Myocardial infarction (MI) is one of the leading
causes of mortality worldwide and occurs due to the acute occlusion
of the coronary arteries (1).
Although revascularization treatment has conferred proven efficacy
for patients with MI, it also causes undesired ischemia/reperfusion
(I/R) injury following the restoration of epicardial blood flow
(2,3). I/R injury is defined as tissue injury
that occurs when the blood supply to organs is interrupted and then
returns (4). To the frustration of
interventional cardiologists and other health professionals, the
desire of whom is the fast restoration of blood flow to the heart
muscle, successful therapeutic strategies that can prevent I/R
injury in the clinic have yet to be established (5).
The endoplasmic reticulum (ER) is an important
organelle for eukaryotic cell survival and development (6,7). It is
responsible for the biosynthesis, folding, assembly and
modification of most secreted and transmembrane proteins.
Furthermore, it serves a role in cellular lipid and steroid
synthesis (8). Approximately 33% of
cellular protein production and folding occurs in the ER (9). Excessive protein synthesis, beyond the
capacity of the folding mechanism in cells, or excess accumulation
of unfolded/misfolded proteins in the ER lumen will disrupt ER
homeostasis and trigger the unfolded protein response (UPR),
eventually leading to ER stress (ERS) (10). Events in the process of I/R can
alter ER function and consequently influence the accumulation of
unfolded/misfolded proteins. The resulting ERS then induces the
activation of three signal transduction pathways, including the
protein kinase R-like endoplasmic reticulum kinase
(PERK)-eukaryotic translation initiation factor 2A
(eIF2a)-activating transcription factor (ATF) 4-C/EBP homologous
protein (CHOP) pathway, pro-ATF6-ATF6-CHOP pathway and inositol
requiring enzyme 1 (IRE1)-X-box binding protein 1 (XBP1) pathway,
which in turn promote the development of I/R injury (11).
The aim of the present review was to summarize
current understanding of the multifactorial mechanisms that
contribute to the genesis of I/R injury, and the relationship
between I/R and ERS. In addition, possible future targets of
therapeutic interventions to enhance recovery after I/R were
discussed.
During cardiac ischemia, myocardial cells are in a
state of hypoxia, where mitochondrial electron transfer chain
complexes are significantly reduced and SOD anions are produced
(34). During reperfusion, ROS
levels are increased significantly due to the reduction in electron
leakage and mitochondrial detoxification (34), causing oxidative stress. Free
radical explosion and oxidative stress are important mechanisms of
myocardial I/R injury. Mitochondrial electron transfer chain
complex I is inactivated during myocardial ischemia because of the
highly reductive environment with low PO2 and low ADP
(35). After reperfusion, the
impaired activity of complex I can also lead to ROS-induced damage
to the mitochondrial phospholipids and respiratory chain super
complex, potentiating the electron leakage of complex I further.
This process promotes a vicious cycle of oxidative stress that
ultimately leads to mitochondrial dysfunction (36). Under these conditions, excessive
mitochondrial ROS cause oxidative damage to proteins, lipids and
DNA, as well as excitation-contraction uncoupling, arrhythmia,
cardiac hypertrophy, apoptosis, necrosis and fibrosis (37). However, low levels of ROS attenuate
myocardial I/R injury through ischemic preconditioning. Recent
evidence has suggested that short-term intermittent hypoxia (IH),
similar to ischemic preconditioning, can serve a cardioprotective
role (38). A previous study
demonstrated that IH increased mitochondrial tolerance to
Ca2+ overload and delayed MPTP opening induced by
oxidative stress (39). In
addition, a previous study has shown that IH may increase the
expression of SOD and glutathione peroxidase (40).
Long-term ischemia can lead to irreversible cellular
necrosis, which triggers the release of a variety of
pro-inflammatory mediators, including cytokines and growth factors,
leading to inflammatory cell infiltration (41). During late stage I/R, genes
associated with inflammation are activated to produce mediators,
including IL-1, IL-6, TNF-α IFN-regulating factor and NF-κB, all of
which promote neutrophil adhesion and transmembrane migration,
leukocyte infiltration, and cytokine and chemokine release,
eventually leading to cell death. I/R can activate the inflammation
cascade to cause further tissue damage (42). TNF-α participates in the development
of myocardial injury during I/R injury, during which its expression
level is increased, promoting adhesion and interaction between
leukocytes and endothelial cells (43). This increases the infiltration of
granulocytes into the I/R region to mediate myocardial cell damage
(43). IL-1 is secreted by
activated monocytes and macrophages, and its expression is also
significantly increased during I/R. Intercellular adhesion molecule
1 (ICAM-1) participates in the adhesion of leukocytes to vascular
endothelial cells and induces cytotoxicity by adhering to
cardiomyocytes (44). Enhanced
ICAM-1 binding can feed back to endothelial cells and macrophages
to promote the expression of inflammatory mediators or cytokines
(44). During I/R, the release of
inflammatory cytokines and chemokines leads to the activation of
neutrophils and macrophages, which promotes tissue damage (45). Neutrophil infiltration serves an
important role in myocardial I/R injury. This step is initiated by
the binding of vascular endothelial adhesion molecules with
corresponding ligands on neutrophils to mediate the adhesion of
neutrophils to endothelial cells. Mast cells serve an important
role in stimulating the inflammatory response by releasing
regulators that can trigger the cascade of cytokine release
(46). Cyclic inflammatory markers,
including C-reactive protein, IL-6 and IL-1, are associated with
increased infarct size and poor prognosis. In recent years,
pro-inflammatory cells such as monocytes and macrophages have been
documented to be a potential cause of MI using a number of cell
tracking and molecular imaging techniques (47).
During I/R, different gene families can also serve
distinct roles in apoptosis. Apoptosis is a tightly controlled
process that is conserved among species and involves the Bcl-2 and
caspase families of proteins in addition to oncogenes, such as
c-Myc and p53(48). The activation,
upregulation, translocation and integration of precursor Bcl-2
proteins, including Bax, BH3 interacting-domain death agonist, p53
upregulated modulator of apoptosis (PUMA) and Bcl-2 interacting
protein 3 (BNIP3), into the mitochondrial membrane within ischemic
injury tissues has been previously reported (49,50).
In addition, pro-apoptotic and anti-apoptotic Bcl-2 proteins have
been found to regulate Ca2+ homeostasis, which is an
important mechanism of I/R injury (51). The caspase family also serves a key
role in I/R-induced cell death. Pan-cysteine aspartase inhibitors,
including zVAD-FMK and MX1013, can attenuate apoptosis and cell
death induced by I/R (52,53). However, it has been previously
reported that caspase inhibition may instead drive the cell towards
necrotic death (54). A number of
studies have demonstrated that the overexpression of BNIP3 in HL-1
myocardial cells can activate Bax to promote the opening of the
MPTP and increase cell death in response to I/R injury (55,56).
miRNAs are short, single-stranded non-coding RNAs
that are 21-23 nucleotides in length and regulate gene expression
by inhibiting translation or promoting the degradation of RNA
(57). Mature miRNAs are processed
from primary miRNA, which is cleaved by a microprocessor complex
that consists of the RNase-III endonuclease Drosha, RNA-binding
protein DiGeorge syndrome critical region gene 8 and other
cofactors, to produce 70-100 nucleotide hairpin precursor small
RNAs. Following export to the cytoplasm by the nuclear export
protein exportin-5, they are trimmed by the RNase III ribonuclease
dicer to produce a mature miRNA duplex that is ~21 nucleotides in
length (58).
Several studies have demonstrated that miRNA
function is closely associated with cardiovascular disease, and a
number of non-cardiac miRNAs are reported to be biomarkers of
myocardial injury and predictors of clinical outcomes after acute
MI. miR-633b and miR-1291 have been documented to indicate MI with
high specificity and sensitivity (59), whereas miR-150 and miR-486
expression levels could be used to distinguish between patients
with and without ST-elevation MI (60). A previous study revealed that heart
biopsies from patients with heart failure demonstrated a
significant increase in miR-377 expression compared with that in
normal control hearts (61). In a
mouse cardiac I/R model, human CD34+ cells in
immune-deficient mice were silenced following the intra-myocardial
transplantation of miR-377, which promoted neovascularization and
reduced interstitial fibrosis 28 days after I/R induction to
improve left ventricular function (61).
MiRNAs serve significant roles in cardiac I/R injury
and function by a wide range of different mechanisms. A previous
study demonstrated that miR-1 and miR-133 mediated opposite effects
when regulating myocyte survival in I/R models, where miR-1 was
pro-apoptotic and miR-133 was anti-apoptotic (62). This difference may be due to their
respective downstream targets. Increased miR-1 expression resulted
in the downregulation of several anti-apoptotic genes, including
heat shock protein (hsp)60, hsp70, insulin-like growth factor-1 and
Bcl-2, whereas miR-133 negatively regulated the expression of
pro-apoptotic genes, such as caspase-9 and caspase-3 (62-64).
Another study revealed that miR-133 overexpression reduced cardiac
fibrosis after transverse aortic banding compared with that in
normal controls, implicating the cardioprotective effects of
miR-133a on I/R-triggered cardiac remodeling (65). miR-21 has been demonstrated to
protect cardiomyocytes from I/R injury by targeting several
apoptotic genes, including phosphatase and tensin homolog, cell
death 4 and Fas ligand (66-68).
Furthermore, miR-21 has been reported to inhibit the proliferation,
migration and tubulogenesis of endothelial cells, and promote the
survival of cardiomyocytes and cardiac fibroblasts after myocardial
I/R (69). miR-25 and miR-145 can
reduce mitochondrial ROS stress and Ca2+ overload by
inhibiting the expression of mitochondrial Ca2+
uniporter and Ca2+/calmodulin-dependent protein kinase
II (CaMKII) (70). miR-214 may
protect cardiomyocytes from oxidative damage induced by ROS
formation initiated by Ca2+ overload by inhibiting
sodium/Ca2+ exchanger 1 (71-73).
From the aforementioned studies, it can be concluded
that miRNAs regulate the expression of pro-apoptotic/anti-apoptotic
genes to regulate cardiac fibrosis, inflammation, ROS generation
and Ca2+ homeostasis. A number of studies have
demonstrated that various miRNAs, including miR-21(67), miR-144/451(74), miR-192(75) and miR-199a (76), are associated with ischemic
preconditioning. Additionally, some experimental studies have
revealed that inhibition of miR-15 and miR-92a in pig models of
acute myocardial I/R, especially at the beginning of reperfusion,
can reduce the size of the MI (77,78).
This suggests that miRNA treatment may be a feasible therapeutic
approach (75).
ERS is an evolutionarily conserved cell stress
response that is associated with numerous diseases, including
cardiovascular, Alzheimer's and Parkinson's diseases, and diabetes,
renal failure, osteosarcoma and pancreatic ductal adenocarcinoma
(79-85).
Under physiological conditions, the ER is an important organelle
that serves a key role in cellular processes, including protein
folding, assembly, modification and secretion, lipid synthesis and
Ca2+ storage. However, when the ER is exposed to stress
stimuli, such as ROS exposure and Ca2+ overload,
homeostasis is impaired, which results in the accumulation of
unfolded/misfolded proteins (86).
These changes may eventually lead to ER dysfunction, collectively
known as ERS (86).
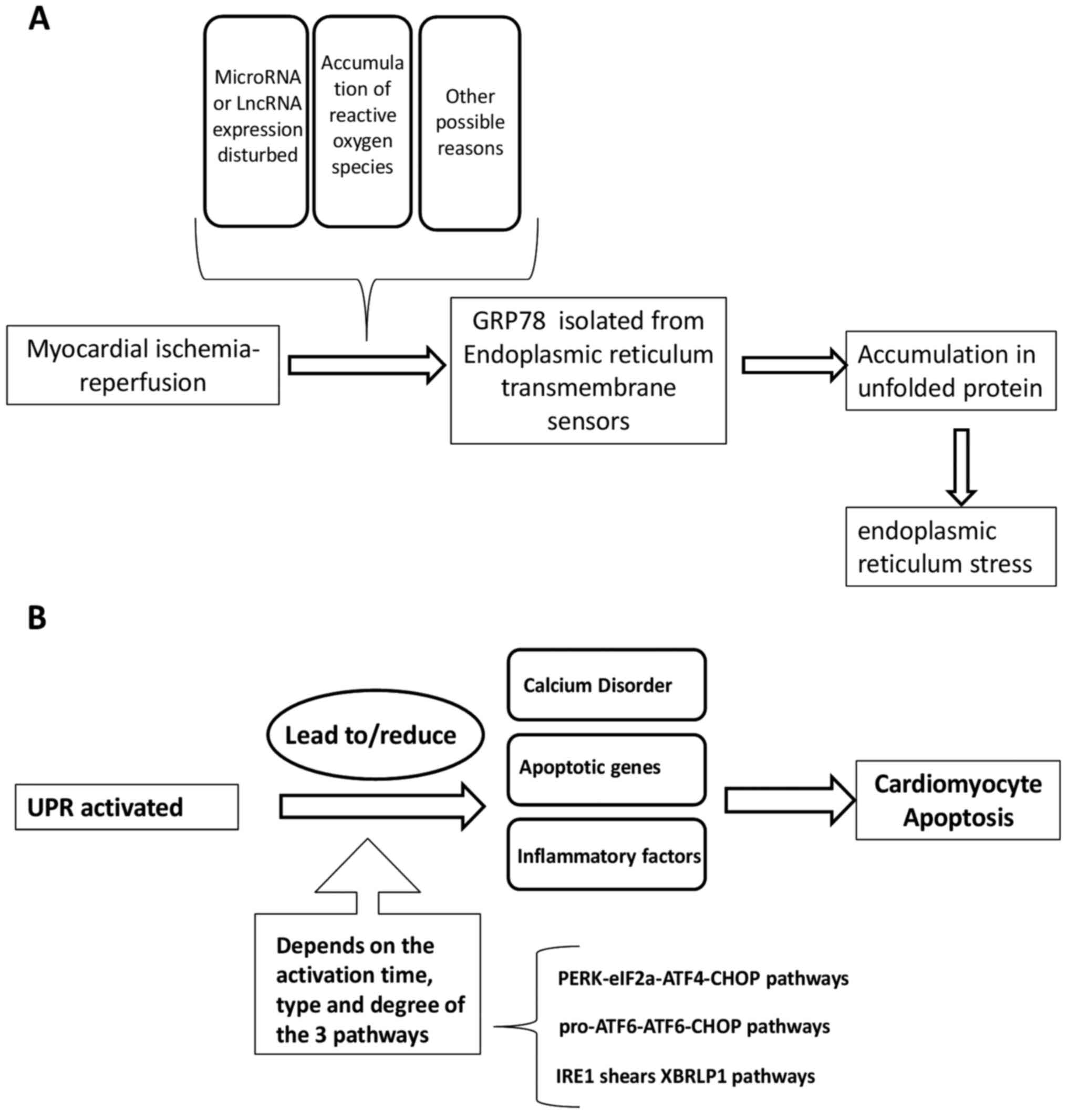
Several ER transmembrane sensors are expressed to
detect the accumulation of unfolded proteins, including PERK, ATF6
and IRE1, which activate the signal pathways of
(eIF2α-ATF4-DNA-damage-inducible transcript/CHOP, pro-ATF6-cleaved
ATF6-CHOP, and IRE1-spliced (Xbp1), respectively (79). This upregulates the expression of ER
chaperones and ER-related degradation components (6). The UPR can activate the ER chaperone
glucose-regulated protein 78 (GRP78) following isolation by any of
the three ER sensors (PERK, ATF6 and IRE1). In the absence of ERS,
binding to GRP78 results in the inactivation of these sensors.
GRP78 is released from the sensors, where they can interact with
misfolded and unfolded proteins when ERS occurs. Ultimately, the
UPR is triggered by the transcription of genes encoding proteins
involved in this process, leading to a reduction in global protein
synthesis (87). The ultimate
purpose of the UPR is to restore normal ER function, the failure of
which results in apoptosis (88,89).
The three main ERS pathways are described in the following
sections.
A previous study indicated that the PERK signaling
pathway serves an essential role in preventing the abnormal
accumulation of unfolded proteins in the ER to promote cell
survival (90). PERK is a type I
transmembrane ER protein that has a ligand-independent dimerization
domain at the N-terminus, which is concealed by binding
immunoglobulin protein (BIP)/GRP78 in the absence of ERS, and a
serine/threonine protein kinase domain at the C-terminus without
endonuclease activity (91). PERK
can block the translation of most proteins, leaving only a specific
few, such as ATF4 and CHOP, to be translated (92,93).
Translation of ATF4 activates the expression of CHOP by directly
interacting with its 5'-untranslated region (92).
Activation of PERK leads to eIF2α phosphorylation.
In addition, it promotes caspase-12 and CHOP overexpression, which
can direct ERS towards cell apoptosis (94). CHOP can in turn activate downstream
targets during ERS, resulting in apoptotic cell death (95).
One arm of the UPR is the activation of the ER
membrane protein ATF6, a fragment of which is translocated into the
nucleus to activate the transcription of genes that mediate protein
folding (96). ATF6 has two
subtypes: ATF6α and ATF6β (96).
The accumulation of misfolded proteins causes ATF6α to be
transported to the Golgi apparatus (97,98).
There, it is sheared and the N-terminal fragment, p50-ATF6a, is
transferred to the nucleus, where it regulates the transcription of
genes associated with protein quality control, translocation,
folding and degradation (99). ERS
leads to the vesicular exit of ER ATF6, which is subsequently
degraded by site-1 and site-2 proteases (S1P and S2P) in the Golgi
complex. This cleavage cuts off the cytoplasmic domain of ATF6 from
its transmembrane anchorage and intraluminal domain, following
which the cytoplasmic ATF6 domain enters the nucleus to
transcriptionally upregulate UPR target genes (100).
IRE1 is a type I transmembrane glycoprotein that can
be divided into two categories: IRE1α and IRE1β. IRE1α is widely
expressed in different tissues, whereas IRE1β is only expressed in
intestinal epithelial cells (101). IRE1α senses the accumulation of
unfolded proteins and is activated by dissociation with the ER
chaperone GRP78/BIP (102-104).
IRE1 then dimerizes and trans-autophosphorylates itself to activate
its endonuclease domain under ERS. This endonuclease domain then
acts on the Xbp1 gene and performs an unconventional splicing.
After 26 nucleotides are removed, a spliced mRNA is produced, which
increases the transcription of UPR target genes (105). Activation of the ER splicing
factor IRE1α and the splicing transcription factor Xbp1 can induce
the transcription of chaperones, which are necessary for
facilitating protein folding (105). A previous study reported that the
activation of the PERK-eIF2α and IRE1α-Xbp1 signaling pathways
inhibited apoptosis and promoted proliferation without affecting
ERK and AKT signaling activation (93). The UPR has been associated with a
number of diseases, including cardiovascular, Alzheimer's and
Parkinson's diseases, and IRE1 has been the focus of several drug
discovery projects such as ligands that interact with IREα's kinase
and pre-emptive activation of IRE1α's homeostatic mode (79,80,106).
The UPR is associated with numerous pathological
processes, including cardiovascular disease, I/R injury,
neurodegenerative diseases, diabetes mellitus, viral infection and
cancer (107). Some of the
earliest studies on the effects of I/R on the UPR were conducted in
the brain (81,108). A previous study has demonstrated
that several pathways of the UPR are activated in the ischemic
rabbit brain such as that of PERK-Xbp1-eIF2α, leading to
translation arrest (108). Several
studies have demonstrated that Xpb1, genetic markers of GRP78 and
the UPR are activated in hypoxic cultured ventricular myocytes or
HL-1 atrial myocytes from neonatal rats or adult mice (109-111).
Therefore, ischemia and I/R can activate numerous components of the
UPR in cardiomyocytes both in vivo and in vitro
(112). In a neuronal studie ERS
has been reported to be associated with neuronal cell death
following ischemia (113). A study
demonstrated that global cerebral I/R induced time-dependent
differences in ER gene expression at both mRNA and protein levels,
which was affected by pre-ischemic therapy (114).
The ER serves a pivotal role in cardiomyocytes, as
the correct synthesis and folding of proteins in the ER is
indispensable for the normal functioning of the heart (123). However, although ERS and the UPR
have been extensively studied in non-muscle ER, there remains an
insufficient number of studies on ERS and the UPR in the
cardiovascular field (124).
If the UPR signal activation during the early stages
of ERS is not sufficient in resolving stress, the persistent
activation of proximal effectors (PRRK, ATF6 and IRE1) will result
in the appearance of a distinct UPR-induced protein setup, where
other signaling pathways are activated, all of which combine to
promote cell death (125,126). Notably, a previous study indicated
that pre-activation of ATF6 in the hearts of transgenic mice
conferred protective effects against I/R injury (127). In addition, a study indicated that
the upregulation of GRP78 during ischemic preconditioning protected
cultured cardiomyocytes from further ischemic damage (128). These studies suggested that when
the UPR is activated in the heart during ischemia or I/R, it may
exert protective effects against the stress response in myocardial
cells. By contrast, several studies have demonstrated that UPR may
lead to I/R injury of the heart. A previous study demonstrated that
overexpression of the ERS response protein PUMA potentiated
apoptosis in cultured cardiomyocytes via the UPR (129). Another study revealed that UPR
activation promoted the activation of caspase-3, JNK and p53, which
contributed to cardiomyocyte apoptosis (130). Additionally, in cultured cardiac
myocytes, UPR mediated protective effects against ischemia
activation in the early stages of ischemia, whereas the same
response resulted in predominantly apoptotic characteristics in the
latter stages (131). The distinct
functions of the UPR may be dependent on the degree of ATF6, PERK
and IRE-1 activation, and the nature of the ERS. ATF6 may mediate
the activation of mostly protective proteins, whilst PERK may
induce the activation of apoptotic genes (127). Therefore, brief ischemic stress
may lead to changes in the proteome under the regulation of the UPR
to promote protective effects, whereas prolonged ischemia may lead
to changes in the proteome leading to cellular damage.
It has previously been reported that pathological
ERS is relevant in a variety of physiological outcomes, including
impaired Ca2+ homeostasis, increased apoptotic
signaling, disrupted protein secretion and increased apoptotic
signaling (132-134).
During ERS, CHOP has been demonstrated to induce the
expression of ER oxidase 1, which activates inositol triphosphate
receptor-mediated Ca2+ release into the cytosol and
activates CaMKII to induce apoptosis (135-137).
It has been revealed that Xbp1 and ATF6 may mediate the
overexpression of GRP94, which could attenuate myocardial cell
necrosis induced by Ca2+ overload or ischemia (138).
The UPR can regulate a number of mitochondrial
functions, including bioenergetics, membrane potential and the
degree of cytochrome c release (142). The UPR can also serve a role in
immune function. A previous study revealed that cathepsin-induced
ERS enhanced the recruitment of IFN regulatory factor-3 and cAMP
response element binding protein (CREB/CBP)/p300 to the murine
IFNB1 promoter during lipopolysaccharide stimulation. ERS-related
inflammation occurred through Xbp1 binding to a potential enhancer
element 6 kb distal to the IFNB1 gene, which may enhance the
recruitment of CBP/p300 and IFN regulatory transcription factor to
the IFNB1 enhanceosome (143).
This observation indicated a novel role of UPR-dependent
transcription in the regulation of inflammatory cytokines, which
may be of significance to the pathogenesis of diseases involving
ERS and type I IFN. One potential avenue of study may be the
relationship among viral infection, I/R injury and inflammatory
diseases (143). ERS can activate
nucleotide binding oligomerization domain-like receptor protein 1
(NLRP1) inflammatory bodies by activating the NF-κB signaling
pathway, which may then promote myocardial I/R injury (144). NLRPs are classified as typical
inflammasomes that include NLRP1 and NLRP3 inflammatory bodies.
They can activate caspase-1, resulting in the maturation and
secretion of pro-inflammatory cytokines IL-1β and IL-18(145). How the UPR in turn mediates I/R
damage is summarized in Fig.
1B.
There are several important proteins that are
activated by ERS, including ATF6, Xbp1, ATF4, CHOP and IRE1. ATF6
normally functions in the adaptive UPR to accelerate the remodeling
of cellular physiology and recovery following acute physiological
and pathological injury (146).
ATF6 can dimerize with UPR-regulated basic leucine zipper
transcription factors, such as Xbp1, by S1P/S2P-dependent
proteolysis, or associate with other stress-responsive signaling
pathways such as mTOR signaling (147,148). In addition, ATF6 has been reported
to induce the expression of the Ca2 + pump SERCA2a and
the expression of several antioxidant genes (149,150).
Xbp1 has been revealed to exert protective effects
against I/R injury in the heart and the brain (133,151-153),
as overexpression of Xbp1 can inhibit cell death induced by oxygen
glucose deprivation/reoxygenation (OGD/R These findings suggested
that inhibiting Xbp1 activation may accelerate neuronal cell death
after I/R, which can be exploited as a therapeutic strategy for
brain I/R injury (154).
Accumulating evidence has demonstrated that ERS serves a key role
in I/R-induced cell dysfunction (155), where destruction of the ER pathway
can result in neuronal cell death. ERS is associated with the
pathology of brain I/R injury. OGD/R stress temporarily inactivates
Xbp1 splicing, resulting in accelerated neuronal death due to ER
dysfunction. Subsequent Xbp1 reactivation may be neuroprotective
against OGD/R stress (154).
ATF4 induces the expression of CHOP under mild ERS.
However, under chronic ERS, PERK then significantly increases CHOP
expression, in turn suppressing the expression of Bcl-2 to increase
cell death (156). In addition,
PERK phosphorylates Kelch-like Ech-related protein 1, which
releases Nrf2 from inhibition and translocates into the nucleus to
activate the expression of antioxidant and detoxifying enzymes
(157). CHOP has also been
reported to upregulate the expression of PUMA and the pro-apoptotic
protein Bim, thereby inducing mitochondrial-dependent apoptosis
(158,159). IRE1 is associated with autophagy
activation, which is an important pro-survival defense mechanism
against cardiac pathology, including hypertrophy and I/R (160).
In the present article, numerous possible causes of
myocardial I/R injury, including Ca2+ overload, ROS
accumulation, increase in expression of inflammatory cytokines and
apoptotic factors, miRNA change and ERS were described. These
factors not only lead to secondary cardiac injury but can also
hinder the reconstruction of blood vessels after clinical treatment
(161). Cardiac I/R injury induces
changes of ERS in a process that is mainly mediated by three
pathways involved in the accumulation of unfolded proteins, which
causes cell damage. At the beginning of the response, a cellular
protective response ensues, which then becomes apoptotic in the
latter stages. However, to understand the specific mechanism
underlying these processes, further study is required. Appropriate
intervention in the ERS process may serve as a potential
therapeutic strategy for heart I/R injury, including intervention
in the expression of ligands and their receptors in the ERS
pathways. With further study of cardiac ERS and I/R injury,
strengthening the understanding of the mechanism underlying I/R
injury will facilitate the optimization of treatment regimens. If
the occurrence and development of myocardial cell apoptosis can be
prevented, it may become possible to alleviate I/R injury, which
will facilitate the development of treatment strategies and drug
discovery for myocardial I/R.
Not applicable.
The current study was supported by grants from the
National Natural Science Foundation of China (grant no. 81770292),
Key Workstation Projects of He Lin (grant no. 18331101) and Wenzhou
Science and Technology Major Projects (grant no. 2018ZY007).
Not applicable.
YR and LL conceived and designed the review. YR, JZ
and QJ collected the related literature. MC, KJ and ZW analyzed the
related papers. YR wrote the manuscript. All authors read and
approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Baulina N, Osmak G, Kiselev I, Matveeva N,
Kukava N, Shakhnovich R, Kulakova O and Favorova O: NGS-identified
circulating miR-375 as a potential regulating component of
myocardial infarction associated network. J Mol Cell Cardiol.
121:173–179. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Nunez-Gomez E, Pericacho M, Ollauri-Ibanez
C, Bernabeu C and Lopez-Novoa JM: The role of endoglin in
post-ischemic revascularization. Angiogenesis. 20:1–24.
2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zhou H, Ma Q, Zhu P, Ren J, Reiter RJ and
Chen Y: Protective role of melatonin in cardiac
ischemia-reperfusion injury: From pathogenesis to targeted therapy.
J Pineal Res. 64(e12471)2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Jennings RB, Sommers HM, Smyth GA, Flack
HA and Linn H: Myocardial necrosis induced by temporary occlusion
of a coronary artery in the dog. Arch Pathol. 70:68–78.
1960.PubMed/NCBI
|
|
5
|
Davidson SM, Ferdinandy P, Andreadou I,
Bøtker HE, Heusch G, Ibáñez B, Ovize M, Schulz R, Yellon DM,
Hausenloy DJ, et al: Multitarget strategies to reduce myocardial
ischemia/reperfusion injury: JACC review topic of the week. J Am
Coll Cardiol. 73:89–99. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Xu C, Bailly-Maitre B and Reed JC:
Endoplasmic reticulum stress: Cell life and death decisions. J Clin
Invest. 115:2656–2664. 2005.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhao S, Liu Y, Wang F, Xu D and Xie P:
N-acetylcysteine protects against microcystin-LR-induced
endoplasmic reticulum stress and germ cell apoptosis in zebrafish
testes. Chemosphere. 204:463–473. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Liu X, Jin X, Su R and Li Z: The
reproductive toxicology of male SD rats after PM2.5 exposure
mediated by the stimulation of endoplasmic reticulum stress.
Chemosphere. 189:547–555. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Almanza A, Carlesso A, Chintha C,
Creedican S, Doultsinos D, Leuzzi B, Luís A, McCarthy N,
Montibeller L, More S, et al: Endoplasmic reticulum stress
signalling-from basic mechanisms to clinical applications. FEBS J.
286:241–278. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Guzel E, Arlier S, Guzeloglu-Kayisli O,
Tabak MS, Ekiz T, Semerci N, Larsen K, Schatz F, Lockwood CJ and
Kayisli UA: Endoplasmic reticulum stress and homeostasis in
reproductive physiology and pathology. Int J Mol Sci.
18(792)2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Xin W, Li X, Lu X, Niu K and Cai J:
Involvement of endoplasmic reticulum stress-associated apoptosis in
a heart failure model induced by chronic myocardial ischemia. Int J
Mol Med. 27:503–509. 2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sanada S, Komuro I and Kitakaze M:
Pathophysiology of myocardial reperfusion injury: Preconditioning,
postconditioning, and translational aspects of protective measures.
Am J Physiol Heart Circ Physiol. 301:H1723–H1741. 2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Marban E, Kitakaze M, Kusuoka H,
Porterfield JK, Yue DT and Chacko VP: Intracellular free calcium
concentration measured with 19F NMR spectroscopy in intact ferret
hearts. Proc Natl Acad Sci USA. 84:6005–6009. 1987.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kalogeris T, Baines CP, Krenz M and
Korthuis RJ: Cell biology of ischemia/reperfusion injury. Int Rev
Cell Mol Biol. 298:229–317. 2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Szydlowska K and Tymianski M: Calcium,
ischemia and excitotoxicity. Cell Calcium. 47:122–129.
2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Jakob R, Beutner G, Sharma VK, Duan Y,
Gross RA, Hurst S, Jhun BS, O-Uchi J and Sheu SS: Molecular and
functional identification of a mitochondrial ryanodine receptor in
neurons. Neurosci Lett. 575:7–12. 2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Peracchia C: Chemical gating of gap
junction channels; roles of calcium, pH and calmodulin. Biochim
Biophysica Acta. 1662:61–80. 2004.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Tribulova N, Knezl V, Szeiffova Bacova B,
Egan Benova T, Viczenczova C, Gonçalvesova E and Slezak J:
Disordered myocardial Ca(2+) homeostasis results in substructural
alterations that may promote occurrence of malignant arrhythmias.
Physiol Res. 65 (Suppl 1):S139–S148. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Javadov S, Hunter JC, Barreto-Torres G and
Parodi-Rullan R: Targeting the mitochondrial permeability
transition: Cardiac ischemia-reperfusion versus carcinogenesis.
Cell Physiol Biochem. 27:179–190. 2011.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Abdallah Y, Gkatzoflia A, Gligorievski D,
Kasseckert S, Euler G, Schlüter KD, Schäfer M, Piper HM and Schäfer
C: Insulin protects cardiomyocytes against reoxygenation-induced
hypercontracture by a survival pathway targeting SR Ca2+
storage. Cardiovasc Res. 70:346–353. 2006.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wu H, Yang H, Rhee JW, Zhang JZ, Lam CK,
Sallam K, Chang ACY, Ma N, Lee J, Zhang H, et al: Modelling
diastolic dysfunction in induced pluripotent stem cell-derived
cardiomyocytes from hypertrophic cardiomyopathy patients. Eur Heart
J. 40:3685–3695. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Inserte J, Hernando V and Garcia-Dorado D:
Contribution of calpains to myocardial ischaemia/reperfusion
injury. Cardiovasc Res. 96:23–31. 2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Croall DE and Ersfeld K: The calpains:
Modular designs and functional diversity. Genome Biol.
8(218)2007.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Commoner B, Townsend J and Pake GE: Free
radicals in biological materials. Nature. 174:689–691.
1954.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Cadenas S: Mitochondrial uncoupling, ROS
generation and cardioprotection. Biochim Biophys Acta Bioenerg.
1859:940–950. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Meitzler JL, Antony S, Wu Y, Juhasz A, Liu
H, Jiang G, Lu J, Roy K and Doroshow JH: NADPH oxidases: A
perspective on reactive oxygen species production in tumor biology.
Antioxid Redox Signal. 20:2873–2889. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Ziech D, Franco R, Pappa A and
Panayiotidis MI: Reactive oxygen species (ROS)-induced genetic and
epigenetic alterations in human carcinogenesis. Mutation Res.
711:167–173. 2011.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Brand MD: The sites and topology of
mitochondrial superoxide production. Exp Gerontol. 45:466–472.
2010.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Lambeth JD: NOX enzymes and the biology of
reactive oxygen. Nat Rev Immunol. 4:181–189. 2004.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Huang P, Feng L, Oldham EA, Keating MJ and
Plunkett W: Superoxide dismutase as a target for the selective
killing of cancer cells. Nature. 407:390–395. 2000.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Srinivas US, Tan BWQ Vellayappan BA and
Jeyasekharan AD: ROS and the DNA damage response in cancer. Redox
Biol. 25(101084)2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Azevedo PS, Polegato BF, Minicucci MF,
Paiva SA and Zornoff LA: Cardiac remodeling: Concepts, clinical
impact, pathophysiological mechanisms and pharmacologic treatment.
Arq Bras Cardiol. 106:62–69. 2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Moris D, Spartalis M, Spartalis E,
Karachaliou GS, Karaolanis GI, Tsourouflis G, Tsilimigras DI,
Tzatzaki E and Theocharis S: The role of reactive oxygen species in
the pathophysiology of cardiovascular diseases and the clinical
significance of myocardial redox. Ann Transl Med.
5(326)2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Bartz RR, Suliman HB and Piantadosi CA:
Redox mechanisms of cardiomyocyte mitochondrial protection. Front
Physiol. 6(291)2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Lee HL, Chen CL, Yeh ST, Zweier JL and
Chen YR: Biphasic modulation of the mitochondrial electron
transport chain in myocardial ischemia and reperfusion. Am J
Physiol Heart Circ Physiol. 302:H1410–H1422. 2012.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Chen YR and Zweier JL: Cardiac
mitochondria and reactive oxygen species generation. Circ Res.
114:524–537. 2014.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Angelova PR and Abramov AY: Functional
role of mitochondrial reactive oxygen species in physiology. Free
Radic Biol Med. 100:81–85. 2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Chang JC, Lien CF, Lee WS, Chang HR, Hsu
YC, Luo YP, Jeng JR, Hsieh JC and Yang KT: Intermittent hypoxia
prevents myocardial mitochondrial Ca2+ overload and cell
death during ischemia/reperfusion: The role of reactive oxygen
species. Cells. 8(564)2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Zhu WZ, Xie Y, Chen L, Yang HT and Zhou
ZN: Intermittent high altitude hypoxia inhibits opening of
mitochondrial permeability transition pores against reperfusion
injury. J Mol Cell Cardiol. 40:96–106. 2006.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Aguilar M, Gonzalez-Candia A, Rodríguez J,
Carrasco-Pozo C, Cañas D, García-Herrera C, Herrera EA and Castillo
RL: Mechanisms of cardiovascular protection associated with
intermittent hypobaric hypoxia exposure in a rat model: Role of
Oxidative Stress. Int J Mol Sci. 19(366)2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Jordan JE, Zhao ZQ and Vinten-Johansen J:
The role of neutrophils in myocardial ischemia-reperfusion injury.
Cardiovasc Res. 43:860–878. 1999.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Chandrasekar B, Colston JT, Geimer J,
Cortez D and Freeman GL: Induction of nuclear factor kappaB but not
kappaB-responsive cytokine expression during myocardial reperfusion
injury after neutropenia. Free Radic Biol Med. 28:1579–1588.
2000.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Sugano M, Hata T, Tsuchida K, Suematsu N,
Oyama J, Satoh S and Makino N: Local delivery of soluble TNF-alpha
receptor 1 gene reduces infarct size following ischemia/reperfusion
injury in rats. Mol Cell Biochem. 266:127–132. 2004.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Merchant SH, Gurule DM and Larson RS:
Amelioration of ischemia-reperfusion injury with cyclic peptide
blockade of ICAM-1. Am J Physiol Heart Circ Physiol.
284:H1260–H1268. 2003.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Dar WA, Sullivan E, Bynon JS, Eltzschig H
and Ju C: Ischaemia reperfusion injury in liver transplantation:
Cellular and molecular mechanisms. Liver Int. 39:788–801.
2019.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Bhattacharya K, Farwell K, Huang M,
Kempuraj D, Donelan J, Papaliodis D, Vasiadi M and Theoharides TC:
Mast cell deficient W/Wv mice have lower serum IL-6 and less
cardiac tissue necrosis than their normal littermates following
myocardial ischemia-reperfusion. Int J Immunopathol Pharmacol.
20:69–74. 2007.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Yap ML and Peter K: Molecular positron
emission tomography in cardiac ischemia/reperfusion. Circ Res.
124:827–829. 2019.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Valente M and Calabrese F: Liver and
apoptosis. Ital J Gastroenterol Hepatol. 31:73–77. 1999.PubMed/NCBI
|
|
49
|
Metukuri MR, Beer-Stolz D, Namas RA,
Dhupar R, Torres A, Loughran PA, Jefferson BS, Tsung A, Billiar TR,
Vodovotz Y and Zamora R: Expression and subcellular localization of
BNIP3 in hypoxic hepatocytes and liver stress. Am J Physiol
Gastrointest Liver Physiol. 296:G499–G509. 2009.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Wu B, Qiu W, Wang P, Yu H, Cheng T,
Zambetti GP, Zhang L and Yu J: p53 independent induction of PUMA
mediates intestinal apoptosis in response to ischaemia-reperfusion.
Gut. 56:645–654. 2007.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Scorrano L, Oakes SA, Opferman JT, Cheng
EH, Sorcinelli MD, Pozzan T and Korsmeyer SJ: BAX and BAK
regulation of endoplasmic reticulum Ca2+: A control
point for apoptosis. Science. 300:135–139. 2003.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Kobayashi A, Imamura H, Isobe M, Matsuyama
Y, Soeda J, Matsunaga K and Kawasaki S: Mac-1 (CD11b/CD18) and
intercellular adhesion molecule-1 in ischemia-reperfusion injury of
rat liver. Am J Physiol Gastrointest Liver Physiol. 281:G577–G585.
2001.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Yang W, Guastella J, Huang JC, Wang Y,
Zhang L, Xue D, Tran M, Woodward R, Kasibhatla S, Tseng B, et al:
MX1013, a dipeptide caspase inhibitor with potent in vivo
antiapoptotic activity. Br J Pharmacol. 140:402–412.
2003.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Vandenabeele P, Declercq W, Van Herreweghe
F and Vanden Berghe T: The role of the kinases RIP1 and RIP3 in
TNF-induced necrosis. Sci Signal. 3(re4)2010.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Kubli DA, Ycaza JE and Gustafsson AB:
Bnip3 mediates mitochondrial dysfunction and cell death through Bax
and Bak. Biochem J. 405:407–415. 2007.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Kubli DA, Quinsay MN, Huang C, Lee Y and
Gustafsson AB: Bnip3 functions as a mitochondrial sensor of
oxidative stress during myocardial ischemia and reperfusion. Am J
Physiol Heart Circ Physiol. 295:H2025–H2031. 2008.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Poller W, Dimmeler S, Heymans S, Zeller T,
Haas J, Karakas M, Leistner DM, Jakob P, Nakagawa S, Blankenberg S,
et al: Non-coding RNAs in cardiovascular diseases: Diagnostic and
therapeutic perspectives. Eur Heart J. 39:2704–2716.
2018.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Winter J, Jung S, Keller S, Gregory RI and
Diederichs S: Many roads to maturity: microRNA biogenesis pathways
and their regulation. Nat Cell Biol. 11:228–234. 2009.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Peng L, Chun-Guang Q, Bei-Fang L, Xue-Zhi
D, Zi-Hao W, Yun-Fu L, Yan-Ping D, Yang-Gui L, Wei-Guo L, Tian-Yong
H and Zhen-Wen H: Clinical impact of circulating miR-133, miR-1291
and miR-663b in plasma of patients with acute myocardial
infarction. Diagn Pathol. 9(89)2014.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Zhang R, Lan C, Pei H, Duan G, Huang L and
Li L: Expression of circulating miR-486 and miR-150 in patients
with acute myocardial infarction. BMC Cardiovasc Disord.
15(51)2015.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Joladarashi D, Garikipati VNS,
Thandavarayan RA, Verma SK, Mackie AR, Khan M, Gumpert AM, Bhimaraj
A, Youker KA, Uribe C, et al: Enhanced cardiac regenerative ability
of stem cells after ischemia-reperfusion injury: Role of human
CD34+ cells deficient in MicroRNA-377. J Am Coll
Cardiol. 66:2214–2226. 2015.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Xu C, Lu Y, Pan Z, Chu W, Luo X, Lin H,
Xiao J, Shan H, Wang Z and Yang B: The muscle-specific microRNAs
miR-1 and miR-133 produce opposing effects on apoptosis by
targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J Cell Sci.
120:3045–3052. 2007.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Tang Y, Zheng J, Sun Y, Wu Z, Liu Z and
Huang G: MicroRNA-1 regulates cardiomyocyte apoptosis by targeting
Bcl-2. Int Heart J. 50:377–387. 2009.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Yu XY, Song YH, Geng YJ, Lin QX, Shan ZX,
Lin SG and Li Y: Glucose induces apoptosis of cardiomyocytes via
microRNA-1 and IGF-1. Biochem Biophys Res Commun. 376:548–552.
2008.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Matkovich SJ, Wang W, Tu Y, Eschenbacher
WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM and Dorn GW II:
MicroRNA-133a protects against myocardial fibrosis and modulates
electrical repolarization without affecting hypertrophy in
pressure-overloaded adult hearts. Circ Res. 106:166–175.
2010.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Cheng Y, Liu X, Zhang S, Lin Y, Yang J and
Zhang C: MicroRNA-21 protects against the H(2)O(2)-induced injury
on cardiac myocytes via its target gene PDCD4. J Mol Cell Cardiol.
47:5–14. 2009.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Cheng Y, Zhu P, Yang J, Liu X, Dong S,
Wang X, Chun B, Zhuang J and Zhang C: Ischaemic
preconditioning-regulated miR-21 protects heart against
ischaemia/reperfusion injury via anti-apoptosis through its target
PDCD4. Cardiovas Res. 87:431–439. 2010.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Sayed D, He M, Hong C, Gao S, Rane S, Yang
Z and Abdellatif M: MicroRNA-21 is a downstream effector of AKT
that mediates its antiapoptotic effects via suppression of Fas
ligand. J Biol Chem. 285:20281–20290. 2010.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Zhu H and Fan GC: Role of microRNAs in the
reperfused myocardium towards post-infarct remodelling. Cardiovasc
Res. 94:284–292. 2012.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Magenta A, Dellambra E, Ciarapica R and
Capogrossi MC: Oxidative stress, microRNAs and cytosolic calcium
homeostasis. Cell Calcium. 60:207–217. 2016.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Cha MJ, Jang JK, Ham O, Song BW, Lee SY,
Lee CY, Park JH, Lee J, Seo HH, Choi E, et al: MicroRNA-145
suppresses ROS-induced Ca2+ overload of cardiomyocytes
by targeting CaMKIIδ. Biochem Biophys Res Commun. 435:720–726.
2013.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Pan L, Huang BJ, Ma XE, Wang SY, Feng J,
Lv F, Liu Y, Liu Y, Li CM, Liang DD, et al: MiR-25 protects
cardiomyocytes against oxidative damage by targeting the
mitochondrial calcium uniporter. Int J Mol Sci. 16:5420–5433.
2015.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Aurora AB, Mahmoud AI, Luo X, Johnson BA,
van Rooij E, Matsuzaki S, Humphries KM, Hill JA, Bassel-Duby R,
Sadek HA and Olson EN: MicroRNA-214 protects the mouse heart from
ischemic injury by controlling Ca2+ overload and cell
death. J Clin Invest. 122:1222–1232. 2012.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Wang X, Zhu H, Zhang X, Liu Y, Chen J,
Medvedovic M, Li H, Weiss MJ, Ren X and Fan GC: Loss of the
miR-144/451 cluster impairs ischaemic preconditioning-mediated
cardioprotection by targeting Rac-1. Cardiovasc Res. 94:379–390.
2012.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Ong SB, Katwadi K, Kwek XY, Ismail NI,
Chinda K, Ong SG and Hausenloy DJ: Non-coding RNAs as therapeutic
targets for preventing myocardial ischemia-reperfusion injury.
Expert Opin Ther Targets. 22:247–261. 2018.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Ong SG and Hausenloy DJ: Hypoxia-inducible
factor as a therapeutic target for cardioprotection. Pharmacol
Ther. 136:69–81. 2012.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Hinkel R, Penzkofer D, Zühlke S, Fischer
A, Husada W, Xu QF, Baloch E, van Rooij E, Zeiher AM, Kupatt C and
Dimmeler S: Inhibition of microRNA-92a protects against
ischemia/reperfusion injury in a large-animal model. Circulation.
128:1066–1075. 2013.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Hullinger TG, Montgomery RL, Seto AG,
Dickinson BA, Semus HM, Lynch JM, Dalby CM, Robinson K, Stack C,
Latimer PA, et al: Inhibition of miR-15 protects against cardiac
ischemic injury. Circ Res. 110:71–81. 2012.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Minamino T and Kitakaze M: ER stress in
cardiovascular disease. J Mol Cell Cardiol. 48:1105–1110.
2010.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Lindholm D, Wootz H and Korhonen L: ER
stress and neurodegenerative diseases. Cell Death Differ.
13:385–392. 2006.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Matus S, Glimcher LH and Hetz C: Protein
folding stress in neurodegenerative diseases: A glimpse into the
ER. Curr Opin Cell Biol. 23:239–252. 2011.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Hotamisligil GS: Endoplasmic reticulum
stress and the inflammatory basis of metabolic disease. Cell.
140:900–917. 2010.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Chiang CK, Hsu SP, Wu CT, Huang JW, Cheng
HT, Chang YW, Hung KY, Wu KD and Liu SH: Endoplasmic reticulum
stress implicated in the development of renal fibrosis. Mol Med.
17:1295–1305. 2011.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Zhang L, Wang Y, Zhang L, Xia X, Chao Y,
He R, Han C and Zhao W: ZBTB7A, a miR-663a target gene, protects
osteosarcoma from endoplasmic reticulum stress-induced apoptosis by
suppressing LncRNA GAS5 expression. Cancer Lett. 448:105–116.
2019.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Wang EM, Akasaka H, Zhao J, Varadhachary
GR, Lee JE, Maitra A, Fleming JB, Hung MC, Wang H and Katz MH:
Expression and clinical significance of protein kinase RNA-like
endoplasmic reticulum kinase and phosphorylated eukaryotic
initiation factor 2α in pancreatic ductal adenocarcinoma. Pancreas.
48:323–328. 2019.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Yu LM, Dong X, Zhang J, Li Z, Xue XD, Wu
HJ, Yang ZL, Yang Y and Wang HS: Naringenin attenuates myocardial
ischemia-reperfusion injury via cGMP-PKGIα signaling and in vivo
and in vitro studies. Oxid Med Cell Longev.
2019(7670854)2019.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Peñaranda Fajardo NM, Meijer C and Kruyt
FA: The endoplasmic reticulum stress/unfolded protein response in
gliomagenesis, tumor progression and as a therapeutic target in
glioblastoma. Biochem Pharmacol. 118:1–8. 2016.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Beck D, Niessner H, Smalley KS, Flaherty
K, Paraiso KH, Busch C, Sinnberg T, Vasseur S, Iovanna JL, Drießen
S, et al: Vemurafenib potently induces endoplasmic reticulum
stress-mediated apoptosis in BRAFV600E melanoma cells. Sci Signal.
6(ra7)2013.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Lee JH, Kwon EJ and Kim DH: Calumenin has
a role in the alleviation of ER stress in neonatal rat
cardiomyocytes. Biochem Biophys Res Commun. 439:327–332.
2013.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Brewer JW and Diehl JA: PERK mediates
cell-cycle exit during the mammalian unfolded protein response.
Proc Natl Acad Sci USA. 97:12625–12630. 2000.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Zhang M, Han N, Jiang Y, Wang J, Li G, Lv
X, Li G and Qiao Q: EGFR confers radioresistance in human
oropharyngeal carcinoma by activating endoplasmic reticulum stress
signaling PERK-eIF2α-GRP94 and IRE1α-XBP1-GRP78. Cancer Med.
7:6234–6246. 2018.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Palam LR, Baird TD and Wek RC:
Phosphorylation of eIF2 facilitates ribosomal bypass of an
inhibitory upstream ORF to enhance CHOP translation. J Biol Chem.
286:10939–10949. 2011.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Oyadomari S, Koizumi A, Takeda K, Gotoh T,
Akira S, Araki E and Mori M: Targeted disruption of the Chop gene
delays endoplasmic reticulum stress-mediated diabetes. J Clin
Invest. 109:525–532. 2002.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Yao Y, Lu Q, Hu Z, Yu Y, Chen Q and Wang
QK: A non-canonical pathway regulates ER stress signaling and
blocks ER stress-induced apoptosis and heart failure. Nat Commun.
8(133)2017.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Correll RN, Grimes KM, Prasad V, Lynch JM,
Khalil H and Molkentin JD: Overlapping and differential functions
of ATF6α versus ATF6β in the mouse heart. Sci Rep.
9(2059)2019.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Okada T, Yoshida H, Akazawa R, Negishi M
and Mori K: Distinct roles of activating transcription factor 6
(ATF6) and double-stranded RNA-activated protein kinase-like
endoplasmic reticulum kinase (PERK) in transcription during the
mammalian unfolded protein response. Biochem J. 366:585–594.
2002.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Yoshida H, Okada T, Haze K, Yanagi H, Yura
T, Negishi M and Mori K: ATF6 activated by proteolysis binds in the
presence of NF-Y (CBF) directly to the cis-acting element
responsible for the mammalian unfolded protein response. Mol Cell
Biol. 20:6755–6767. 2000.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Xiang C, Wang Y, Zhang H and Han F: The
role of endoplasmic reticulum stress in neurodegenerative disease.
Apoptosis. 22:1–26. 2017.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Korennykh A and Walter P: Structural basis
of the unfolded protein response. Annu Rev Cell Dev Biol.
28:251–277. 2012.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Tsuru A, Fujimoto N, Takahashi S, Saito M,
Nakamura D, Iwano M, Iwawaki T, Kadokura H, Ron D and Kohno K:
Negative feedback by IRE1β optimizes mucin production in goblet
cells. Proc Natl Acad Sci USA. 110:2864–2869. 2013.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Pavitt GD and Ron D: New insights into
translational regulation in the endoplasmic reticulum unfolded
protein response. Cold Spring Harbor Perspect Biol.
4(a012278)2012.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Han F, Yan S and Shi Y: Single-prolonged
stress induces endoplasmic reticulum-dependent apoptosis in the
hippocampus in a rat model of Post-traumatic stress disorder. PLoS
One. 8(e69340)2013.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Yu B, Wen L, Xiao B, Han F and Shi Y:
Single prolonged stress induces ATF6 alpha-dependent Endoplasmic
reticulum stress and the apoptotic process in medial Frontal Cortex
neurons. BMC Neurosci. 15(115)2014.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Walter P and Ron D: The unfolded protein
response: From stress pathway to homeostatic regulation. Science.
334:1081–1086. 2011.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Maly DJ and Papa FR: Druggable sensors of
the unfolded protein response. Nat Chem Biol. 10:892–901.
2014.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Yoshida H: ER stress and diseases. FEBS J.
274:630–658. 2007.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Paschen W: Endoplasmic reticulum
dysfunction in brain pathology: Critical role of protein synthesis.
Curr Neurovasc Res. 1:173–181. 2004.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Thuerauf DJ, Marcinko M, Gude N, Rubio M,
Sussman MA and Glembotski CC: Activation of the unfolded protein
response in infarcted mouse heart and hypoxic cultured cardiac
myocytes. Circ Res. 99:275–282. 2006.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Severino A, Campioni M, Straino S, Salloum
FN, Schmidt N, Herbrand U, Frede S, Toietta G, Di Rocco G, Bussani
R, et al: Identification of protein disulfide isomerase as a
cardiomyocyte survival factor in ischemic cardiomyopathy. J Am Coll
Cardiol. 50:1029–1037. 2007.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Terai K, Hiramoto Y, Masaki M, Sugiyama S,
Kuroda T, Hori M, Kawase I and Hirota H: AMP-activated protein
kinase protects cardiomyocytes against hypoxic injury through
attenuation of endoplasmic reticulum stress. Mol Cell Biol.
25:9554–9575. 2005.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Glembotski CC: The role of the unfolded
protein response in the heart. J Mol Cell Cardiol. 44:453–459.
2008.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Hadley G, Neuhaus AA, Couch Y, Beard DJ,
Adriaanse BA, Vekrellis K, DeLuca GC, Papadakis M, Sutherland BA
and Buchan AM: The role of the endoplasmic reticulum stress
response following cerebral ischemia. Int J Stroke. 13:379–390.
2018.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Lehotský J, Urban P, Pavlíková M,
Tatarková Z, Kaminska B and Lehotský J: Molecular mechanisms
leading to neuroprotection/ischemic tolerance: Effect of
preconditioning on the stress reaction of endoplasmic reticulum.
Cell Mol Neurobiol. 29:917–925. 2009.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Eijkelenboom A and Burgering BM: FOXOs:
Signalling integrators for homeostasis maintenance. Nat Rev Mol
Cell Biol. 14:83–97. 2013.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Huang H and Tindall DJ: Dynamic FoxO
transcription factors. J Cell Sci. 120:2479–2487. 2007.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Chien CT, Lee PH, Chen CF, Ma MC, Lai MK
and Hsu SM: De novo demonstration and co-localization of
free-radical production and apoptosis formation in rat kidney
subjected to ischemia/reperfusion. J Am Soc Nephrol. 12:973–982.
2001.PubMed/NCBI
|
|
118
|
Liu H, Wang L, Weng X, Chen H, Du Y, Diao
C, Chen Z and Liu X: Inhibition of Brd4 alleviates renal
ischemia/reperfusion injury-induced apoptosis and endoplasmic
reticulum stress by blocking FoxO4-mediated oxidative stress. Redox
Biol. 24(101195)2019.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Ren L, Wang Q, Chen Y, Ma Y and Wang D:
Involvement of MicroRNA-133a in the protective effect of hydrogen
sulfide against ischemia/Reperfusion-induced endoplasmic reticulum
stress and cardiomyocyte apoptosis. Pharmacology. 103:1–9.
2019.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Yan Y, Zhang B, Liu N, Qi C, Xiao Y, Tian
X, Li T and Liu B: Circulating long noncoding RNA UCA1 as a novel
biomarker of acute myocardial infarction. BioMed Res Int.
2016(8079372)2016.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Chen J, Hu Q, Zhang BF, Liu XP, Yang S and
Jiang H: Long noncoding RNA UCA1 inhibits ischaemia/reperfusion
injury induced cardiomyocytes apoptosis via suppression of
endoplasmic reticulum stress. Genes Genomics. 41:803–810.
2019.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Guo R, Ma H, Gao F, Zhong L and Ren J:
Metallothionein alleviates oxidative stress-induced endoplasmic
reticulum stress and myocardial dysfunction. J Mol Cell Cardiol.
47:228–237. 2009.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Minamino T, Komuro I and Kitakaze M:
Endoplasmic reticulum stress as a therapeutic target in
cardiovascular disease. Circ Res. 107:1071–1082. 2010.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Glembotski CC: Endoplasmic reticulum
stress in the heart. Circ Res. 101:975–984. 2007.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Urano F, Wang X, Bertolotti A, Zhang Y,
Chung P, Harding HP and Ron D: Coupling of stress in the ER to
activation of JNK protein kinases by transmembrane protein kinase
IRE1. Science. 287:664–666. 2000.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Yoneda T, Imaizumi K, Oono K, Yui D, Gomi
F, Katayama T and Tohyama M: Activation of caspase-12, an
endoplastic reticulum (ER) resident caspase, through tumor necrosis
factor receptor-associated factor 2-dependent mechanism in response
to the ER stress. J Biol Chem. 276:13935–13940. 2001.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Martindale JJ, Fernandez R, Thuerauf D,
Whittaker R, Gude N, Sussman MA and Glembotski CC: Endoplasmic
reticulum stress gene induction and protection from
ischemia/reperfusion injury in the hearts of transgenic mice with a
tamoxifen-regulated form of ATF6. Circ Res. 98:1186–1193.
2006.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Shintani-Ishida K, Nakajima M, Uemura K
and Yoshida K: Ischemic preconditioning protects cardiomyocytes
against ischemic injury by inducing GRP78. Biochem Biophys Res
Commun. 345:1600–1605. 2006.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Weigand K, Brost S, Steinebrunner N,
Buchler M, Schemmer P and Muller M: Ischemia/Reperfusion injury in
liver surgery and transplantation: Pathophysiology. HPB Surg.
2012(176723)2012.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Hartley T, Siva M, Lai E, Teodoro T, Zhang
L and Volchuk A: Endoplasmic reticulum stress response in an INS-1
pancreatic beta-cell line with inducible expression of a
folding-deficient proinsulin. BMC Cell Biol. 11(59)2010.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Szegezdi E, Duffy A, O'Mahoney ME, Logue
SE, Mylotte LA, O'brien T and Samali A: ER stress contributes to
ischemia-induced cardiomyocyte apoptosis. Biochem Biophys Res
Commun. 349:1406–1411. 2006.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Murphy E and Steenbergen C: Mechanisms
underlying acute protection from cardiac ischemia-reperfusion
injury. Physiol Rev. 88:581–609. 2008.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Wang X, Xu L, Gillette TG, Jiang X and
Wang ZV: The unfolded protein response in ischemic heart disease. J
Mol Cell Cardiol. 117:19–25. 2018.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Yang W and Paschen W: Unfolded protein
response in brain ischemia: A timely update. J Cereb Blood Flow
Metab. 36:2044–2050. 2016.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Li G, Mongillo M, Chin KT, Harding H, Ron
D, Marks AR and Tabas I: Role of ERO1-alpha-mediated stimulation of
inositol 1,4,5-triphosphate receptor activity in endoplasmic
reticulum stress-induced apoptosis. J Cell Biol. 186:783–792.
2009.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Timmins JM, Ozcan L, Seimon TA, Li G,
Malagelada C, Backs J, Backs T, Bassel-Duby R, Olson EN, Anderson
ME and Tabas I: Calcium/calmodulin-dependent protein kinase II
links ER stress with Fas and mitochondrial apoptosis pathways. J
Clin Invest. 119:2925–2941. 2009.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Vitadello M, Penzo D, Petronilli V,
Michieli G, Gomirato S, Menabò R, Di Lisa F and Gorza L:
Overexpression of the stress protein Grp94 reduces cardiomyocyte
necrosis due to calcium overload and simulated ischemia. FASEB J.
17:923–925. 2003.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Ikeda Y, Young LH and Lefer AM:
Attenuation of neutrophil-mediated myocardial ischemia-reperfusion
injury by a calpain inhibitor. Am J Physiol Heart Circ Physiol.
282:H1421–H1426. 2002.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Hernando V, Inserte J, Sartorio CL, Parra
VM, Poncelas-Nozal M and Garcia-Dorado D: Calpain translocation and
activation as pharmacological targets during myocardial
ischemia/reperfusion. J Mol Cell Cardiol. 49:271–279.
2010.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Zheng D, Wang G, Li S, Fan GC and Peng T:
Calpain-1 induces endoplasmic reticulum stress in promoting
cardiomyocyte apoptosis following hypoxia/reoxygenation. Biochim
Biophys Acta. 1852:882–892. 2015.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Munoz JP, Ivanova S, Sanchez-Wandelmer J,
Martínez-Cristóbal P, Noguera E, Sancho A, Díaz-Ramos A,
Hernández-Alvarez MI, Sebastián D, Mauvezin C, et al: Mfn2
modulates the UPR and mitochondrial function via repression of
PERK. EMBO J. 32:2348–2361. 2013.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Zeng L, Liu YP, Sha H, Chen H, Qi L and
Smith JA: XBP-1 couples endoplasmic reticulum stress to augmented
IFN-beta induction via a cis-acting enhancer in macrophages. J
Immunol. 185:2324–2330. 2010.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Cao L, Chen Y, Zhang Z, Li Y and Zhao P:
Endoplasmic reticulum stress-induced NLRP1 inflammasome activation
contributes to myocardial ischemia/reperfusion injury. Shock.
51:511–518. 2019.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Yi YS: Role of inflammasomes in
inflammatory autoimmune rheumatic diseases. Korean J Physiol
Pharmacol. 22:1–15. 2018.PubMed/NCBI View Article : Google Scholar
|
|
146
|
Zhang G, Wang X, Gillette TG, Deng Y and
Wang ZV: Unfolded protein response as a therapeutic target in
cardiovascular disease. Curr Top Med Chem. 19:1902–1917.
2019.PubMed/NCBI View Article : Google Scholar
|
|
147
|
Asada R, Kanemoto S, Kondo S, Saito A and
Imaizumi K: The signalling from endoplasmic reticulum-resident bZIP
transcription factors involved in diverse cellular physiology. J
Biochem. 149:507–518. 2011.PubMed/NCBI View Article : Google Scholar
|
|
148
|
Zhang K, Shen X, Wu J, Sakaki K, Saunders
T, Rutkowski DT, Back SH and Kaufman RJ: Endoplasmic reticulum
stress activates cleavage of CREBH to induce a systemic
inflammatory response. Cell. 124:587–599. 2006.PubMed/NCBI View Article : Google Scholar
|
|
149
|
Jin JK, Blackwood EA, Azizi K, Thuerauf
DJ, Fahem AG, Hofmann C, Kaufman RJ, Doroudgar S and Glembotski CC:
ATF6 decreases myocardial ischemia/reperfusion damage and links ER
stress and oxidative stress signaling pathways in the heart. Circ
Res. 120:862–875. 2017.PubMed/NCBI View Article : Google Scholar
|
|
150
|
Thuerauf DJ, Hoover H, Meller J, Hernandez
J, Su L, Andrews C, Dillmann WH, McDonough PM and Glembotski CC:
Sarco/endoplasmic reticulum calcium ATPase-2 expression is
regulated by ATF6 during the endoplasmic reticulum stress response:
Intracellular signaling of calcium stress in a cardiac myocyte
model system. J Biol Chem. 276:48309–48317. 2001.PubMed/NCBI View Article : Google Scholar
|
|
151
|
Zhang C, Tang Y, Li Y, Xie L, Zhuang W,
Liu J and Gong J: Unfolded protein response plays a critical role
in heart damage after myocardial ischemia/reperfusion in rats. PLoS
One. 12(e0179042)2017.PubMed/NCBI View Article : Google Scholar
|
|
152
|
Jiang D, Niwa M and Koong AC: Targeting
the IRE1α-XBP1 branch of the unfolded protein response in human
diseases. Semin Cancer Biol. 33:48–56. 2015.PubMed/NCBI View Article : Google Scholar
|
|
153
|
Schmitz ML, Shaban MS, Albert BV, Gökçen A
and Kracht M: The crosstalk of endoplasmic reticulum (ER) stress
pathways with NF-κB: Complex mechanisms relevant for cancer,
inflammation and infection. Biomedicines. 6(58)2018.PubMed/NCBI View Article : Google Scholar
|
|
154
|
Ibuki T, Yamasaki Y, Mizuguchi H and
Sokabe M: Protective effects of XBP1 against oxygen and glucose
deprivation/reoxygenation injury in rat primary hippocampal
neurons. Neurosci Lett. 518:45–48. 2012.PubMed/NCBI View Article : Google Scholar
|
|
155
|
DeGracia DJ and Montie HL: Cerebral
ischemia and the unfolded protein response. J Neurochem. 91:1–8.
2004.PubMed/NCBI View Article : Google Scholar
|
|
156
|
McCullough KD, Martindale JL, Klotz LO, Aw
TY and Holbrook NJ: Gadd153 sensitizes cells to endoplasmic
reticulum stress by down-regulating Bcl2 and perturbing the
cellular redox state. Mol Cell Biol. 21:1249–1259. 2001.PubMed/NCBI View Article : Google Scholar
|
|
157
|
Cullinan SB, Zhang D, Hannink M, Arvisais
E, Kaufman RJ and Diehl JA: Nrf2 is a direct PERK substrate and
effector of PERK-dependent cell survival. Mol Cell Biol.
23:7198–7209. 2003.PubMed/NCBI View Article : Google Scholar
|
|
158
|
Puthalakath H, O'Reilly LA, Gunn P, Lee L,
Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin
J, Motoyama N, et al: ER stress triggers apoptosis by activating
BH3-only protein Bim. Cell. 129:1337–1349. 2007.PubMed/NCBI View Article : Google Scholar
|
|
159
|
Ghosh AP, Klocke BJ, Ballestas ME and Roth
KA: CHOP potentially co-operates with FOXO3a in neuronal cells to
regulate PUMA and BIM expression in response to ER stress. PLoS
One. 7(e39586)2012.PubMed/NCBI View Article : Google Scholar
|
|
160
|
De Meyer GR and Martinet W: Autophagy in
the cardiovascular system. Biochim Biophy Acta. 1793:1485–1495.
2009.PubMed/NCBI View Article : Google Scholar
|
|
161
|
Wei L, Zhang Y, Qi X, Sun X, Li Y and Xu
Y: Ubiquitin-proteasomes are the dominant mediators of the
regulatory effect of microRNA-1 on cardiac remodeling after
myocardial infarction. Int J Mol Med. 44:1899–1907. 2019.PubMed/NCBI View Article : Google Scholar
|