1. Introduction
Diabetes mellitus (DM), is a chronic endocrine
disease that is characterized by peripheral insulin resistance and
eventual pancreatic islet β-cell failure, which poses a major
threat to public health (1). The
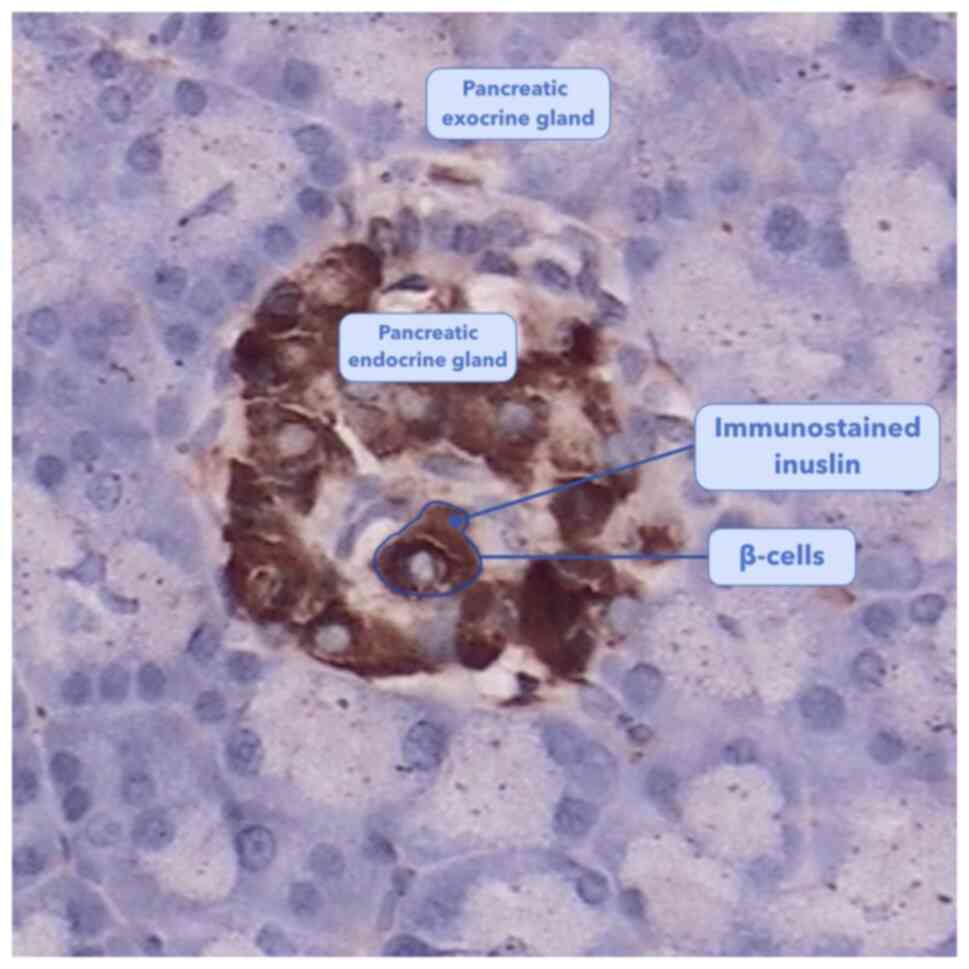
pancreas consists of endocrine glands surrounded by exocrine
glands. β-cells are located within the pancreatic endocrine
section, which comprise ~1.5% total pancreatic volume (2). Pancreatic endocrine glands, also
called pancreatic islets, mainly contain four types of cells: α, β,
δ and pancreatic polypeptide cells (3). β-cells comprise ~54% of endocrine
pancreatic cells and are the sole source of insulin in the body
(Fig. 1) (3,4). In
total, ~425 million people have been diagnosed with DM worldwide
(2017) (5) and >90% of DM were
type 2 DM in the UK (2015) (6). The
primary causes of DM-associated mortality involve complications
leading to cardiovascular disease, diabetic retinopathy,
nephropathy, neuropathy and lower extremity amputation (7,8).
The pathogenesis of DM involves numerous genetic and
environmental factors. Over the past decade, the role of
mitochondria in DM has been extensively investigated (7,9,10).
Mitochondria serve roles in diverse cellular signaling and
metabolic pathways in addition to producing ATP (11). During the process of insulin
secretion, ATP generated from oxidative phosphorylation (OXPHOS)
contributes to the closure of ATP-sensitive K+ channels
and subsequent opening of voltage-dependent Ca2+
channels on the plasma membranes of β-cells (12). This increased intracellular
concentration of Ca2+ ultimately initiates insulin
exocytosis in a process known as glucose-stimulated insulin
secretion (GSIS) (12).
Furthermore, completed cycles of the tricarboxylic acid (TCA) cycle
generates one GTP molecule, which serves no direct influence on
GSIS unless transformed to ATP (13,14).
However, GTP is an essential signal in insulin secretion in β-cells
(13,14). Numerous studies have investigated
the association between mitochondrial dysfunction and β-cell
failure, in addition to the pathogenesis of DM (7,15-18).
Protecting mitochondrial function and the antioxidant ability of
pancreatic β-cells may prove to be a beneficial future therapeutic
target for type-2 DM, consistent with the previous reports
(9,19). The present review provides an
overview of recent reports regarding the association between
mitochondrial dysfunction and β-cell failure.
2. Mitochondrial dysfunction
Mitochondrial function
Mitochondria serve a number of roles in different
cellular processes, with the most significant one of which is that
of energetic powerhouses for cellular activities (20). Mitochondria produce ATP
predominantly through OXPHOS, during which protons flow through ATP
synthase located in the inner mitochondrial membrane (IMM) to drive
ATP synthesis (20,21). Specifically, electrons are
transferred from NADH to oxygen through a chain of electron
carriers collectively called the electron transport chain (ETC)
(20). The four major protein
complexes located in the IMM within the ETC are NADH-Q reductase
(complex I), succinate-Q reductase (complex II), Q-cytochrome
c reductase (complex III) and cytochrome c oxidase
(complex IV). Complexes I, III and IV pump H+ ions into
the mitochondrial intermembranous space from the mitochondrial
matrix to produce a gradient of H+ ions when electrons
are transported down the ETC (20).
This generates a positive H+ gradient on the side of the
IMM that faces the intermembranous space. Subsequently, protons
flow back into the mitochondrial matrix via ATP synthase, also
called complex V, to produce ATP (20). Although the reduced form of flavin
adenine dinucleotide (FADH2) also produces ATP, the
level of production is less than that of NADH, since
FADH2 oxidation induces H+ ions to be pumped
out by complexes III and IV (20).
Of note, mitochondria also serve as the main source of reactive
oxygen species (ROS) in cells, which engage in ROS-mediated
signaling and can activate pathways such as calcium signaling
(22-24).
Furthermore, mitochondria participate in intracellular metabolic
processes, including the TCA cycle (25), synthesis of heme (26,27)
and certain steroids, including adrenal hormone (28,29),
calcium storage in conjunction with the endoplasmic reticulum (ER)
(30) and apoptotic regulation via
cytochrome c (31).
Therefore, when mitochondria become dysfunctional, concurrent
alterations in cellular phenotypes will occur.
Mitochondria-related genomic and
proteomic abnormalities
Mitochondrial DNA (mtDNA) is a multi-copy, 16.6-kb
circular double-stranded DNA molecule (32). The mitochondrial genome is most
probably a remnant of an archaeon, α-proteobacterium (33). However, most archaeon genes have
been transmitted from the mitochondria into the nucleus and
function as nuclear genes (33).
The remaining mtDNA is comprised of 37 genes encoding 13 proteins,
22 transfer RNAs (tRNAs) and two ribosomal RNAs (rRNAs) (32). There are two main categories of
mitochondrial genomic abnormalities: Maternally inherited genetic
defects and acquired mutations. Due to the existence of
mitochondrial genetic heteroplasmy, the precise association between
mtDNA defects and disease remains unclear (34). For instance, based on the prevalence
of mtDNA-related diseases in 2017, ≥1 in 10,000-15,000 individuals
in the general population is influenced by mtDNA-related
cardiomyopathy worldwide, though the exact prevalence remains
unknown (34). Accumulating
evidence has demonstrated that in cases of mtDNA defects and
mutations caused by pathogenic changes, including the
overproduction of ROS, diseases emerge directly or indirectly due
to these defects/mutations (15,16,34-36).
In particular, DM, neuromuscular degenerative disease and perinatal
lethality have all been previously associated with increased
heteroplasmy levels of the mtDNA tRNALeu nucleotide
mutation (15,16,35,36).
Mitochondrial rRNAs are regulated by transcription
factors (TFs) B1 (TFB1M) and B2 (TFB2M), both of which are
essential mitochondrial TFs (37).
Overexpression or 1555A>G mutation in TFB1M results in the
over-production of methyltransferases, which in turn lead to the
excessive methylation of mitochondrial rRNAs (17,37).
In human mitochondria, the 5'-processing of several mitochondrial
tRNAs is completed by the RNase P complex, consisting of
mitochondrial RNase P Proteins (MRPP)1, MRRP2 and MRPP3(38). MRPP1 and MRPP2 comprise a strong
protein complex known as MRPP1/2(39), where abnormal expression of MRPP1/2,
caused by missense variants in tRNA methyltransferase 10 homolog C,
which encodes MRPP1, results in impaired mitochondrial RNA
processing and defective mitochondrial protein synthesis (39,40).
In addition, other factors, such as phosphodiesterase 12, have been
previously demonstrated to regulate mitochondrial non-coding RNAs
(41). Since mitochondrial mRNAs,
tRNAs and non-coding RNAs are critical to mitochondrial biogenesis,
abnormalities can lead to mitochondrial dysfunction (17,18,37,39,41).
Proteins that participate in the ETC and OXPHOS,
including complexes I-V, directly serve crucial roles in
mitochondrial function and dysfunction (20,42,43).
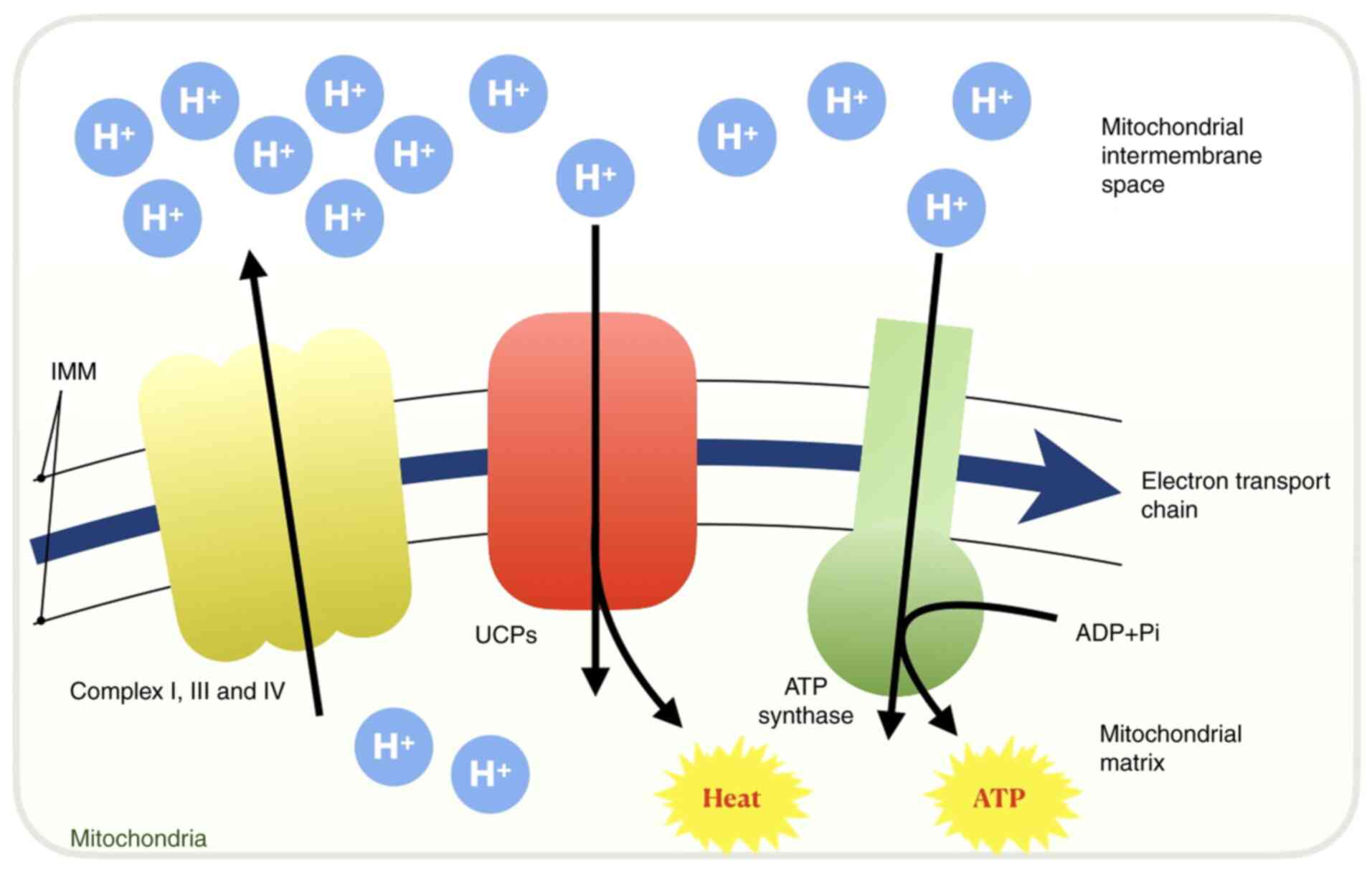
Uncoupling proteins (UCPs) are closely associated with IMM
potential (44,45). Complexes I-IV in the ETC pump
protons from the mitochondrial matrix into the IMM and the
intermembranous space to establish a H+ gradient, whilst
UCPs (particularly UCP-1) in the IMM facilitate proton leakage to
reduce the IMM potential, uncoupling the respiratory process from
ADP phosphorylation (42,46). During this process, UCPs render the
final products derived from electron transport to be heat instead
of ATP (Fig. 2) (42). This process is indispensable in
brown adipose tissues for heat generation, particularly in newborn
infants (47). However, in other
cells, overexpression of UCPs results in the increased uncoupling
of electron transport from ATP synthesis, thereby causing
mitochondrial dysfunction (48,49).
The following sections discuss the association between these
regulatory proteins and β-cell failure.
Mitochondrial impairment induced by
ROS-induced metabolic stress
Mitochondria utilize large quantities of
O2 for energy production. However, the ETC exhibits
reduced efficiency due to proton leakage (50). When leakage occurs at the IMM
between coenzyme Q and semi-ubiquinone, one electron instead of the
usual two is passed onto one molecule of O2 to form
superoxide anion radicals as the byproduct (50,51). A
free radical is an uncharged molecule that has an unpaired valence
electron (52). Such O2
free radicals, including superoxide anions
(O2·-), are referred to as ROS and are strong
oxidants (53).
When a large amount of ROS is produced,
mitochondrial manganese superoxide dismutase (Mn-SOD) cannot
scavenge oxidants at a sufficient rate (54). As a result, oxidative stress occurs
and causes damage to mitochondrial components, such that
degradative processes, including apoptosis, are initiated (51). Excessive ROS can create a vicious
cycle in which ROS continues to induce mitochondrial damage via a
positive-feedback mechanism that results in increased ROS
production (55). Extensive
investigation has previously revealed the mechanism by which ROS
causes damage to DNA, proteins, cellular lipids and organs,
including the kidneys and blood vessels (56-58).
Increased levels of O2·- in afferent
arterioles have been found to decrease nitric oxide NO production,
resulting in afferent arteriole vasoconstriction and low glomerular
filtration rate (58).
During hyperglycemia, which is caused by a
high-sugar diet or genetic mutations, the concentration of
intracellular glucose is elevated and glycolysis substrates become
more available in β-cells, which then accelerates glycolysis, TCA
cycle and pyruvate oxidation (59,60).
This metabolic stress causes an increased flux via OXPHOS,
increasing the mitochondrial membrane potential beyond a critical
threshold, resulting in blockage of the ETC at complex III
(61,62). Electrons trapped in complex III may
leak, reducing O2 to form ROS (61,62).
Although the number of mitochondria can increase to partially
compensate for this metabolic stress, their functionalities or
efficiencies do not seem to correspondingly increase, which was
previously demonstrated by a mouse model (63-65).
By contrast, increased mitochondrial density has actually been
observed to increase ROS production (66).
The IMM is rich in cardiolipin, a phospholipid which
has a high affinity for ROS and is particularly susceptible to ROS
attack (67). When a cardiolipin is
oxidized by ROS, excessive cytochrome c attached to the
cardiolipin is released into the cytosol, inducing apoptosis
(67). Additionally, the products
of phospholipid peroxidation may undergo fragmentation to form
reactive intermediates, including malondialdehyde and
4-hydroxy-trans-2-nonenal (68).
These substances can interfere with the ETC and inactivate ETC
components by forming adducts, causing mitochondrial dysfunction
(67,68). Consequently, the
mitochondria-dependent pathway of apoptosis may be activated by the
release of mitochondrial content (e.g., cytochrome c)
through the IMM (69). Therefore,
whilst ROS held at tightly regulated levels serves as a signaling
molecule that regulates cellular metabolism and apoptosis,
excessive ROS can mediate destructive processes (22,55).
Specific phenomena emerging following
mitochondrial dysfunction
The consequences of mitochondrial dysfunction can
vary according to specific conditions. Under the same pathogenic
conditions, mitochondria with functional deficiencies exhibit
diverse behaviors (16). Most
commonly, dysfunctional mitochondria with morphological
abnormalities such as hypertrophy frequently exhibit a rounded
instead of an elliptical shape (66). Numerous studies have reported an
increase in mitochondrial density during hyperglycemia (7,66,70).
However, earlier studies have reported a reduction in mitochondrial
density during hyperglycemia (71,72).
Hence, this phenomenon appears to depend on the specific
experimental models, subjects, tissues, cell types and examined
pathological states. The microstructures of proteins within
mitochondria can also change following mitochondrial dysfunction
(73,74). Complexes I and III in the ETC
produce large amounts of ROS and, therefore, are particularly
vulnerable to ROS damage among all mitochondrial proteins (73,74).
Complexes I and III have been observed to alter their conformation
following ROS damage (73,74). Furthermore, mitophagy and the
mitochondrial-dependent pathway of apoptosis may eventually occur
due to mitochondrial dysfunction (67,75).
Mitophagy is a specialized form of autophagy that regulates damaged
and dysfunctional mitochondria, where damaged mitochondria are
scavenged or all mitochondria are eliminated during specific
developmental stages, including fertilization, blood-cell
maturation and starvation (75).
During mitophagy, autophagosomes are usually formed through the
phosphatase and tensin homolog-induced kinase 1/Parkin pathway
(75). Following this, the
autophagic lysosomal degradative pathway is activated (76). Exosomes may have a close association
with mitophagy (10,77). Recent studies have demonstrated that
exosomes can carry intracellular components, including proteins,
mRNAs, microRNAs and lipids, across the extracellular environment
(10,78). In total, ≤10% exosomal proteins are
derived from mitochondria (77).
Exosomes containing mitochondrial proteins can either be
transferred from an unhealthy cell with mitochondrial dysfunction
to a healthy cell to interfere with metabolism, or from a healthy
cell to an unhealthy cell with mitochondrial dysfunction to
facilitate metabolism (10).
Therefore, exosomes may enable cells to remove peroxidated
cardiolipins to maintain a healthy mitochondrial population
(10). However, the definitive
mechanism and consequence of exosomal transfer of mitochondrial
proteins remains poorly understood and requires further study.
When the IMM is damaged by oxidative stress, certain
proapoptotic proteins are exported into the cytosol (69). These proapoptotic proteins hinder
ATP synthesis by disturbing the mitochondrial transmembrane
potential, oxidizing NADH, NADPH and glutathione (GSH) (69). This deteriorates mitochondrial
metabolism and induces a vicious cycle of ROS damage (69). These proapoptotic proteins,
including cytochrome c, ultimately initiate a cascade of
caspase-dependent apoptosis (67).
Cytochrome c becomes unchained to cardiolipin in the IMM
when a cardiolipin is oxidized by ROS (69). This release of cytochrome c
is then transported from the intermembranous space into the cytosol
through membranous pores in the outer mitochondrial membrane (OMM).
In response to cytochrome c release, apoptotic protease
activating factor-1 (Apaf-1) oligomerizes (79). Following this, cytochrome c,
Apaf-1 and procaspase-9 form the multiprotein apoptosome, which
activates caspase-9 and initiates the mitochondria-dependent
pathway of apoptosis (69).
3. Association between mitochondrial
dysfunction and islet β-cell failure
The mechanism of GSIS
Insulin secretion is regulated by numerous metabolic
processes, including glycolysis and dehydrogenation, and depends on
the level of blood sugar (13).
GSIS is the principle mechanism by which insulin is secreted and it
involves several processes, including the glycolysis of glucose,
the transformation of pyruvate, the production of ATP and finally
the opening of voltage-dependent Ca2+ channels (80-82).
β-cells intake glucose into the cytosol via glucose transporter 2
on the plasma membrane (80).
Cytosolic glucose undergoes glycolysis to produce pyruvate
following phosphorylation by glucokinase (13). This pyruvate is then dehydrogenized
into acetyl-coenzyme A by the pyruvate dehydrogenase complex and is
transported into the mitochondria, where it generates ATP through
the TCA cycle and OXPHOS to increase cytosolic ATP concentration
(13). This results in the closure
of ATP-sensitive K+ channels and the opening of
voltage-dependent Ca2+ channels on the plasma membrane
to allow Ca2+ entry into the cytosol, which induces
insulin exocytosis (13).
Therefore, mitochondrial OXPHOS in islet β-cells is crucial during
GSIS (81,82).
Abnormalities in mitochondrial genes
and proteins leading to β-cell dysfunction
It is beyond the scope of the present review to
discuss all of the mitochondrial genes and proteins. Previous
reviews have comprehensively described how these genes and proteins
are associated with DM. In subsequent sections, various examples of
abnormalities in mitochondrial genes and proteins leading to β-cell
dysfunction, including TFs, are discussed (16,83,84).
Previous studies have demonstrated that the
m.3243A>G mutation in the mitochondrial gene tRNALeu
causes maternally inherited diabetes and deafness (MIDD) syndrome
(85,86). This mutation can interfere with the
expression or function of other mitochondrial genes (for instance,
m.16093T>C) to induce mitochondrial dysfunction (85). Dysfunctional mitochondria produce
less ATP, which further impairs insulin secretion, since the
K+ channels required for insulin release from β-cells
are ATP-sensitive (13). A previous
study demonstrated that human patients with this mutation develop
increased gluconeogenesis which, alongside insulin resistance,
results in increased ROS production by dysfunctional mitochondria
in tissues such as skeletal muscle to induce DM (87).
Nicotinamide nucleotide transhydrogenase (NNT)
catalyzes the reversible conversion of NADP+ and NADH
into NADPH and NAD+, respectively, in mitochondria
(88). Primitive studies in mice
have previously suggested that a lack of NNT is associated with
impaired insulin secretion (88).
Another previous study revealed that a lack of NNT hinders GSIS by
altering Ca2+-induced exocytosis and its metabolic
amplification instead of altering glucose-mediated stimulation of
Ca2+ influx or other mitochondrial events further
upstream (89). In summary,
dysfunctional mitochondria lacking NNTs lead to the failure of
Ca2+-induced exocytosis of insulin from β-cells.
Cytochrome c serves a pivotal role in the
mitochondria-dependent pathway of apoptosis in β-cells (69). It is widely acknowledged that a high
rate of β-cell apoptosis due to increased cytoplasmic levels of
cytochrome c occurs prior to DM (90). Upregulated expression of cytochrome
c and apoptosis have been previously observed in muscle
biopsies from patients with DM (91). Cyclin dependent kinase 11A (CDC2L2)
is a gene that is a risk factor for type-2 DM (92,93).
High expression of p58, which is encoded by CDC2L2 in β-cells,
increases OMM permeability to enable cytochrome c leakage
into the cytosol to exacerbate ER stress, ultimately leading to
apoptosis (92,93). However, the mechanism of how
upregulated cytochrome c induces β-cell failure in the
cytosol remains unclear.
The potential role of UCPs in the modulation of
insulin secretion in response to glucose has been the focus of
research attention. UCP-2 is a negative regulator during pancreatic
development (46) and silences
glucose oxidation in β-cells, which is the initial process of GSIS
(94). Previous studies have
demonstrated that upregulated UCP-2 and downregulated UCP-3 under
chronic high glucose conditions impaired GSIS in β-cells (43,95).
However, how UCP-2 is involved in DM remains unclear. However,
using the same experimental model of streptozotocin-induced
diabetic mice, a previous study demonstrated that the destruction
of β-cells during autoimmune DM is more severe in
UCP-2-/- mice (96), while another study has hypothesized
that enhanced β-cell function is associated with higher basal ROS
levels constitutively activating the ROS-signaling pathway in
UCP-2-/- mice, resulting in
significantly reduced severity of hyperglycemia (97). Collectively, these findings
indicated that the mitochondrial protein UCP-2 is has multiple
roles β-cell failure.
TFs A, B1M and B2M mediate the transcription of
mtDNA, where TFB1M mainly serves as a dimethyl transferase
(37). Reduced TFB1M expression,
damaged islets and dysfunctional mitochondria have all been
previously observed in heterozygous
TFB1M+/- mice (98). As a result, β-cells in islets
release less insulin in response to glucose, since less ATP is
produced by these dysfunctional mitochondria (16,83,98).
Additionally, previous studies have demonstrated that GSIS is
impaired and β-cell mass is reduced during mitochondrial
dysfunction due to a lack of TFB2M in mitochondria (17,99).
To preserve cell viability, impaired mitochondria
can be removed by exosomes that carry mitochondrial fragments out
of the cells (100). If exosomes
transporting unwanted or harmful fragments from unhealthy β-cells
are received by healthy β-cells nearby, the metabolism of recipient
healthy β-cells may become disturbed. Dysfunctional mitochondria
within one β-cell can induce β-cell failure in adjacent cells
(10). However, further research is
required to investigate these possibilities further (10).
ROS: The initial destroyer of islet
β-cells
Continuous deposition of glucose into the plasma
from a constant high-carbohydrate diet results in a high basal
level of plasma glucose (101).
Additionally, since the rate of glucose transportation across the
β-cell membrane is higher compared with the rate of glucose
phosphorylation, glucose then accumulates within the cytosol of
β-cells (102). Increased glucose
influx into β-cells increases the activity of the TCA cycle,
generating more ATP by OXPHOS to raise the level of Ca2+
in the β-cell cytosol to stimulate insulin secretion via GSIS
(13). Mitochondrial oxidative
activity then reaches its maximum capacity and may produce more ROS
(60,103,104). Furthermore, at the end of the GSIS
signaling pathway, elevated intracellular Ca2+ serves as
a signal to stimulate mitochondrial ROS generation (105) and activate protein kinase C, which
initiates the NADPH oxidase-dependent generation of superoxides and
other free radicals (60,106). Previous studies have demonstrated
that ROS production increases with the degree of obesity and
hyperglycemia (107,108).
Mitochondria are the major source of ROS in islet β
cells (109). Following
dismutation by Mn-SOD, O2·- is converted into
the less reactive H2O2, which can diffuse
through aquaporins located in mitochondrial membranes (22). If H2O2 cannot
be removed in a timely manner, excessive H2O2
will be converted into the highly reactive hydroxyl radical HO
(109). The capacity of scavenging
oxidants in β-cells is different from that in other cells. An
earlier study has indicated that β-cells express lower levels of
antioxidant enzymes, including Mn-SODs, glutathione peroxidases
(GPXs) and catalases (110). As a
compensatory mechanism, β-cells are rich in other peroxidase-based
antioxidant enzymes, including glutaredoxin and thioredoxin
(110,111). However, once the levels of
oxidative species exceed the levels of antioxidants, abnormal
accumulation of ROS results in pro-oxidative conditions (104). O2·- can
oxidize NO to produce peroxinitrite (ONOO-) and initiate
an oxidation cascade, which severely oxidizes proteins, lipids and
carbohydrates (112).
Additionally, other types of ROS can damage cellular components
through nitration, carbonylation, peroxidation and nitrosylation
modifications (113). Ultimately,
defective gene expression, reduced glucose secretion and increased
β-cell apoptosis occur due to prolonged exposure to ROS (54).
Excessive ROS such as O2·-
and H2O2, can alter the structure and
function of proteins and lipids, particularly in the mitochondria,
leading to a chain reaction of impaired mitochondrial function,
reduced ATP production and defective insulin secretion (103). Additionally, these oxidants break
single-strand DNAs and initiate apoptosis through caspase
activation (69). A key mediator of
apoptosis may be ONOO-, which can concomitantly trigger
nitrosative stress (114).
Therefore, mitochondria are the source and target of these reactive
species (69,103,109,114). Oxidative damage contributes to the
difficulty of scavenging ROS (115-118).
Under conditions of excessive ROS, protective antioxidant
mechanisms deteriorate due to the oxidative destruction of
antioxidant enzymes, which exaggerates the imbalance between
oxidants and antioxidants (116-118).
When the intracellular concentration of glucose is high, glucose
can be transformed into sorbitol through the polyol pathway
(115). Sorbitol consumes
antioxidants, including NADPH and GSH, resulting in increased ROS
production (115). This type of
ROS accumulation then inhibits glucose-6-phosphate dehydrogenase
(G6PDH), which is crucial for reducing intracellular glucose levels
(115). Additionally, since the
production of NADPH involves the pentose phosphate pathway, where
G6PDH catalyzes glucose-6-phosphate to generate ribose 5-phosphate
and NADPH, the inhibition of G6PDH reduces NADPH levels to
consequently decrease those of GSH, further aggravating oxidative
stress (116-118).
UCPs maintain glucose homeostasis by regulating
insulin secretion (119). Although
UCP-1 has been reported to downregulate ROS generation, its
mechanism of action remains unclear (120,121). Increased ROS, in particular
O2·-, activates UCP-2 by generating carbon
radicals, which peroxidize unsaturated side chains of fatty acid
substituents in mitochondrial phospholipids such as cardiolipin
(67). Subsequently, UCP-2
overexpression results in β-cell failure due to damaged GSIS
(122). Nonetheless, the mechanism
underlying the activation of UCP-2 by ROS and the precise effects
of UCP-2 on insulin secretion remain unclear (123). Numerous studies on UCP-3 have
focused on insulin resistance or insulin sensitivity instead of
β-cell insulin secretion (124,125). A previous study has indicated that
the progressive reduction in UCP-3 levels results in insulin
resistance (124), though the
function of UCP-3 remain unclear (126).
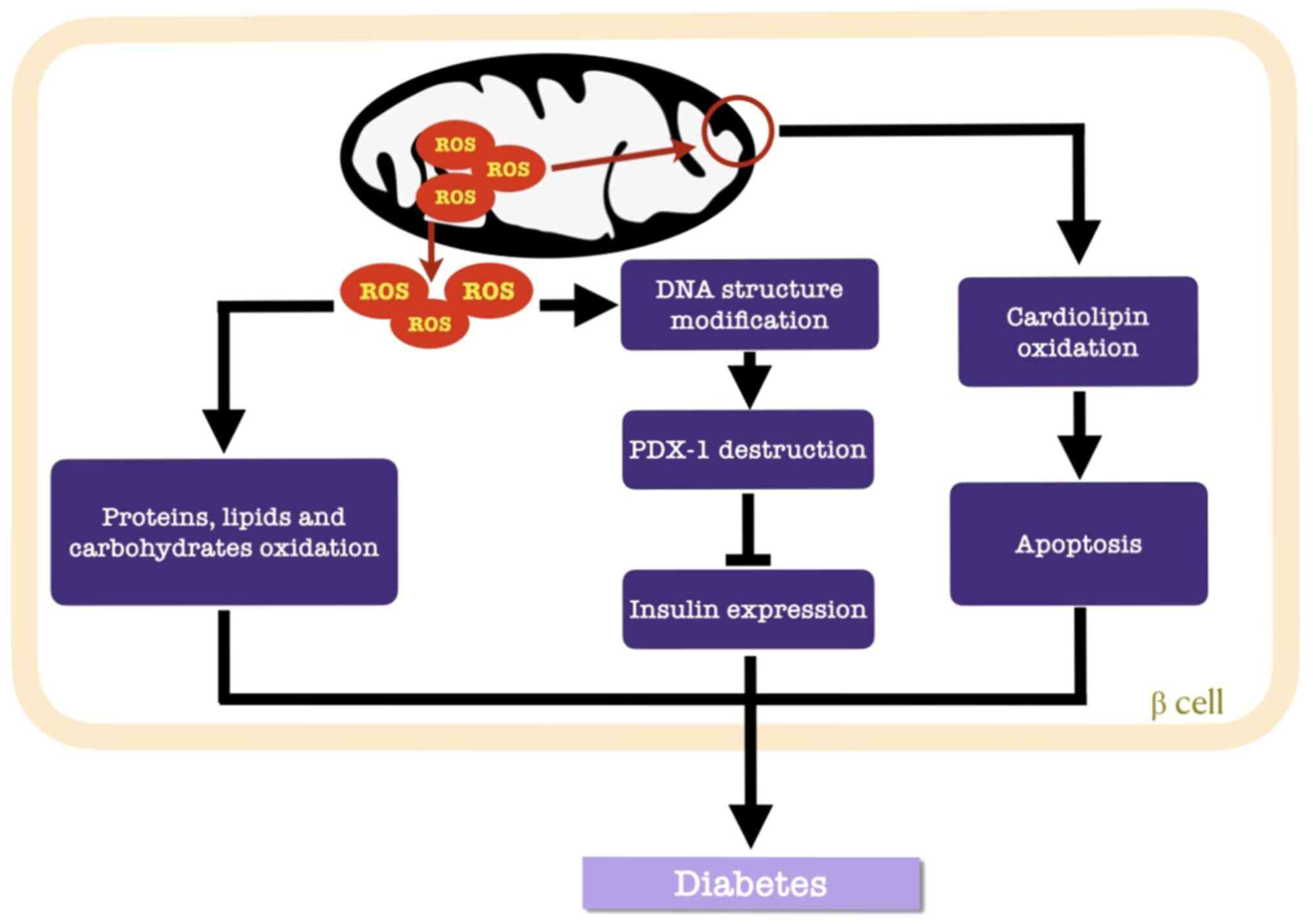
Cardiolipin is a constructive phospholipid in the
IMM that is vulnerable to ROS (67). Once the IMM is perforated,
cytochrome c-mediated apoptosis is initiated (69). Additionally, the destructive nature
of ROS involves structural modifications in DNA (127) and alterations in gene expression
(54). Pancreatic duodenal homeobox
1 (PDX-1) is a target of ROS in β cells, where
H2O2 suppresses the DNA binding activity of
PDX-1 to consequently downregulate insulin gene expression
(Fig. 3) (60).
Based on these aforementioned observations, DM
therapies aimed at reducing ROS levels or increasing the
antioxidant defenses of β-cells have become a focus of interest
(9,128,129). GPX4 is a GPX selenoprotein that
has been reported to repair oxidized mitochondrial membranes
(128,130). The phospholipase A2
family catalyzes the hydrolysis of SN-2 fatty acyl bond of
phospholipids, which repair oxidized membranous phospholipids
(9). Defective GSIS can be
ameliorated by fatty-acid-oxidation inhibitors or glucose-oxidation
promoters (9,128-131).
Gene therapy that upregulate the expression levels of glutaredoxin
and thioredoxin can potentially improve the capacity of scavenging
the excess ROS in β-cells (131).
Furthermore, dietary interventions and exercise help mitigate the
progression of DM (129). Reduced
intake and increased output of glucose can also alleviate the
pressure of oxidative stress within β-cell mitochondria, whilst
exercise strengthens mitochondrial function and increase peripheral
insulin sensitivity (129).
Inflammatory attack against islet
β-cells
Unhealthy, dying or damaged β-cells transmit
immunogenic signals to activate immune cells in pancreatic islets
(132). Reduced UCP-2 gene
expression in mononuclear cells of individuals who are obese and
diabetic may contribute to metabolic disorders due to immunological
abnormalities (133). These
activated immune cells release inflammatory cytokines and
chemokines, which activate macrophages and T cells within and
around pancreatic islets, amplifying inflammation (134). Cytokines such as IL-1β have been
documented to inhibit insulin secretion (134). These metabolic perturbations
increase the number of dysfunctional or apoptotic β-cells (134). Cytokine-induced β-cell dysfunction
or apoptosis is associated with complex gene networks that are
controlled by transcription factors, including NF-κB and those that
belong to the STAT family (135).
NF-κB is derived from ER stress signals, whilst members of the STAT
family serve as signal transducers and activators (135). Parkin is a RING-between-RING-type
E3 ubiquitin ligase that modulates the K63 ubiquitination status of
receptor-interacting serine/threonine-protein kinase 1 to promote
NF-κB activation (136). These
inflammation-stimulating reactions result in the activation of
NF-κB, which then triggers the transcription of genes associated
with pro-inflammatory responses (66). Myeloid differentiation primary
response gene 88 (MyD88) and interleukin-1 receptor-associated
kinase (IRAK) are regulated by NF-κB (137). MyD88 and IRAK serve indispensable
roles in the pathogenesis of type-1 DM (138). NF-κB activation enhances the
transcription of proapoptotic proteins whilst inhibiting that of
antiapoptotic proteins in β-cells, leading to higher permeability
of mitochondrial membranes (103,139). Additionally, cytochrome c
may be released through the highly permeable mitochondrial
membranes, resulting in apoptosis (71). High quantities of Ca2+
released into β-cell cytosol from the damaged ER during
inflammation can also contribute to β-cell apoptosis (60,105,140). Angiotensin II, which induces
IL-1-mediated inflammation, has been previously found to enhance
β-cell apoptosis, weaken mitochondrial function and insulin
secretion (141).
Clinical trials, in vivo and in vitro
studies, have provided useful evidence indicating that monotherapy
or adjuvant therapy targeting IL-1β in type-2 DM can confer
long-term benefits against blood sugar and mitigating diabetic
complications (134,142). Furthermore, sulforaphane is
currently being developed as a nutraceutical to preserve β-cell
function due to its reported antioxidative and anti-inflammatory
properties, along with its abilities to preserve and improve
mitochondrial bioenergetic function (143). Therefore, inflammation modulation
may prove to be a useful future therapeutic target for type-2 DM
(144,145).
4. Conclusion
Mitochondrial dysfunction in β-cells serves a
central role in the pathogenesis of DM. Specifically, timely
insulin secretion in response to increased serum glucose depends on
whether OXPHOS in mitochondria functions sufficiently to increase
ATP concentrations in islet β-cells, leading to increased cytosolic
Ca2+ and insulin exocytosis. Accumulating data have
indicated that genetic defects such as those in MIDD, mutations
such as those in TFB1 and TFB2M, dysregulated gene expression such
as that of UCPs in mitochondria, excessive ROS, cytokine impairment
and unwanted exosomes received from other mitochondria, can all
lead to mitochondrial dysfunction. Nevertheless, the mechanism of
these pathogenic processes remain unclear. Further research into
the association between dysfunctional mitochondria and β-cell
failure may lead to breakthroughs in the treatment of DM. Use of
insulin injections to downregulate blood sugar cannot fully prevent
or treat diabetic complications, since this therapy does not target
DM pathogenesis. In summary, previous research has indicated that
protection of mitochondrial function, enhancement of antioxidants
and regulation of transcription factors in islet β-cells may be
potential therapeutic targets for DM.
Acknowledgements
Not applicable.
Funding
The current review was supported by the Ningbo
Science and Technology Innovation Team Program (grant no.
2014B82002), the Ningbo 3315 Program (grant no. 2013A-10-G) and the
National Natural Science Foundation of China (grant no.
81370165).
Availability of data and materials
Not applicable.
Authors' contributions
SB designed the present study. WS prepared and
wrote the manuscript. FH performed literature research and selected
the included studies. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Schmidt AM: Highlighting diabetes
mellitus: The epidemic continues. Arterioscler Thromb Vasc Biol.
38:e1–e8. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Wang YJ, Schug J, Won KJ, Liu C, Naji A,
Avrahami D, Golson ML and Kaestner KH: Single-cell transcriptomics
of the human endocrine pancreas. Diabetes. 65:3028–3038.
2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lawlor N, George J, Bolisetty M, Kursawe
R, Sun L, Sivakamasundari V, Kycia I, Robson P and Stitzel ML:
Single-cell transcriptomes identify human islet cell signatures and
reveal cell-type-specific expression changes in type 2 diabetes.
Genome Res. 27:208–222. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Huang Q, Bu S, Yu Y, Guo Z, Ghatnekar G,
Bu M, Yang L, Lu B, Feng Z, Liu S and Wang F: Diazoxide prevents
diabetes through inhibiting pancreatic beta-cells from apoptosis
via Bcl-2/Bax rate and p38-beta mitogen-activated protein kinase.
Endocrinology. 148:81–91. 2007.PubMed/NCBI View Article : Google Scholar
|
|
5
|
International Diabetes Federation. IDF
Diabetes Atlas, 8th edition. International Diabetes Federation,
Brussels 2017. Available from: urihttp://www.diabetesatlas.orgsimplehttp://www.diabetesatlas.org.
|
|
6
|
Holman N, Young B and Gadsby R: Current
prevalence of type 1 and type 2 diabetes in adults and children in
the UK. Diabet Med. 32:1119–1120. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Blake R and Trounce IA: Mitochondrial
dysfunction and complications associated with diabetes. Biochim
Biophys Acta. 1840:1404–1412. 2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ma RCW: Epidemiology of diabetes and
diabetic complications in China. Diabetologia. 61:1249–1260.
2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ma ZA, Zhao Z and Turk J: Mitochondrial
dysfunction and β-cell failure in type 2 diabetes mellitus. Exp
Diabetes Res. 2012(703538)2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Montgomery MK: Mitochondrial dysfunction
and diabetes: Is mitochondrial transfer a friend or foe? Biology
(Basel). 8(33)2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
van der Bliek AM, Sedensky MM and Morgan
PG: Cell biology of the mitochondrion. Genetics. 207:843–871.
2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Henquin JC: Triggering and amplifying
pathways of regulation of insulin secretion by glucose. Diabetes.
49:1751–1760. 2000.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Komatsu M, Takei M, Ishii H and Sato Y:
Glucose-stimulated insulin secretion: A newer perspective. J
Diabetes Investig. 4:511–516. 2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kibbey RG, Pongratz RL, Romanelli AJ,
Wollheim CB, Cline GW and Shulman GI: Mitochondrial GTP regulates
glucose-stimulated insulin secretion. Cell Metab. 5:253–264.
2007.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Molnar MJ and Kovacs GG: Mitochondrial
diseases. Handb Clin Neurol. 145:147–155. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Fex M, Nicholas LM, Vishnu N, Medina A,
Sharoyko VV, Nicholls DG, Spégel P and Mulder H: The pathogenetic
role of β-cell mitochondria in type 2 diabetes. J Endocrinol.
236:R145–R159. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
O'Sullivan M, Rutland P, Lucas D, Ashton
E, Hendricks S, Rahman S and Bitner-Glindzicz M: Mitochondrial
m.1584A 12S m62A rRNA methylation in families with m.1555A>G
associated hearing loss. Hum Mol Genet. 24:1036–1044.
2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Bohnsack MT and Sloan KE: The
mitochondrial epitranscriptome: The roles of RNA modifications in
mitochondrial translation and human disease. Cell Mol Life Sci.
75:241–260. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Subramanian S, Kalyanaraman B and Migrino
RQ: Mitochondrially targeted antioxidants for the treatment of
cardiovascular diseases. Recent Pat Cardiovasc Drug Discov.
5:54–65. 2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Papa S, Martino PL, Capitanio G, Gaballo
A, De Rasmo D, Signorile A and Petruzzella V: The oxidative
phosphorylation system in mammalian mitochondria. Adv Exp Med Biol.
942:3–37. 2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Conley KE: Mitochondria to motion:
Optimizing oxidative phosphorylation to improve exercise
performance. J Exp Biol. 219:243–249. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Waypa GB, Smith KA and Schumacker PT: O2
sensing, mitochondria and ROS signaling: The fog is lifting. Mol
Aspects Med. 47-48:76–89. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Angelova PR and Abramov AY: Functional
role of mitochondrial reactive oxygen species in physiology. Free
Radic Biol Med. 100:81–85. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kausar S, Wang F and Cui H: The role of
mitochondria in reactive oxygen species generation and its
implications for neurodegenerative diseases. Cells.
7(274)2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Akram M: Citric acid cycle and role of its
intermediates in metabolism. Cell Biochem Biophys. 68:475–478.
2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Chiabrando D, Mercurio S and Tolosano E:
Heme and erythropoieis: More than a structural role. Haematologica.
99:973–983. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Moreno-Navarrete JM, Rodríguez A, Ortega
F, Becerril S, Girones J, Sabater-Masdeu M, Latorre J, Ricart W,
Frühbeck G and Fernández-Real JM: Heme biosynthetic pathway is
functionally linked to adipogenesis via mitochondrial respiratory
activity. Obesity (Silver Spring). 25:1723–1733. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Elustondo P, Martin LA and Karten B:
Mitochondrial cholesterol import. Biochim Biophys Acta Mol Cell
Biol Lipids. 1862:90–101. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Martin LA, Kennedy BE and Karten B:
Mitochondrial cholesterol: Mechanisms of import and effects on
mitochondrial function. J Bioenerg Biomembr. 48:137–151.
2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Bravo-Sagua R, Parra V, López-Crisosto C,
Díaz P, Quest AF and Lavandero S: Calcium transport and signaling
in mitochondria. Compr Physiol. 7:623–634. 2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Wang C, Du J, Du S, Liu Y, Li D, Zhu X and
Ni X: Endogenous H2S resists mitochondria-mediated apoptosis in the
adrenal glands via ATP5A1 S-sulfhydration in male mice. Mol Cell
Endocrinol. 474:65–73. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yan C, Duanmu X, Zeng L, Liu B and Song Z:
Mitochondrial DNA: Distribution, mutations, and elimination. Cells.
8(379)2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Roger AJ, Muñoz-Gómez SA and Kamikawa R:
The origin and diversification of mitochondria. Curr Biol.
27:R1177–R1192. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Stefano GB, Bjenning C, Wang F, Wang N and
Kream RM: Mitochondrial heteroplasmy. Adv Exp Med Biol.
982:577–594. 2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Saneto RP: Genetics of mitochondrial
disease. Adv Genet. 98:63–116. 2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Kopinski PK, Janssen KA, Schaefer PM,
Trefely S, Perry CE, Potluri P, Tintos-Hernandez JA, Singh LN,
Karch KR, Campbell SL, et al: Regulation of nuclear epigenome by
mitochondrial DNA heteroplasmy. Proc Natl Acad Sci USA.
116:16028–16035. 2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Cotney J, McKay SE and Shadel GS:
Elucidation of separate, but collaborative functions of the rRNA
methyltransferase-related human mitochondrial transcription factors
B1 and B2 in mitochondrial biogenesis reveals new insight into
maternally inherited deafness. Hum Mol Genet. 18:2670–2682.
2009.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Karasik A, Fierke CA and Koutmos M:
Interplay between substrate recognition, 5'end tRNA processing and
methylation activity of human mitochondrial RNase P. RNA.
25:1646–1660. 2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Reinhard L, Sridhara S and Hallberg BM:
The MRPP1/MRPP2 complex is a tRNA-maturation platform in human
mitochondria. Nucleic Acids Res. 45:12469–12480. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Metodiev MD, Thompson K, Alston CL, Morris
AAM, He L, Assouline Z, Rio M, Bahi-Buisson N, Pyle A, Griffin H,
et al: Recessive mutations in TRMT10C cause defects in
mitochondrial RNA processing and multiple respiratory chain
deficiencies. Am J Hum Genet. 98:993–1000. 2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Pearce SF, Rorbach J, Van Haute L, D'Souza
AR, Rebelo-Guiomar P, Powell CA, Brierley I, Firth AE and Minczuk
M: Maturation of selected human mitochondrial tRNAs requires
deadenylation. Elife. 6(e27596)2017.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Ricquier D: UCP1, the mitochondrial
uncoupling protein of brown adipocyte: A personal contribution and
a historical perspective. Biochimie. 134:3–8. 2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Li Y, Maedler K, Shu L and Haataja L:
UCP-2 and UCP-3 proteins are differentially regulated in pancreatic
beta-cells. PLoS One. 3(e1397)2008.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Pitt MA: Overexpression of uncoupling
protein-2 in cancer: Metabolic and heat changes, inhibition and
effects on drug resistance. Inflammopharmacology. 23:365–369.
2015.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Chan SHH and Chan JYH: Mitochondria and
reactive oxygen species contribute to neurogenic hypertension.
Physiology (Bethesda). 32:308–321. 2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Broche B, Ben Fradj S, Aguilar E, Sancerni
T, Bénard M, Makaci F, Berthault C, Scharfmann R, Alves-Guerra MC
and Duvillié B: Mitochondrial protein UCP2 controls pancreas
development. Diabetes. 67:78–84. 2018.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Oelkrug R, Polymeropoulos ET and Jastroch
M: Brown adipose tissue: Physiological function and evolutionary
significance. J Comp Physiol B. 185:587–606. 2015.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Giralt M and Villarroya F: Mitochondrial
uncoupling and the regulation of glucose homeostasis. Curr Diabetes
Rev. 13:386–394. 2017.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Hu M, Lin H, Yang L, Cheng Y and Zhang H:
Interleukin-22 restored mitochondrial damage and impaired
glucose-stimulated insulin secretion through down-regulation of
uncoupling protein-2 in INS-1 cells. J Biochem. 161:433–439.
2017.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Nanayakkara GK, Wang H and Yang X: Proton
leak regulates mitochondrial reactive oxygen species generation in
endothelial cell activation and inflammation-A novel concept. Arch
Biochem Biophys. 662:68–74. 2019.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Rugarli E and Trifunovic A: Is
mitochondrial free radical theory of aging getting old? Biochim
Biophys Acta. 1847:1345–1346. 2015.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Cheeseman KH and Slater TF: An
introduction to free radical biochemistry. Br Med Bull. 49:481–493.
1993.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Brieger K, Schiavone S, Miller FJ Jr and
Krause KH: Reactive oxygen species: From health to disease. Swiss
Med Wkly. 142(w13659)2012.PubMed/NCBI View Article : Google Scholar
|
|
54
|
He L, He T, Farrar S, Ji L, Liu T and Ma
X: Antioxidants maintain cellular redox homeostasis by elimination
of reactive oxygen species. Cell Physiol Biochem. 44:532–553.
2017.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Vallabh NA, Romano V and Willoughby CE:
Mitochondrial dysfunction and oxidative stress in corneal disease.
Mitochondrion. 36:103–113. 2017.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Panieri E and Santoro MM: ROS homeostasis
and metabolism: A dangerous liason in cancer cells. Cell Death Dis.
7(e2253)2016.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Loperena R and Harrison DG: Oxidative
stress and hypertensive diseases. Med Clin North Am. 101:169–193.
2017.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Rani V, Deep G, Singh RK, Palle K and
Yadav UC: Oxidative stress and metabolic disorders: Pathogenesis
and therapeutic strategies. Life Sci. 148:183–193. 2016.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Gerber PA and Rutter GA: The role of
oxidative stress and hypoxia in pancreatic beta-cell dysfunction in
diabetes mellitus. Antioxid Redox Signal. 26:501–518.
2017.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Pramanik KC, Boreddy SR and Srivastava SK:
Role of mitochondrial electron transport chain complexes in
capsaicin mediated oxidative stress leading to apoptosis in
pancreatic cancer cells. PLoS One. 6(e20151)2011.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Sena LA and Chandel NS: Physiological
roles of mitochondrial reactive oxygen species. Mol Cell.
48:158–167. 2012.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Bugger H, Chen D, Riehle C, Soto J,
Theobald HA, Hu XX, Ganesan B, Weimer BC and Abel ED:
Tissue-specific remodeling of the mitochondrial proteome in type 1
diabetic akita mice. Diabetes. 58:1986–1997. 2009.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Makino A, Scott BT and Dillmann WH:
Mitochondrial fragmentation and superoxide anion production in
coronary endothelial cells from a mouse model of type 1 diabetes.
Diabetologia. 53:1783–1794. 2010.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Broderick TL: ATP production and TCA
activity are stimulated by propionyl-L-carnitine in the diabetic
rat heart. Drugs R D. 9:83–91. 2008.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Anello M, Lupi R, Spampinato D, Piro S,
Masini M, Boggi U, Del Prato S, Rabuazzo AM, Purrello F and
Marchetti P: Functional and morphological alterations of
mitochondria in pancreatic beta cells from type 2 diabetic
patients. Diabetologia. 48:282–289. 2005.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Paradies G, Paradies V, Ruggiero FM and
Petrosillo G: Oxidative stress, cardiolipin and mitochondrial
dysfunction in nonalcoholic fatty liver disease. World J
Gastroenterol. 20:14205–14218. 2014.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Musatov A, Carroll CA, Liu YC, Henderson
GI, Weintraub ST and Robinson NC: Identification of bovine heart
cytochrome c oxidase subunits modified by the lipid peroxidation
product 4-hydroxy-2-nonenal. Biochemistry. 41:8212–8220.
2002.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Sinha K, Das J, Pal PB and Sil PC:
Oxidative stress: The mitochondria-dependent and
mitochondria-independent pathways of apoptosis. Arch Toxicol.
87:1157–1180. 2013.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Molina AJ, Wikstrom JD, Stiles L, Las G,
Mohamed H, Elorza A, Walzer G, Twig G, Katz S, Corkey BE and
Shirihai OS: Mitochondrial networking protects beta-cells from
nutrient-induced apoptosis. Diabetes. 58:2303–2315. 2009.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Morino K, Petersen KF, Dufour S, Befroy D,
Frattini J, Shatzkes N, Neschen S, White MF, Bilz S, Sono S, et al:
Reduced mitochondrial density and increased IRS-1 serine
phosphorylation in muscle of insulin-resistant offspring of type 2
diabetic parents. J Clin Invest. 115:3587–3593. 2005.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Petersen KF, Dufour S, Befroy D, Garcia R
and Shulman GI: Impaired mitochondrial activity in the
insulin-resistant offspring of patients with type 2 diabetes. N
Engl J Med. 350:664–671. 2004.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Dan Dunn J, Alvarez LA, Zhang X and
Soldati T: Reactive oxygen species and mitochondria: A nexus of
cellular homeostasis. Redox Biol. 6:472–485. 2015.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Kauppila TES, Kauppila JHK and Larsson NG:
Mammalian mitochondria and aging: An update. Cell Metab. 25:57–71.
2017.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Ye X, Sun X, Starovoytov V and Cai Q:
Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer's
disease patient brains. Hum Mol Genet. 24:2938–2951.
2015.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Fivenson EM, Lautrup S, Sun N,
Scheibye-Knudsen M, Stevnsner T, Nilsen H, Bohr VA and Fang EF:
Mitophagy in neurodegeneration and aging. Neurochem Int.
109:202–209. 2017.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Choi DS, Kim DK, Kim YK and Gho YS:
Proteomics, transcriptomics and lipidomics of exosomes and
ectosomes. Proteomics. 13:1554–1571. 2013.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Alenquer M and Amorim MJ: Exosome
biogenesis, regulation, and function in viral infection. Viruses.
7:5066–5083. 2015.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Shakeri R, Kheirollahi A and Davoodi J:
Apaf-1: Regulation and function in cell death. Biochimie.
135:111–125. 2017.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Thorens B: GLUT2, glucose sensing and
glucose homeostasis. Diabetologia. 58:221–232. 2015.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Nicholls DG: The pancreatic β-cell: A
bioenergetic perspective. Physiol Rev. 96:1385–1447.
2016.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Ježek P and Dlasková A: Dynamic of
mitochondrial network, cristae, and mitochondrial nucleoids in
pancreatic β-cells. Mitochondrion. 49:245–258. 2019.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Mulder H: Transcribing β-cell mitochondria
in health and disease. Mol Metab. 6:1040–1051. 2017.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Kwak SH and Park KS: Role of mitochondrial
DNA variation in the pathogenesis of diabetes mellitus. Front
Biosci (Landmark Ed). 21:1151–1167. 2016.PubMed/NCBI View
Article : Google Scholar
|
|
85
|
Jiang Z, Zhang Y, Yan J, Li F, Geng X, Lu
H, Wei X, Feng Y, Wang C and Jia W: De novo mutation of
m.3243A>G together with m.16093T>C associated with atypical
clinical features in a pedigree with MIDD syndrome. J Diabetes Res.
2019(5184647)2019.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Alves D, Calmeiro ME, Macário C and Silva
R: Family phenotypic heterogeneity caused by mitochondrial DNA
mutation A3243G. Acta Med Port. 30:581–585. 2017.PubMed/NCBI View Article : Google Scholar
|
|
87
|
El-Hattab AW, Emrick LT, Hsu JW,
Chanprasert S, Jahoor F, Scaglia F and Craigen WJ: Glucose
metabolism derangements in adults with the MELAS m.3243A>G
mutation. Mitochondrion. 18:63–69. 2014.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Meimaridou E, Goldsworthy M, Chortis V,
Fragouli E, Foster PA, Arlt W, Cox R and Metherell LA: NNT is a key
regulator of adrenal redox homeostasis and steroidogenesis in male
mice. J Endocrinol. 236:13–28. 2018.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Santos LRB, Muller C, de Souza AH,
Takahashi HK, Spégel P, Sweet IR, Chae H, Mulder H and Jonas JC:
NNT reverse mode of operation mediates glucose control of
mitochondrial NADPH and glutathione redox state in mouse pancreatic
β-cells. Mol Metab. 6:535–547. 2017.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Dutta P, Ma L, Ali Y, Sloot PMA and Zheng
J: Boolean network modeling of β-cell apoptosis and insulin
resistance in type 2 diabetes mellitus. BMC Syst Biol. 13 (Suppl
2)(S36)2019.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Tabebi M, Khabou B, Boukadi H, Ben Hamad
M, Ben Rhouma B, Tounsi S, Maalej A, Kamoun H, Keskes-Ammar L, Abid
M, et al: Association study of apoptosis gene polymorphisms in
mitochondrial diabetes: A potential role in the pathogenicity of
MD. Gene. 639:18–26. 2018.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Zhang J, Liu Y, Yang HW, Xu HY and Meng Y:
Molecular mechanism of beta cell apoptosis induced by p58 in high
glucose medium. Sheng Li Xue Bao. 61:379–385. 2009.(In Chinese).
PubMed/NCBI
|
|
93
|
Han J, Song B, Kim J, Kodali VK, Pottekat
A, Wang M, Hassler J, Wang S, Pennathur S, Back SH, et al:
Antioxidants complement the requirement for protein chaperone
function to maintain β-cell function and glucose homeostasis.
Diabetes. 64:2892–2904. 2015.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Vozza A, Parisi G, De Leonardis F, Lasorsa
FM, Castegna A, Amorese D, Marmo R, Calcagnile VM, Palmieri L,
Ricquier D, et al: UCP2 transports C4 metabolites out of
mitochondria, regulating glucose and glutamine oxidation. Proc Natl
Acad Sci USA. 111:960–965. 2014.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Collins S, Pi J and Yehuda-Shnaidman E:
Uncoupling and reactive oxygen species (ROS)-a double-edged sword
for β-cell function? ‘Moderation in all things’. Best Pract Res
Clin Endocrinol Metab. 26:753–758. 2012.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Emre Y, Hurtaud C, Karaca M, Nubel T,
Zavala F and Ricquier D: Role of uncoupling protein UCP2 in
cell-mediated immunity: How macrophage-mediated insulitis is
accelerated in a model of autoimmune diabetes. Proc Natl Acad Sci
USA. 104:19085–19090. 2007.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Lee SC, Robson-Doucette CA and Wheeler MB:
Uncoupling protein 2 regulates reactive oxygen species formation in
islets and influences susceptibility to diabetogenic action of
streptozotocin. J Endocrinol. 203:33–43. 2009.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Sharoyko VV, Abels M, Sun J, Nicholas LM,
Mollet IG, Stamenkovic JA, Göhring I, Malmgren S, Storm P, Fadista
J, et al: Loss of TFB1M results in mitochondrial dysfunction that
leads to impaired insulin secretion and diabetes. Hum Mol Genet.
23:5733–5749. 2014.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Nicholas LM, Valtat B, Medina A, Andersson
L, Abels M, Mollet IG, Jain D, Eliasson L, Wierup N, Fex M and
Mulder H: Mitochondrial transcription factor B2 is essential for
mitochondrial and cellular function in pancreatic β-cells. Mol
Metab. 6:651–663. 2017.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Baixauli F, López-Otín C and Mittelbrunn
M: Exosomes and autophagy: Coordinated mechanisms for the
maintenance of cellular fitness. Front Immunol.
5(403)2014.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Wong SK, Chin KY, Suhaimi FH, Ahmad F and
Ima-Nirwana S: The effects of a modified high-carbohydrate high-fat
diet on metabolic syndrome parameters in male rats. Exp Clin
Endocrinol Diabetes. 126:205–212. 2018.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Rutter GA, Pullen TJ, Hodson DJ and
Martinez-Sanchez A: Pancreatic β-cell identity, glucose sensing and
the control of insulin secretion. Biochem J. 466:203–218.
2015.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Newsholme P, Cruzat VF, Keane KN, Carlessi
R and de Bittencourt PI Jr: Molecular mechanisms of ROS production
and oxidative stress in diabetes. Biochem J. 473:4527–4550.
2016.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Rehman K and Akash MSH: Mechanism of
generation of oxidative stress and pathophysiology of type 2
diabetes mellitus: How are they interlinked? J Cell Biochem.
118:3577–3585. 2017.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Rharass T, Lemcke H, Lantow M, Kuznetsov
SA, Weiss DG and Panáková D: Ca2+-mediated mitochondrial reactive
oxygen species metabolism augments Wnt/beta-catenin pathway
activation to facilitate cell differentiation. J Biol Chem.
289:27937–27951. 2014.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Sarre A, Gabrielli J, Vial G, Leverve XM
and Assimacopoulos-Jeannet F: Reactive oxygen species are produced
at low glucose and contribute to the activation of AMPK in
insulin-secreting cells. Free Radic Biol Med. 52:142–150.
2012.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Furukawa S, Fujita T, Shimabukuro M, Iwaki
M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M and
Shimomura I: Increased oxidative stress in obesity and its impact
on metabolic syndrome. J Clin Invest. 114:1752–1761.
2004.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Nowotny K, Jung T, Höhn A, Weber D and
Grune T: Advanced glycation end products and oxidative stress in
type 2 diabetes mellitus. Biomolecules. 5:194–222. 2015.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Turrens JF: Mitochondrial formation of
reactive oxygen species. J Physiol. 552:335–344. 2003.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Wang J and Wang H: Oxidative stress in
pancreatic beta cell regeneration. Oxid Med Cell Longev.
2017(1930261)2017.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Ivarsson R, Quintens R, Dejonghe S,
Tsukamoto K, in 't Veld P, Renström E and Schuit FC: Redox control
of exocytosis: Regulatory role of NADPH, thioredoxin, and
glutaredoxin. Diabetes. 54:2132–2142. 2005.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Hopps E, Noto D, Caimi G and Averna MR: A
novel component of the metabolic syndrome: The oxidative stress.
Nutr Metab Cardiovasc Dis. 20:72–77. 2010.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Rao R: Oxidative stress-induced disruption
of epithelial and endothelial tight junctions. Front Biosci.
13:7210–7226. 2008.PubMed/NCBI View
Article : Google Scholar
|
|
114
|
Newsholme P, Rebelato E, Abdulkader F,
Krause M, Carpinelli A and Curi R: Reactive oxygen and nitrogen
species generation, antioxidant defenses, and β-cell function: A
critical role for amino acids. J Endocrinol. 214:11–20.
2012.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Fiorentino TV, Prioletta A, Zuo P and
Folli F: Hyperglycemia-induced oxidative stress and its role in
diabetes mellitus related cardiovascular diseases. Curr Pharm Des.
19:5695–5703. 2013.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Koehler A and Van Noorden CJ: Reduced
nicotinamide adenine dinucleotide phosphate and the higher
incidence of pollution-induced liver cancer in female flounder.
Environ Toxicol Chem. 22:2703–2710. 2003.PubMed/NCBI View
Article : Google Scholar
|
|
117
|
Baldewpersad Tewarie NM, Burgers IA,
Dawood Y, den Boon HC, den Brok MG, Klunder JH, Koopmans KB,
Rademaker E, van den Broek HB, van den Bersselaar SM, et al:
NADP+-dependent IDH1 R132 mutation and its relevance for
glioma patient survival. Med Hypotheses. 80:728–731.
2013.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Atai NA, Renkema-Mills NA, Bosman J,
Schmidt N, Rijkeboer D, Tigchelaar W, Bosch KS, Troost D, Jonker A,
Bleeker FE, et al: Differential activity of NADPH-producing
dehydrogenases renders rodents unsuitable models to study IDH1R132
mutation effects in human glioblastoma. J Histochem Cytochem.
59:489–503. 2011.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Pan HC, Lee CC, Chou KM, Lu SC and Sun CY:
Serum levels of uncoupling proteins in patients with differential
insulin resistance: A community-based cohort study. Medicine
(Baltimore). 96(e8053)2017.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Brondani LA, Assmann TS, Duarte GC, Gross
JL, Canani LH and Crispim D: The role of the uncoupling protein 1
(UCP1) on the development of obesity and type 2 diabetes mellitus.
Arq Bras Endocrinol Metabol. 56:215–225. 2012.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Oelkrug R, Goetze N, Meyer CW and Jastroch
M: Antioxidant properties of UCP1 are evolutionarily conserved in
mammals and buffer mitochondrial reactive oxygen species. Free
Radic Biol Med. 77:210–216. 2014.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Sreedhar A and Zhao Y: Uncoupling protein
2 and metabolic diseases. Mitochondrion. 34:135–140.
2017.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Li N, Karaca M and Maechler P:
Upregulation of UCP2 in beta-cells confers partial protection
against both oxidative stress and glucotoxicity. Redox Biol.
13:541–549. 2017.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Senese R, Valli V, Moreno M, Lombardi A,
Busiello RA, Cioffi F, Silvestri E, Goglia F, Lanni A and de Lange
P: Uncoupling protein 3 expression levels influence insulin
sensitivity, fatty acid oxidation, and related signaling pathways.
Pflugers Arch. 461:153–164. 2011.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Edwards KS, Ashraf S, Lomax TM, Wiseman
JM, Hall ME, Gava FN, Hall JE, Hosler JP and Harmancey R:
Uncoupling protein 3 deficiency impairs myocardial fatty acid
oxidation and contractile recovery following ischemia/reperfusion.
Basic Res Cardiol. 113(47)2018.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Chan CB and Harper ME: Uncoupling
proteins: Role in insulin resistance and insulin insufficiency.
Curr Diabetes Rev. 2:271–283. 2006.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Jena NR: DNA damage by reactive species:
Mechanisms, mutation and repair. J Biosci. 37:503–517.
2012.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Borchert A, Kalms J, Roth SR, Rademacher
M, Schmidt A, Holzhutter HG, Kuhn H and Scheerer P: Crystal
structure and functional characterization of
selenocysteine-containing glutathione peroxidase 4 suggests an
alternative mechanism of peroxide reduction. Biochim Biophys Acta
Mol Cell Biol Lipids. 1863:1095–1107. 2018.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Jung CH and Choi KM: Impact of
high-carbohydrate diet on metabolic parameters in patients with
type 2 diabetes. Nutrients. 9(322)2017.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Li C, Deng X, Xie X, Liu Y, Friedmann
Angeli JP and Lai L: Activation of glutathione peroxidase 4 as a
novel anti-inflammatory strategy. Front Pharmacol.
9(1120)2018.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Lillig CH and Holmgren A: Thioredoxin and
related molecules-from biology to health and disease. Antioxid
Redox Signal. 9:25–47. 2007.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Eguchi K and Nagai R: Islet inflammation
in type 2 diabetes and physiology. J Clin Invest. 127:14–23.
2017.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Margaryan S, Witkowicz A, Partyka A,
Yepiskoposyan L, Manukyan G and Karabon L: The mRNA expression
levels of uncoupling proteins 1 and 2 in mononuclear cells from
patients with metabolic disorders: Obesity and type 2 diabetes
mellitus. Postepy Hig Med Dosw (Online). 71:895–900.
2017.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Dalmas E, Venteclef N, Caer C, Poitou C,
Cremer I, Aron-Wisnewsky J, Lacroix-Desmazes S, Bayry J, Kaveri SV,
Clément K, et al: T cell-derived IL-22 amplifies IL-1β-driven
inflammation in human adipose tissue: Relevance to obesity and type
2 diabetes. Diabetes. 63:1966–1977. 2014.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Oh H, Park SH, Kang MK, Kim YH, Lee EJ,
Kim DY, Kim SI, Oh S, Lim SS and Kang YH: Asaronic acid attenuates
macrophage activation toward M1 phenotype through inhibition of
NF-κB pathway and JAK-STAT signaling in glucose-loaded murine
macrophages. J Agric Food Chem, 2019.
|
|
136
|
Wang Y, Shan B, Liang Y, Wei H and Yuan J:
Parkin regulates NF-κB by mediating site-specific ubiquitination of
RIPK1. Cell Death Dis. 9(732)2018.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Kim DH, Lee JC, Kim S, Oh SH, Lee MK, Kim
KW and Lee MS: Inhibition of autoimmune diabetes by TLR2 tolerance.
J Immunol. 187:5211–5220. 2011.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Tan Q, Majewska-Szczepanik M, Zhang X,
Szczepanik M, Zhou Z, Wong FS and Wen L: IRAK-M deficiency promotes
the development of type 1 diabetes in NOD mice. Diabetes.
63:2761–2775. 2014.PubMed/NCBI View Article : Google Scholar
|
|
139
|
QiNan W, XiaGuang G, XiaoTian L, WuQuan D,
Ling Z and Bing C: Par-4/NF-κB mediates the apoptosis of islet β
cells induced by glucolipotoxicity. J Diabetes Res.
2016(4692478)2016.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Cnop M, Toivonen S, Igoillo-Esteve M and
Salpea P: Endoplasmic reticulum stress and eIF2α phosphorylation:
The Achilles heel of pancreatic β cells. Mol Metab. 6:1024–1039.
2017.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Sauter NS, Thienel C, Plutino Y, Kampe K,
Dror E, Traub S, Timper K, Bédat B, Pattou F, Kerr-Conte J, et al:
Angiotensin II induces interleukin-1β-mediated islet inflammation
and β-cell dysfunction independently of vasoconstrictive effects.
Diabetes. 64:1273–1283. 2015.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Dinarello CA, Donath MY and
Mandrup-Poulsen T: Role of IL-1beta in type 2 diabetes. Curr Opin
Endocrinol Diabetes Obes. 17:314–321. 2010.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Carrasco-Pozo C, Tan KN Gotteland M and
Borges K: Sulforaphane protects against high cholesterol-induced
mitochondrial bioenergetics impairments, inflammation, and
oxidative stress and preserves pancreatic β-cells function. Oxid
Med Cell Longev. 2017(3839756)2017.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Donath MY and Shoelson SE: Type 2 diabetes
as an inflammatory disease. Nat Rev Immunol. 11:98–107.
2011.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Gomes BF and Accardo CM:
Immunoinflammatory mediators in the pathogenesis of diabetes
mellitus. Einstein (Sao Paulo). 17(eRB4596)2019.PubMed/NCBI View Article : Google Scholar : (In En,
Portuguese).
|