Epilepsy in Mowat-Wilson syndrome:
Delineation of the electroclinical phenotype. Am J Med Genet A.
161A:273–284. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Coyle D and Puri P: Hirschsprung's disease
in children with Mowat-Wilson syndrome. Pediatr Surg Int.
31:711–717. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Bourchany A, Giurgea I, Thevenon J,
Goldenberg A, Morin G, Bremond-Gignac D, Paillot C, Lafontaine PO,
Thouvenin D, Massy J, et al: Clinical spectrum of eye malformations
in four patients with Mowat-Wilson syndrome. Am J Med Genet A.
167:1587–1592. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Garavelli L, Ivanovski I, Caraffi SG,
Santodirocco D, Pollazzon M, Cordelli DM, Abdalla E, Accorsi P,
Adam MP, Baldo C, et al: Neuroimaging findings in Mowat-Wilson
syndrome: A study of 54 patients. Genet Med. 19:691–700.
2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Mowat DR, Wilson MJ and Goossens M:
Mowat-Wilson syndrome. J Med Genet. 40:305–310. 2003.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Adam MP, Schelley S, Gallagher R, Brady
AN, Barr K, Blumberg B, Shieh JT, Graham J, Slavotinek A, Martin M,
et al: Clinical features and management issues in Mowat-Wilson
syndrome. Am J Med Genet A. 140:2730–2741. 2006.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Cacheux V, Dastot-Le Moal F, Kaariainen H,
Bondurand N, Rintala R, Boissier B, Wilson M, Mowat D and Goossens
M: Loss-of-function mutations in SIP1 Smad interacting protein 1
result in a syndromic Hirschsprung disease. Hum Mol Genet.
10:1503–1510. 2001.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wakamatsu N, Yamada Y, Yamada K, Ono T,
Nomura N, Taniguchi H, Kitoh H, Mutoh N, Yamanaka T, Mushiake K, et
al: Mutations in SIP1, encoding Smad interacting protein-1, cause a
form of Hirschsprung disease. Nat Genet. 27:369–370.
2001.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Postigo AA, Depp JL, Taylor JJ and Kroll
KL: Regulation of Smad signaling through a differential recruitment
of coactivators and corepressors by ZEB proteins. EMBO J.
22:2453–2462. 2003.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Van de Putte T, Maruhashi M, Francis A,
Nelles L, Kondoh H, Huylebroeck D and Higashi Y: Mice lacking
ZFHX1B, the gene that codes for Smad-interacting protein-1, reveal
a role for multiple neural crest cell defects in the etiology of
Hirschsprung disease-mental retardation syndrome. Am J Hum Genet.
72:465–470. 2003.PubMed/NCBI View
Article : Google Scholar
|
|
15
|
Vandewalle C, Van Roy F and Berx G: The
role of the ZEB family of transcription factors in development and
disease. Cell Mol Life Sci. 66:773–787. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Remacle JE, Kraft H, Lerchner W, Wuytens
G, Collart C, Verschueren K, Smith JC and Huylebroeck D: New mode
of DNA binding of multi-zinc finger transcription factors: DeltaEF1
family members bind with two hands to two target sites. EMBO J.
18:5073–5084. 1999.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ghoumid J, Drevillon L, Alavi-Naini SM,
Bondurand N, Rio M, Briand-Suleau A, Nasser M, Goodwin L, Raymond
P, Yanicostas C, et al: ZEB2 zinc-finger missense mutations lead to
hypomorphic alleles and a mild Mowat-Wilson syndrome. Hum Mol
Genet. 22:2652–2661. 2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
HGMD® Professional 2020.2 Trial
version. urihttp://hgmdtrial.biobase-international.com/hgmd/pro/gene.php?gene=ZEB2simplehttp://hgmdtrial.biobase-international.com/hgmd/pro/gene.php?gene=ZEB2.
Accessed September 2, 2020.
|
|
19
|
Cerruti Mainardi P, Pastore G, Zweier C
and Rauch A: Mowat-Wilson syndrome and mutation in the zinc finger
homeo box 1B gene: A well defined clinical entity. J Med Genet.
41(e16)2004.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ishihara N, Yamada K, Yamada Y, Miura K,
Kato J, Kuwabara N, Hara Y, Kobayashi Y, Hoshino K, Nomura Y, et
al: Clinical and molecular analysis of Mowat-Wilson syndrome
associated with ZFHX1B mutations and deletions at 2q22-q24.1. J Med
Genet. 41:387–393. 2004.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Scheffer IE, Berkovic S, Capovilla G,
Connolly MB, French J, Guilhoto L, Hirsch E, Jain S, Mathern GW,
Moshé SL, et al: ILAE classification of the epilepsies: Position
paper of the ILAE commission for classification and terminology.
Epilepsia. 58:512–521. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Emwas AH, Al-Talla ZA, Yang Y and
Kharbatia NM: Gas chromatography-mass spectrometry of biofluids and
extracts. Methods Mol Biol. 1277:91–112. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Yan Y, Kusalik AJ and Wu FX: Recent
developments in computational methods for de novo peptide
sequencing from tandem mass spectrometry (MS/MS). Protein Pept
Lett. 22:983–991. 2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Krumholz A: Which EEG protocol is best in
young adults with possible epilepsy? Nat Clin Pract Neurol.
3:128–129. 2007.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Martinez-Rios C, McAndrews MP, Logan W,
Krings T, Lee D and Widjaja E: MRI in the evaluation of
localization-related epilepsy. J Magn Reson Imaging. 44:12–22.
2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Huang J, Liang X, Xuan Y, Geng C, Li Y, Lu
H, Qu S, Mei X, Chen H, Yu T, et al: A reference human genome
dataset of the BGISEQ-500 sequencer. Gigascience. 6:1–9.
2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Miller NA, Farrow EG, Gibson M, Willig LK,
Twist G, Yoo B, Marrs T, Corder S, Krivohlavek L, Walter A, et al:
A 26-hour system of highly sensitive whole genome sequencing for
emergency management of genetic diseases. Genome Med.
7(100)2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Edico Genomes DRAGEN Platform. urihttp://edicogenome.com/dragen-bioit-platform/simplehttp://edicogenome.com/dragen-bioit-platform/.
Accessed February 5, 2018.
|
|
29
|
Lek M, Karczewski KJ, Minikel EV, Samocha
KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ,
Cummings BB, et al: Analysis of protein-coding genetic variation in
60,706 humans. Nature. 536:285–291. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Karczewski KJ, Franciol LC, Tiao G,
Cummings BB, Alföld J, Wang Q, Collins RL, Laricchia KM, Gann A,
Birnbaum DP, et al: The mutational constraint spectrum quantified
from variation in 141,456 humans. Nature. 581:434–443.
2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
1000 Genomes Project Consortium, Auton A,
Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL,
McCarthy S, McVean GA and Abecasis GR: A global reference for human
genetic variation. Nature 526: 68-74, 2015.
|
|
32
|
Landrum MJ, Lee JM, Benson M, Brown G,
Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al:
ClinVar: Public archive of interpretations of clinically relevant
variants. Nucleic Acids Res. 44 (D1):D862–D868. 2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Solomon BD, Nguyen AD, Bear KA and
Wolfsberg TG: Clinical genomic database. Proc Natl Acad Sci USA.
110:9851–9855. 2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Hamosh A, Scott AF, Amberger JS, Bocchini
CA and McKusick VA: Online Mendelian Inheritance in Man (OMIM), a
knowledgebase of human genes and genetic disorders. Nucleic Acids
Res. 33 (Database Issue):D514–D517. 2005.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Stenson PD, Mort M, Ball EV, Evans K,
Hayden M, Heywood S, Hussain M, Phillips AD and Cooper DN: The
human gene mutation database: Towards a comprehensive repository of
inherited mutation data for medical research, genetic diagnosis and
next-generation sequencing studies. Hum Genet. 136:665–677.
2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Ng PC and Henikoff S: SIFT: Predicting
amino acid changes that affect protein function. Nucleic Acids Res.
31:3812–3814. 2003.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Choi Y, Sims GE, Murphy S, Miller JR and
Chan AP: Predicting the functional effect of amino acid
substitutions and indels. PLoS One. 7(e46688)2012.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Yang H, Robinson PN and Wang K:
Phenolyzer: Phenotype-based prioritization of candidate genes for
human diseases. Nat Methods. 12:841–843. 2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Rath A, Olry A, Dhombres F, Brandt MM,
Urbero B and Ayme S: Representation of rare diseases in health
information systems: The Orphanet approach to serve a wide range of
end users. Hum Mutat. 33:803–808. 2012.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Welter D, MacArthur J, Morales J, Burdett
T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, et
al: The NHGRI GWAS Catalog, a curated resource of SNP-trait
associations. Nucleic Acids Res. 42 (Database Issue):D1001–D1006.
2014.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Notredame C, Higgins DG and Heringa J:
T-Coffee: A novel method for fast and accurate multiple sequence
alignment. J Mol Biol. 302:205–217. 2000.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Yamada Y, Nomura N, Yamada K, Matsuo M,
Suzuki Y, Sameshima K, Kimura R, Yamamoto Y, Fukushi D, Fukuhara Y,
et al: The spectrum of ZEB2 mutations causing the Mowat-Wilson
syndrome in Japanese populations. Am J Med Genet A. 164A:1899–1908.
2014.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Ho S, Luk HM, Chung BH, Fung JL, Mak HH
and Lo IFM: Mowat-Wilson syndrome in a Chinese population: A case
series. Am J Med Genet A. 182:1336–1341. 2020.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Ivanovski I, Djuric O, Caraffi SG,
Santodirocco D, Pollazzon M, Rosato S, Cordelli DM, Abdalla E,
Accorsi P, Adam MP, et al: Phenotype and genotype of 87 patients
with Mowat-Wilson syndrome and recommendations for care. Genet Med.
20:965–975. 2018.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Huang L and Wilkinson MF: Regulation of
nonsense-mediated mRNA decay. Wiley Interdiscip Rev RNA. 3:807–828.
2012.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Sheppard D: Dominant negative mutants:
Tools for the study of protein function in vitro and in vivo. Am J
Respir Cell Mol Biol. 11:1–6. 1994.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Veitia RA, Caburet S and Birchler JA:
Mechanisms of mendelian dominance. Clin Genet. 93:419–428.
2018.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Dastot-Le Moal F, Wilson M, Mowat D,
Collot N, Niel F and Goossens M: ZFHX1B mutations in patients with
Mowat-Wilson syndrome. Hum Mutat. 28:313–321. 2007.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Zweier C, Temple IK, Beemer F, Zackai E,
Lerman-Sagie T, Weschke B, Anderson CE and Rauch A:
Characterisation of deletions of the ZFHX1B region and
genotype-phenotype analysis in Mowat-Wilson syndrome. J Med Genet.
40:601–605. 2003.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Rossi A, Kontarakis Z, Gerri C, Nolte H,
Hölper S, Krüger M and Stainier DY: Genetic compensation induced by
deleterious mutations but not gene knockdowns. Nature. 524:230–233.
2015.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Peng J: Gene redundancy and gene
compensation: An updated view. J Genet Genomics. 46:329–333.
2019.PubMed/NCBI View Article : Google Scholar
|














