Introduction
The tumor suppressor p53 has been identified as an
important mediator of various cell signaling pathways, including
proliferation, differentiation, apoptosis, ferroptosis, DNA damage,
DNA repair, senescence, autophagy, metabolism and angiogenesis
(1,2). p53 functions as the guardian of the
genome, not only in response to DNA damage, which is associated
with tumor suppression, but also by inducing apoptosis in response
to an altered cell cycle status, which is determined by the
homeostasis of key proteins of various signaling pathways,
including NF-κB and Bcl-2 family members (3). Since p53 is the most frequently
mutated gene across various cancer types, such as breast
carcinomas, brain cancers, sarcomas and adrenal cortical carcinomas
(4), and is functionally inactive
in ~50% of human tumors, it is considered as one of the most
important tumor suppressor genes, being able to reduce the
uncontrolled proliferation of cancer cells by promoting apoptosis,
cell cycle arrest and chemosensitivity (5).
Apoptosis, a type of programmed cell death, is an
important regulatory mechanism by which tumor cells die if DNA
damage is not repaired (6).
Additionally, apoptosis serves an important role in tissue
development, homoeostasis and cancer cell death (7). During tumor development, cancer cells
can develop unique mechanisms to evade the immune system, including
increasing tolerance to apoptosis (8). Therefore, designing and identifying
apoptosis-inducing agents is an effective therapeutic strategy in
cancer research (9,10) and antitumor drugs may be identified
by studying the apoptosis-inducing potential of these agents
(11). The mechanisms of action
underlying the apoptosis-induced potential of chemotherapeutic
agents are involved in two major pathways; the death
receptor-mediated pathway and the mitochondria-mediated pathway
(12,13). Tumor necrosis factor-related
apoptosis-inducing ligand-receptor (TRAIL)1 (also known as DR4),
TRAIL2 (also known as DR5) and CD95, with their associated ligands,
are responsible for the initiation of the death receptor-mediated
apoptosis (14).
Mitochondria-mediated apoptosis regulates the homeostatic balance
between pro- and anti-apoptotic protein members, such as p53 and
p53 downstream target factors, including p53-upregulated modulator
of apoptosis (PUMA), phorbol-12-myristate-13-acetate-induced
protein 1 (Noxa) and Bcl-2 family members (15). A cascade of caspases is the final
common convergence of the two pathways. Caspases cleave regulatory
and structural factors, thus inducing apoptosis in tumor cells
(16). Additionally, cell cycle
arrest is frequently induced by p53 activation (17). The p53-mediated regulation of the
cell cycle serves a central role in inhibiting tumor proliferation
and progression (17). Accumulating
evidence has suggested that factors regulated by p53 involved in
the cell cycle and apoptosis represent alternative targets for
cancer therapy and for the development of new targeting therapeutic
agents (2,18-20).
Notably,
3-substituted-2,3-dihydro-1H-isoindolin-1-one is a useful class of
functional pharmacophore because they exert various biological
activities, such as antihypertensive, antipsychotic, anesthetic,
antiulcer, vasodilatory, antiviral, antileukemic, anxiolytic,
antiasthma, anti-obesity, anti-ardiacangionosis and antitumor
properties (21-27).
In previous studies, cell-viability-based screening was performed
using MTT assays in the 3-aryl isoindolinone ring library and the
small molecule
2-[1-(4-(benzyloxy)phenyl)-3-oxoisoindolin-2-yl)-2-(4-methoxyphenyl)]
acetic acid (CDS-3078) significantly increased the inhibitory ratio
on HeLa cells (28,29). In the present study, CDS-3078 was
found to significantly increase p53 transcriptional activity and
induce apoptosis via the p53-mediated cell cycle arrest.
Materials and methods
Compounds and regents
The small organic molecule, CDS-3078, was
synthesized by the Center for Combinatorial Chemistry and Drug
Discovery of Jilin University, according to previous studies
(28,29). DMSO-dissolved CDS-3078 was diluted
in complete media for the following experiments performed in the
present study. Antibodies targeting total caspase-3 (cat. no.
sc-56053), caspase-8 (cat. no. sc-81656), caspase-9 (cat. no.
sc-133109), poly ADP-ribose polymerase (PARP; cat. no. sc-56196),
p53 (cat. no. sc-47698), p21 (cat. no. sc-71811), cytochrome c
(cat. no. sc-13560), cyclin B1 (cat. no. sc-245), CDK1 (cat. no.
sc-5319), checkpoint kinase (CHK) 1 (cat. no. sc-56288), CHK2 (cat.
no. sc-136251), M-phase phosphatase 3 (CDC25C; cat. no. sc-327),
phosphorylated (p)-ataxia telangiectasia and Rad3-related protein
(ATR; cat. no. sc-515173), Bcl-2 (cat. no. sc-7382), Bcl-2
homologous antagonist killer (BAK; cat. no. sc-517390), BAX (cat.
no. sc-7480), PUMA (cat. no. sc-374223), GAPDH (cat. no. sc-47724),
β-actin (cat. no. sc-8432), goat anti-rabbit IgG-HRP (cat. no.
sc-2005) and goat anti-rabbit IgG-HRP (cat. no. sc-2004) were
purchased from Santa Cruz Biotechnology, Inc. Antibodies against
p-CHK1 (cat. no. 12302) and p-CHK2 (cat. no. 2197) were obtained
from Cell Signaling Technology, Inc. Antibodies against the
γ-histone 2A family, member X (H2AX; cat. no. ab26350) was from
purchased from Abcam. PI, DAPI, MTT and other chemical reagents
were obtained from Sigma-Aldrich; Merck KGaA. The FITC-Annexin V
Apoptosis Detection kit I was purchased from Bioteke Corporation.
Caspase-3 (cat. no. BB-4106), Caspase-8 (cat. no. BB-4107) and
Caspase-9 (cat. no. BB-4108) activity assay kits were obtained from
Shanghai bestbio Biotechnology Co., Ltd. (www.bestbio.com.cn).
Cell cultures
Cervical cancer HeLa cells and human umbilical vein
endothelium cells (HUVECs) were purchased from the Chinese Academy
of Sciences Cell Bank. The cells were grown in DMEM (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS (Hyclone; GE
Healthcare Life Sciences), 100 µg/ml streptomycin, 100 U/ml
penicillin and 2 mM L-glutamine at 37˚C in a humidified atmosphere
with 5% CO2.
In vitro cell viability assays
Cell viability or cell growth inhibition of CDS-3078
was performed using an MTT assay. Briefly, HeLa cells
(5x103 cells/well) were plated in a 96-well plate and
incubated overnight. After removal of the media, the cells were
treated with fresh DMEM or indicated concentrations (0.14, 0.37,
1.1, 3.3, 11, 33 or 100 µM) of CDS-3078 for 12, 24 and 48 h at
37˚C. After treatment, 20 µl of MTT (0.5 mg/well) solution was
added and the plates were incubated for an additional 3 h at 37˚C.
After removal of the culture media, 150 µl of DMSO was added to the
culture wells to dissolve the resultant pellet. The quantity of
formazan crystals that were an MTT redox product reduced by the
mitochondrial succinate dehydrogenase in living cells represented
the number of living cells. The absorbance of formazan crystal at
495 nm was measured using a microplate reader.
Nuclear staining with DAPI
To observe the morphology changes, HeLa cells
(5x104 cells/well) were seeded in a 24-well plate and
incubated for 24 h at 37˚C. Subsequently, the cells were treated
with DMEM (control) or 2 µM of CDS-3078 solution for 24 h at 37˚C.
Finally, the cells were rinsed twice with PBS and stained with
fresh media containing DAPI (2.5 µg/ml) for 5 min at room
temperature. The morphology of the nuclei was determined using
fluorescence microscopy.
Reverse transcription-quantitative
PCR
Total cell RNA was purified from HeLa cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and cDNA was prepared using cDNA Synthesis Kit (Thermo Fisher
Scientific, Inc.). Quantitative PCR was conducted using SYBR™-Green
PCR Master Mix (Thermo Fisher Scientific, Inc.) with a 7500 Fast
Real-Time PCR System according to the manufacturer's guidelines.
The thermocycling conditions for real-time PCR were as follows:
Denaturation at 95˚C for 15 sec, annealing at 60˚C for 20 sec and
extension at 72˚C for 1 min, for 40 cycles. Specific primers pairs
were used: p53 forward, 5'-CTGCCTTCCGGGTCACTGC-3' and reverse,
5'-TTGGGACGGCAAGGGGGACA-3'; p21 forward, 5'-GGAAGACCATGTGGACCTGT-3'
and reverse, 5'-GGCGTTTGGAGTGGTAGAAA-3'; PUMA forward,
5'-TAGAGAGAGCGACGTGAC-3' and reverse, 5'-CGGTATCTACAGCAGCGCAT-3';
Noxa forwards, 5'-AGAGCTGGAAGTCGAGTGTG-3' and reverse,
5'-GGAGTCCCCTCATGCAAGTT-3'; and GAPDH forward,
5'-ACCACAGTCCATGCCATCAC-3' and reverse,
5'-TCCACCACC-CTGTTGCTGTA-3'. The experiments were repeated ≥3
times, and mRNA expression levels were standardized by comparison
to the transcript levels of the reference gene, GAPDH, based on the
comparative threshold method (30).
Apoptosis and cell cycle analysis
Apoptosis assay was conducted using a FITC-Annexin V
Apoptosis Detection kit (Bioteke Corporation) according to the
manufacturer's protocol. In total, 3x105 cells were
plated into a 6-well plate and incubated overnight at 37˚C.
Subsequently, the cells were pretreated with fresh DMEM or
different concentrations of CDS-3078 (2, 5 and 10 µM) for 24 h at
37˚C. After treatment, the cells were collected and washed three
times with ice-cold PBS. The resultant cell pellets were incubated
with 5 µl of FITC-conjugated Annexin V and 5 µl of propidium iodide
(PI) for 15 min at room temperature. Fluorescence analysis was
performed using a Beckman Flow Cytometry Analyzer (Beckman
CytoFLEX; Beckman Coulter, Inc.). Cells were classified as early
apoptotic (Annexin V-positive/PI-negative), late apoptotic/necrotic
(Annexin V-positive/PI-positive), necrotic/dead (Annexin
V-negative/PI-positive) and living cells
(annexin-negative/PI-negative).
For cell cycle assays, the collected samples were
fixed in 70% ice-cold ethanol overnight and washed twice in
ice-cold PBS. The resultant cells were stained with in 100 µl PI
solution (25 µg/ml RNase A, 50 µg/ml PI) and incubated at 37˚C for
30 min in the dark. The cell cycle distribution was examined using
a Beckman Flow Cytometry Analyzer (Beckman CytoFLEX; Beckman
Coulter, Inc.) analyzed using Flowjo 10.0.7 software (FlowJo
LLC).
Western blotting analysis
After the indicated drug treatments, cell lysates
were extracted using ice-cold lysis buffer [50 mM Tris (pH 8.0),
150 mM NaCl, 0.1% SDS, 1% NP-40 and 0.5% sodium deoxycholate]
containing protease/phosphatase inhibitors [1% Cocktail
(Sigma-Aldrich; Merck KGaA)] and 1 mM phenylmethylsulfonyl fluoride
at 4˚C for 10 min and protein was quantified using Pierce BCA
Protein Assay Kit (Invitrogen; Thermo Fisher Scientific, Inc.).
Equal amounts of protein (40 µg) were resolved by SDS-PAGE (10%
gel) and blotted to PVDF membranes (EMD Millipore). Subsequently,
the membranes were blocked with 5% non-fat milk in TBS supplemented
with 0.1% Tween-20 (TBST) for 1 h at room temperature and then
incubated with the aforementioned antibodies (dilution ratio,
1:200) overnight at 4˚C. Next, membranes were washed with TBST
three times and incubated with the aforementioned
peroxidase-conjugated secondary antibodies (dilution ratio,
1:2,000) for 1 h at room temperature. Finally, blots were
visualized using the BeyoECL Star kit (Beyotime Institute of
Biotechnology) according to the manufacturer's protocol and GAPDH
or β-actin were used as internal control. The relative density was
determined using grayscale analysis on ImageJ v1.8.0_112 (National
Institutes of Health) and normalized to GAPDH or β-actin.
Caspase activity assay
Caspase-3/8/9 activity assay was performed using
caspase-3/8/9 activity kits by colorimetric assays, according to
the manufacturer's protocol. After treating the cells for the
indicated period, the cells were collected by centrifugation
(10,000 x g, 10 min) at 4˚C. The resultant cell pellets were
incubated in 100 µl of lysis buffer. The suspension was centrifuged
at 10,000 x g for 10 min at 4˚C and an equal amount of supernatant
was incubated with the corresponding substrates [Ac-DEVD-AFC
(caspase-3), Ac-LETD-AFC (caspase-8), Ac-LEHD-AFC (caspase-9)] in
the reaction buffer containing dithiothreitol at 37˚C for 10 min.
The absorbance at 405 nm was examined using a microplate
reader.
Statistical analysis
All quantitative data are expressed as the mean ± SD
of three independent experiments. Statistical differences were
assessed using ANOVAs and post hoc test (Dunnett's test). SPSS 19.0
(IBM Corp.) was used for statistical analysis and P<0.05
represented a statistically significant difference.
Results
Inhibitory effects of CDS-3078 on HeLa
cells
The heterocyclic alkaloid
2,3-dihydro-1H-isoindolin-1-one is widely found in natural products
and has been shown to have a potential activity in cancer therapy
by regulating cell cycle arrest or by inducing apoptosis in various
cancer cells (31,32). Based on the anti-tumor effects of
CDS-3078, the present study synthesized the small molecule
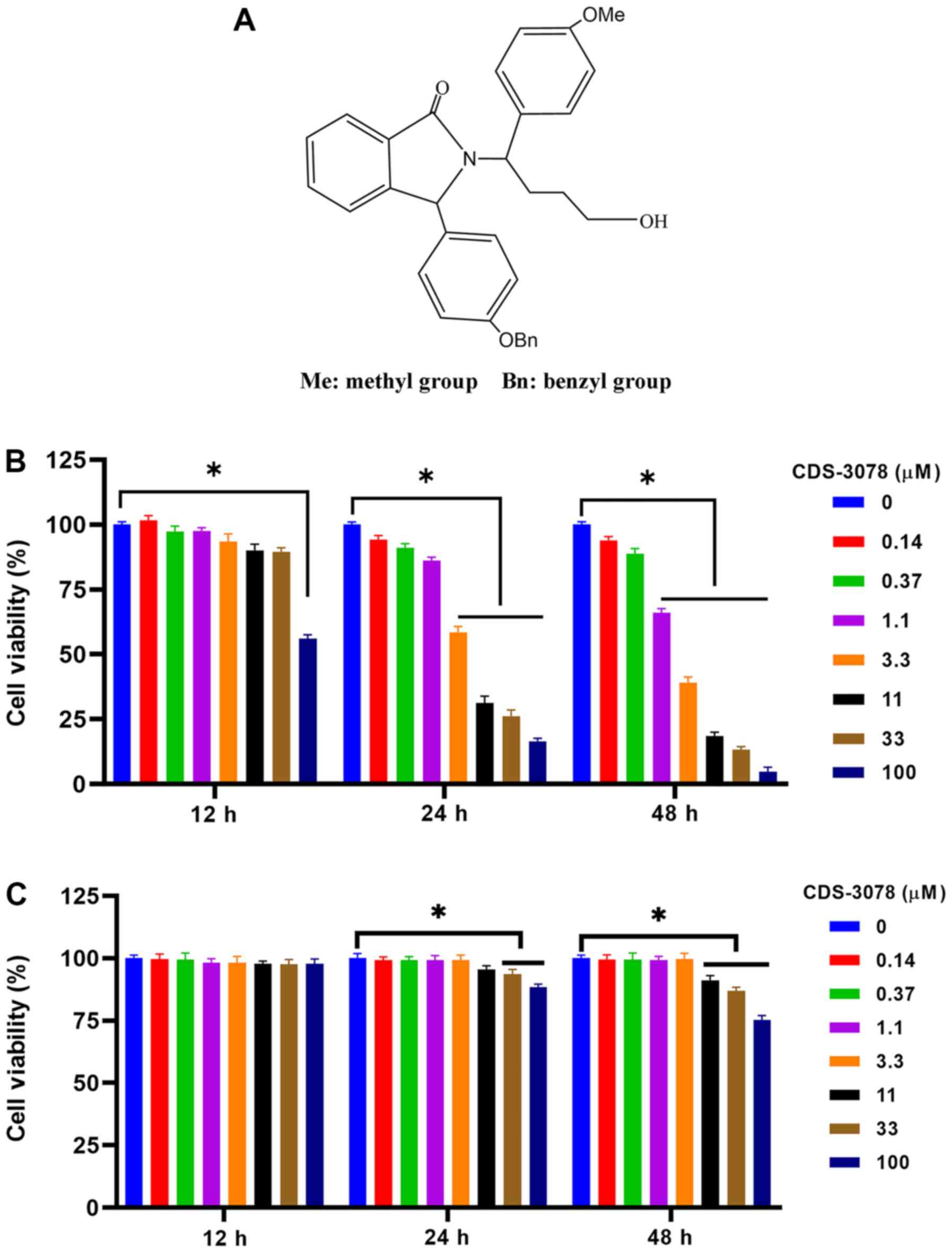
(Fig. 1A). To confirm the
inhibitory effects of CDS-3078 on HeLa and HUVEC cell
proliferation, the cytotoxicity of CDS-3078 was analyzed using MTT
assays. As shown in Fig. 1B, the
inhibitory activity of CDS-3078 significantly increased in a
concentration-dependent manner. The IC50 of CDS-3078
were 3.794 and 1.944 µM after 24 and 48 h, respectively. Moreover,
CDS-3078 did not show an obvious proliferation inhibition on HUVECs
(Fig. 1C). The present results
suggested that CDS-3078 may be a potential anti-cancer agent.
CDS-3078 induces p53-dependent
apoptotic cell death in human cervical cancer cells
p53 is involved in the regulation of the
proliferation of various cancer cells (16). In order to investigate whether the
inhibitory effect of CDS-3078 on HeLa cells is through p53, the RNA
and protein expression levels of p53 and its downstream factors
were examined. As shown in Fig. 2A,
the mRNA expression levels of p53, p21 and PUMA were significantly
increased in a dose-dependent manner following CDS-3078 treatment
(5 µM) in HeLa cells, whereas no changes in Noxa mRNA expression
were detected. In line with these results, CDS-3078 treatment
increased the protein expression levels p53 and p21 in a time- and
dose-dependent manner (Fig. 2B). It
was then examined whether CDS-3078 could induce apoptotic cell
death in p53+/+ HeLa cells. The apoptosis assay
suggested that CDS-3078 treatment markedly induced accumulation of
early apoptotic cells (Annexin V-positive and PI-negative) in a
time- and dose-dependent manner (Fig.
2C and D). The rates of early
apoptotic cells were 5.52, 37.15 and 77.26% following a 24-h
administration with CDS-3078 at 2, 5 and 10 µM, respectively. By
contrast, the apoptotic rates were 7.81, 82.3 and 95.89% following
a 48-h treatment with CDS-3078 at 2, 5 and 10 µM, respectively
(Fig. 2D). Additionally,
microscopic investigations indicated that 5 µM of CDS-3078 induced
a series of morphological changes such as cell shrinkage, chromatin
condensation and nuclear fragmentation (Fig. 2E). The present results suggested
that CDS-3078 administration inhibited the proliferation and
induced apoptotic cell death in HeLa cells.
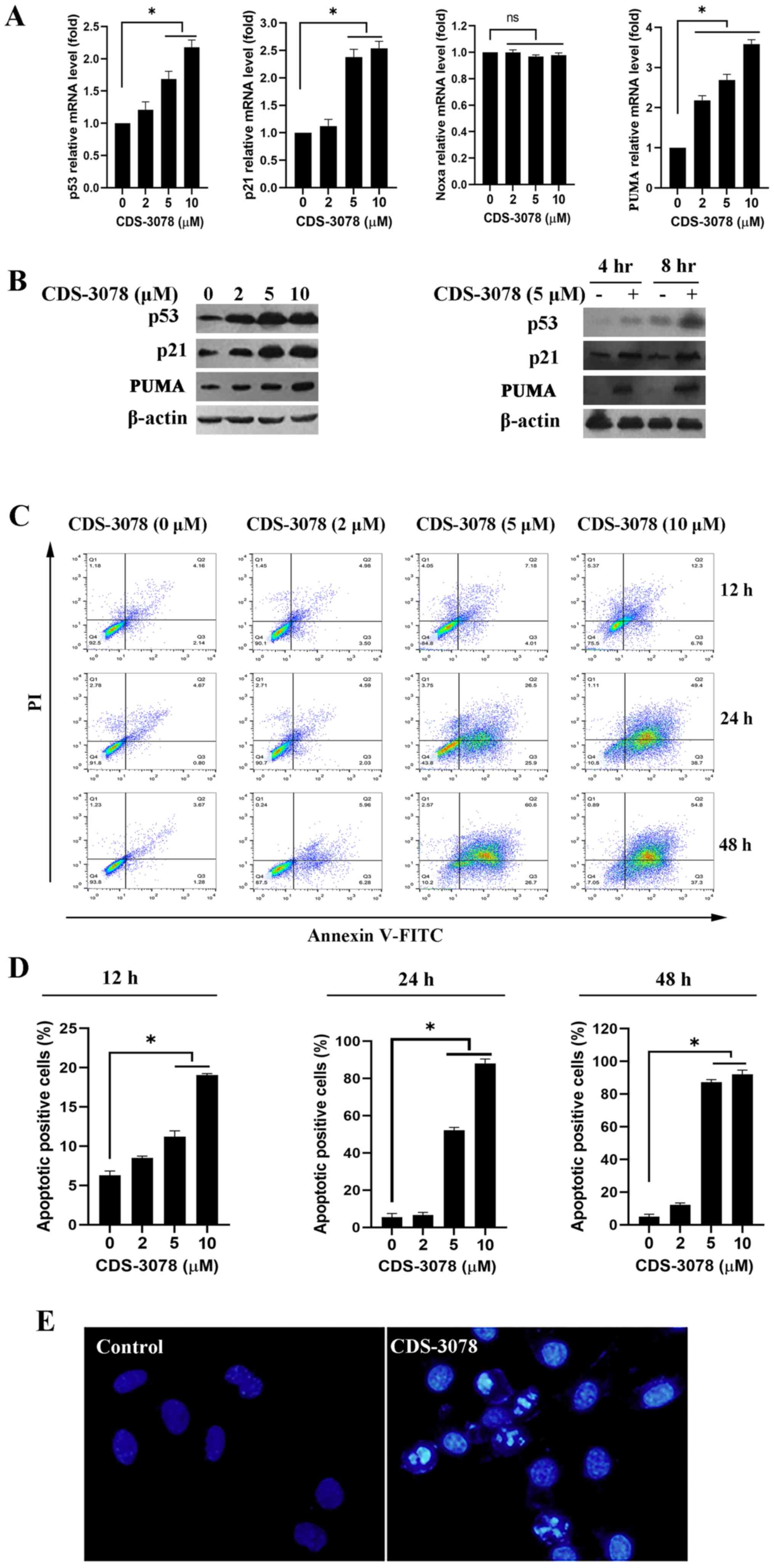 | Figure 2CDS-3078 enhances p53 transcriptional
activity and protein expression, as well as inducing apoptotic
death. (A) HeLa cells were treated with the specific concentrations
of CDS-3078 (2, 5 and 10 µM) for 8 h and then the mRNA expression
levels of p53, p21, Noxa and PUMA were determined using reverse
transcription-quantitative PCR. (B) HeLa cells were treated with
indicated concentrations of CDS-3078 (2, 5 and 10 µM) for 24 h and
then the protein expression levels of p53, p21 and PUMA were
detected using western blotting. β-actin was used as a loading
control. (C) HeLa cells were treated with different concentrations
of CDS-3078 (2, 5 and 10 µM) for 12, 24 or 48 h. Subsequently, the
apoptotic ratio was evaluated using flow cytometry. (D)
Quantification of the percentage of early apoptotic HeLa cells
following the various administration times. (E) Cells were treated
with 5 µM of CDS-3078 for 24 h. The cells were fixed and stained
with DAPI. The chromatin condensation and DNA fragmentation were
then observed under a fluorescent microscope (magnification, x200)
using a blue filter. *P<0.05 vs. vehicle control.
CDS-3078,
2-[1-(4-(Benzyloxy)phenyl)-3-oxoisoindolin-2-yl)-2-(4-methoxyphenyl)]
acetic acid; Noxa, phorbol-12-myristate-13-acetate-induced protein
1; ns, not significant; PUMA, p53 upregulated modulator of
apoptosis. |
CDS-3078 induces apoptosis involving
members of the Bcl-2-family
A previous study has demonstrated that p53-mediated
apoptosis influences the intrinsic mitochondrial pathway (33). In this pathway, mitochondrial
dysfunction is associated with the expression levels of
Bcl-2-family members, thus causing apoptosis (34). The effects of CDS-3078
administration on the mitochondria-mediated apoptotic signaling
pathway were therefore assessed in the present study. As shown in
Fig. 3A and B, CDS-3078 treatment resulted in the
proteolytic cleavage of pro-caspase-3, pro-caspase-9 and PARP, but
did not induce changes in the expression levels of pro-caspase-8,
indicating the activation of the mitochondria-mediated apoptosis
pathway. In line with these results, CDS-3078 administration led to
a 4-fold increase in caspase-3 and 2.7-fold increase in caspase-9
expression levels compared with respective negative controls;
however, without affecting the expression levels of caspase-8
(Fig. 3C). Regarding the
mitochondria-mediated pathway, CDS-3078 treatment resulted in a
decrease in Bcl-2 and an increase in the pro-apoptotic BAK protein
expression levels, but BAX protein expression levels were
unaltered. Moreover, CDS-3078 induced the cytoplasmic release of
cytochrome c in HeLa cells (Fig.
3A). Collectively, the present data showed that CDS-3078
administration downregulated Bcl-2 protein expression levels, but
upregulated BAK expression and led to cytochrome c release
by inducing disruption of the mitochondrial signaling pathway.
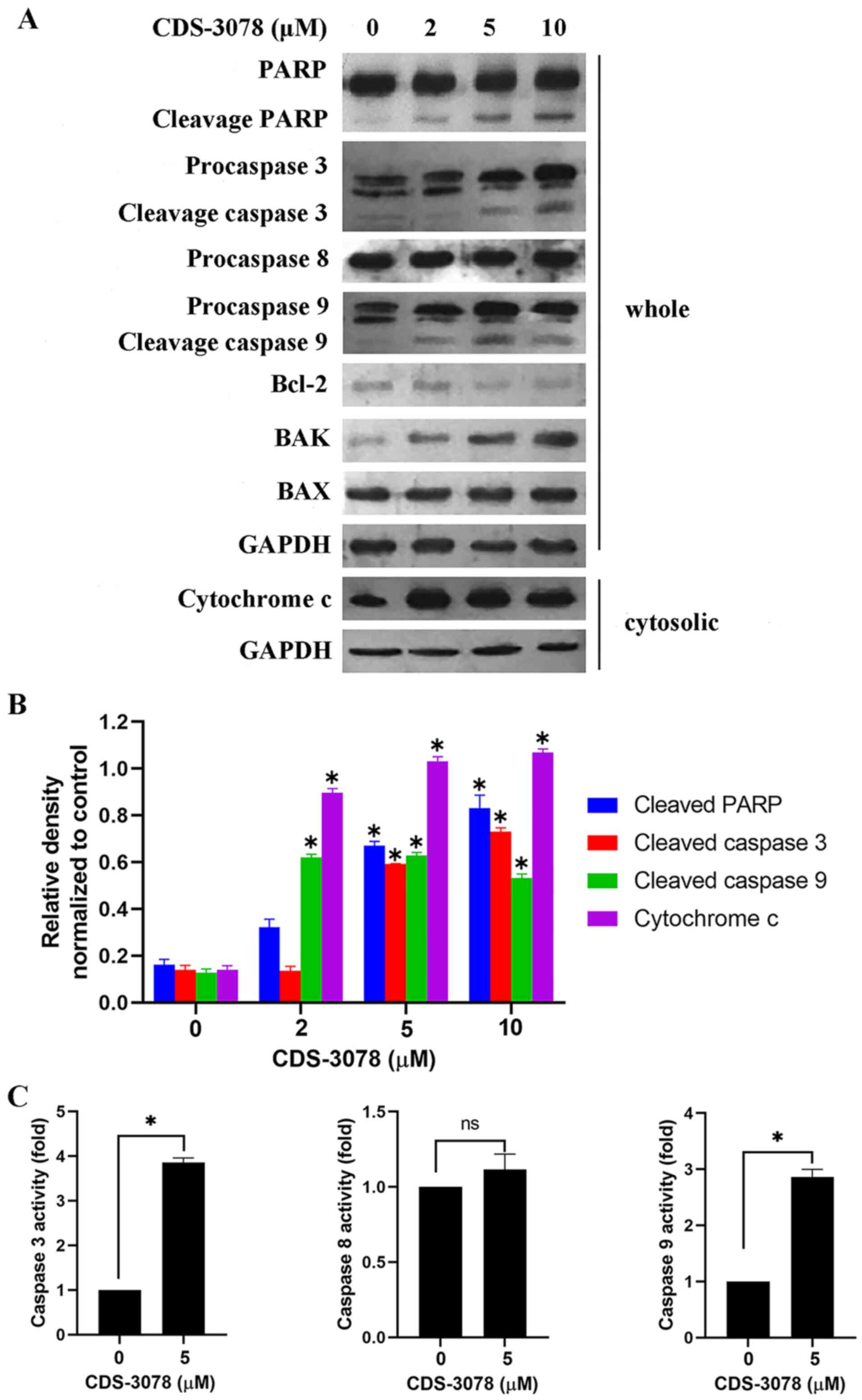 | Figure 3CDS-3078 promotes
mitochondria-mediated dysfunction and regulates the protein
expression of Bcl-2 family. (A) Cells were treated with various
concentrations of CDS-3078 (2, 5 and 10 µM) for 24 h, cytosolic
extracts and whole-cell lysates were detected by immunoblotting to
examine the expression levels of cytochrome c, Bcl-2, BAK,
BAX, PARP and caspase-3/8/9. (B) The relative density was
determined using grayscale analysis on ImageJ v 1.8.0_112 (National
Institutes of Health), normalized to GAPDH. (C) Cells were treated
with 5 µM CDS-3078 for 12 h. Equal amounts of cell lysates were
analyzed for caspase-3, caspase-8 and caspase-9 activity using
Ac-DEVD-AFC, Ac-LETD-AFC, Ac-LEHD-AFC as substrates, respectively.
DMSO treatment was used as the control. The concentration of the
fluorescent products released were then measured. Results represent
the mean ± SD of three independent repeats. *P<0.05
vs. 0 µM CDS-3078 control. CDS-3078,
2-[1-(4-(Benzyloxy)phenyl)-3-oxoisoindolin-2-yl)-2-(4-methoxyphenyl)]
acetic acid; PARP, poly ADP-ribose polymerase. |
CDS-3078 induces cell cycle arrest at
the G2/M phase
G2/M phase arrest in response to
genotoxic stress is associated with the p53-p21 signaling pathway
(17). To examine whether CDS-3078
was able to induce cell-cycle arrest, the DNA distribution ratio of
CDS-3078-treated HeLa cells was investigated after 24 h of
treatment using flow cytometry. CDS-3078 treatment increased the
proportion of cells in the G2/M phase from 7.97±2.45%
(control) to 15.44±2.11% (2 µM), 77.13±4.02% (5 µM) and 99.35±3.71%
(10 µM) in HeLa cells (Fig. 4A and
B). To further investigate the
possible molecular mechanisms underlying G2/M-phase
arrest in CDS-3078-induced HeLa cells, the expression levels of
important regulators of cell cycle progression involved in
G2/M phase arrest were investigated. The activation
and/or deactivation of specific cyclin-CDK complexes regulate
G2/M phase transition (35). The present results suggested that
CDS-3078 administration decreased the expression levels of cyclin
B1 in a time-dependent manner, decreased the expression levels of
CDC25C and significantly upregulated the expression levels of
p-CHK1 (Fig. 4C and D). By contrast, the protein expression
levels of CHK2 and p-CHK2 were not significantly changed following
CDS-3078 treatment in HeLa cells. The activation of CHK1 is
involved in DNA damage (36);
therefore, the expression levels of markers associated with DNA
damage, including p-ATR and γ-H2AX were examined. As shown in
Fig. 4C and D, CDS-3078 treatment increased the protein
expression levels of γ-H2AX and p-ATR. Taken together, the present
results suggested that CDS-3078 was involved in
G2/M-phase arrest through the DNA-damage checkpoint cell
signaling pathways.
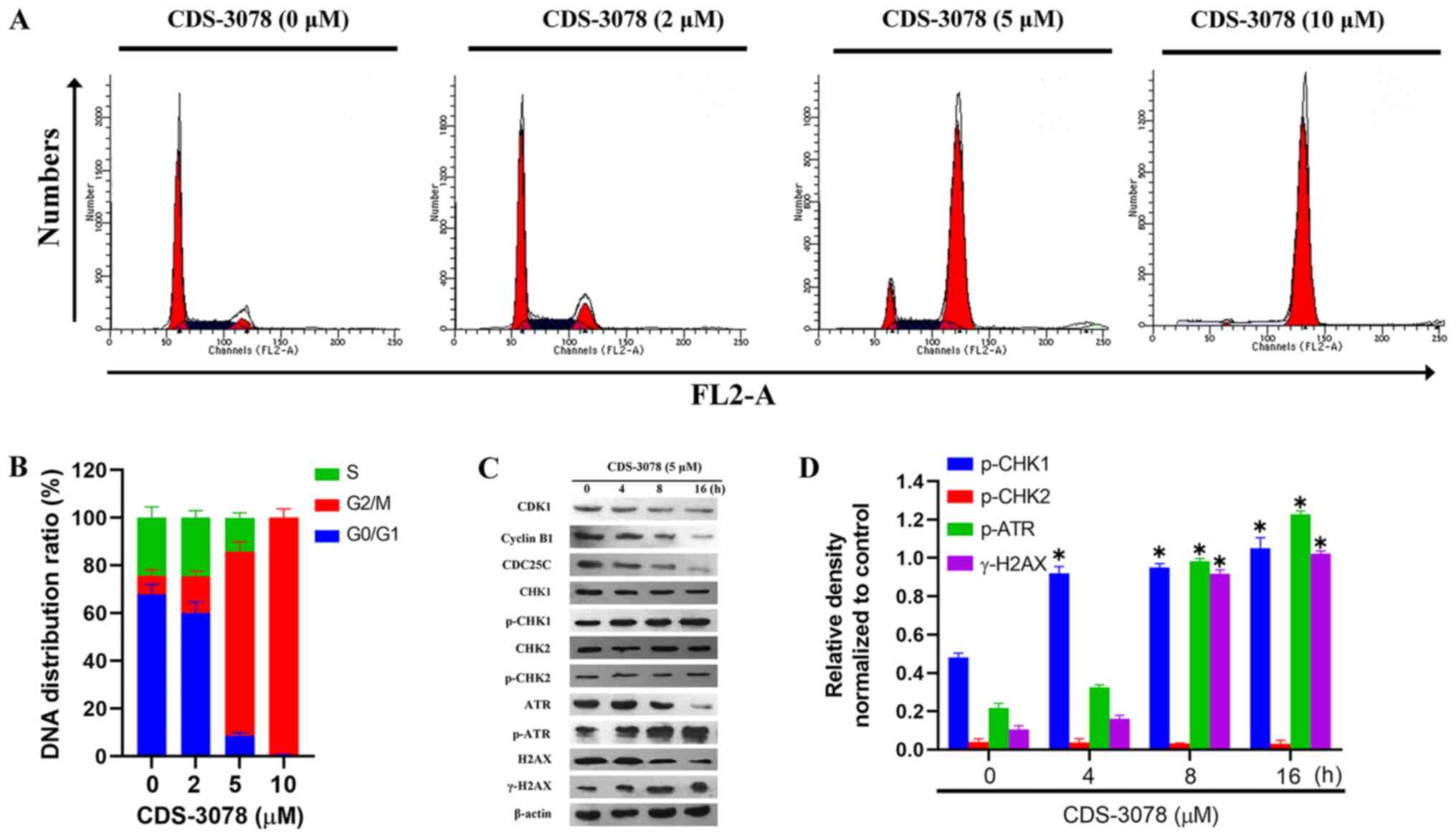 | Figure 4CDS-3078 induces cell cycle arrest at
the G2/M phase. (A) HeLa cells were pretreated with 2, 5
and 10 µM of CDS-3078 for 24 h, stained with PI and then subjected
to DNA content analysis using flow cytometry. Data represents three
independent repeats. (B) Quantification of the percentage of
G0/G1, S and G2/M HeLa cells at
the various administration times. (C) HeLa cells were treated with
CDS-3078 (5 µM) for 4, 8 and 16 h. The expression levels of the
proteins associated with the cell cycle were examined using
immunoblotting to detect expression levels of CDK1, cyclin B1,
CDC25C, CHK1, p-CHK1, CHK2, p-CHK2, ATR, p-ATR, H2AX and γ-H2AX.
(D) The relative density was determined using grayscale analysis on
ImageJ 1.8.0_112 (National Institutes of Health) normalized to
corresponding non-phosphorylated total protein. CDS-3078,
2-[1-(4-(Benzyloxy)phenyl)-3-oxoisoindolin-2-yl)-2-(4-methoxyphenyl)acetic
acid; CHK, checkpoint kinase; CDC25C, M-phase phosphatase 3; H2AX,
H2A histone family, member X; ATR, ataxia telangiectasia and
Rad3-related protein; p-, phosphorylated. *P<0.05 vs.
control. |
Discussion
In the present study, a small-molecule anticancer
agent was identified from a 3-aryl isoindolinone ring library using
a cell viability assay. CDS-3078, a
3-substituted-2,3-dihydro-1H-isoindolin-1-one derivate, was
identified as a p53-activating agent with potential proliferation
inhibitory-effects against tumor cells.
It is well documented that p53 is a widely accepted
cancer driver gene, making it a valuable target for anticancer
therapy (1). After 40 years of
research focused on p53, the United States Food and Drug
Administration approved various p53-specific therapies, including
gene therapy. However, various p53-targeted therapeutic strategies,
including the interaction between p53 and Mouse double minute 2
homolog (MDM2), as well as the pharmacological restoration of
mutant p53, require further investigation (37,38).
The present study suggested that CDS-3078
upregulated the mRNA and protein expression levels of p53 and
p53-target genes. In addition, p21 serves a key role in cell cycle
arrest following DNA damage by inhibiting either CDK activity or
the formation of the cyclin B1-CDK1 complex (39). The expression levels of PUMA, a key
effector of p53-mediated apoptosis (40), was found to be upregulated in the
present study. It was hypothesized that p53- and p53-targeting
factors-mediated signaling pathways may serve important roles in
inducing G2/M cell cycle arrest and apoptosis. The
present results suggested that CDS-3078 administration inhibited
cell proliferation and induced apoptosis in p53+/+ HeLa
cells. Moreover, mitochondrial membrane impairment caused by
CDS-3078 administration is considered a sign of early apoptosis
(41); however, the potential of
this small molecule to affect the mitochondrial membrane requires
further verification. Notably, the drug safety and application
potential of CDS-3078 requires further verification in various
cancer cells types and in vivo experiments.
The activation of cell cycle checkpoint-related
proteins, inducing cell cycle arrest, allows DNA repair in response
to DNA damage. G2/M checkpoints are more sensitive to
chemotherapeutic or radiotherapeutic agents compared with
G1 checkpoints (42,43).
CHK1 and CHK2, effectors of DNA damage checkpoints, serve a central
role in inducing cell cycle arrest and DNA repair in response to
DNA damage (44). The present
results suggested that CDS-3078 treatment induced a
concentration-dependent increase in cells arrested at the
G2 phase due to the decrease in the expression levels of
cyclin B1, CDK1 and CDC25C. As an important kinase associated with
CDC25C regulation, phosphorylation levels of CHK1 were found to be
increased after CDS-3078 administration, indicating the activation
of CHK1(45). Although CHK1 and
CHK2 are functionally associated, no changes in the phosphorylation
and protein expression levels of CHK2 were identified in
CDS-3078-treated HeLa cells. CDS-3078 treatment also increased the
expression levels of ATR and γ-H2AX, which are involved in the
transduction of early signals following DNA double-strand breaks in
response to cytotoxic stresses (46). The present results suggested that
CDS-3078 induced p53-mediated apoptotic cell death through
G2/M phase arrest.
Apoptosis can be induced by various stimuli and is
mediated by two signaling pathways: The intrinsic and extrinsic
pathways. These pathways are involved in the activation of various
effectors and the release of proapoptotic mediators from
mitochondria (47). In the present
study, CDS-3078 treatment resulted in apoptosis via the proteolytic
cleavage of caspase-3 and PARP; downregulation of Bcl-2;
upregulation of BAK; and release of cytochrome c. Based on these
data, it was speculated that CDS-3078 served as a potential small
molecule inhibitor through the caspase-dependent mitochondrial
apoptotic pathway in HeLa cells.
In conclusion, the present results suggested that
the small molecule CDS-3078 exhibited a potential cytotoxic effect
on human cervical cancer cells. Furthermore, CDS-3078
administration triggered mitochondria-mediated apoptotic cell death
and G2/M-phase cell cycle arrest in the
p53+/+ HeLa cells. Therefore, the present results
suggested that CDS-3078 may be a potential chemotherapeutic
candidate to be evaluated in clinical settings for the treatment of
cervical cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Foundation of Education Department of Jilin Province (grant no.
2016133), the Special Foundation for Industry Innovation of
Development and Reform Commission of Jilin (grant no. 2018C049-4),
the Youth Foundation of Jilin Science and Technology Bureau (grant
nos. 20166026 and 201750259) and the Major Programs of the Jilin
Institute of Chemical Technology (grant no. 20180101).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ, XB, and WS conceived the experiments. YZ, JR,
YG, PL and CH conducted the experiments. YZ, WS produced the
manuscript and performed result analysis. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sabapathy K and Lane DP: Therapeutic
targeting of p53: All mutants are equal, but some mutants are more
equal than others. Nat Rev Clin Oncol. 15:13–30. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bykov VJN, Eriksson SE, Bianchi J and
Wiman KG: Targeting mutant p53 for efficient cancer therapy. Nat
Rev Cancer. 18:89–102. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Oren M: Decision making by p53: Life,
death and cancer. Cell Death Differ. 10:431–442. 2003.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Olivier M, Hollstein M and Hainaut P: TP53
mutations in human cancers: Origins, consequences, and clinical
use. Cold Spring Harb Perspect Biol. 2(a001008)2010.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ryu H, Nam KY, Kim JS, Hwang SG, Song JY
and Ahn J: The small molecule AU14022 promotes colorectal cancer
cell death via p53-mediated G2/M-phase arrest and
mitochondria-mediated apoptosis. J Cell Physiol. 233:4666–4676.
2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Su ZY, Yang ZZ, Xu YQ, Chen YB and Yu Q:
Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol
Cancer. 14:48–61. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bremer E, van Dam G, Kroesen BJ, de Leij L
and Helfrich W: Targeted induction of apoptosis for cancer therapy:
Current progress and prospects. Trends Mol Med. 12:382–393.
2006.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Makin G and Hickman JA: Apoptosis and
cancer chemotherapy. Cell Tissue Res. 301:143–152. 2000.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sellers WR and Fisher DE: Apoptosis and
cancer drug targeting. J Clin Invest. 104:1655–1661.
1999.PubMed/NCBI View
Article : Google Scholar
|
|
11
|
Pan ST, Li ZL, He ZX, Qiu JX and Zhou SF:
Molecular mechanisms for tumour resistance to chemotherapy. Clin
Exp Pharmacol Physiol. 43:723–737. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Ichim G and Tait SW: A fate worse than
death: Apoptosis as an oncogenic process. Nat Rev Cancer.
16:539–548. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Koff JL, Ramachandiran S and
Bernal-Mizrachi L: A time to kill: Targeting apoptosis in cancer.
Int J Mol Sci. 16:2942–2955. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Von Karstedt S, Montinaro A and Walczak H:
Exploring the TRAILs less travelled: TRAIL in cancer biology and
therapy. Nat Rev Cancer. 17:352–366. 2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Aubrey BJ, Kelly GL, Janic A, Herold MJ
and Strasser A: How does p53 induce apoptosis and how does this
relate to p53-mediated tumour suppression? Cell Death Differ.
25:104–113. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Singh R, Letai A and Sarosiek K:
Regulation of apoptosis in health and disease: The balancing act of
BCL-2 family proteins. Nat Rev Mol Cell Biol. 20:175–193.
2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Chen JD: The cell-cycle arrest and
apoptotic functions of p53 in tumor initiation and progression.
Cold Spring Harb Perspect Med. 6(a026104)2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Stegh AH: Targeting the p53 signaling
pathway in cancer therapy-the promises, challenges, and perils.
Expert Opin Ther Targets. 16:67–83. 2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Prabhu VV, Allen JE, Hong B, Zhang SL,
Cheng HR and EI-Deiry WS: Therapeutic targeting of the p53 pathway
in cancer stem cells. Expert Opin Ther Targest. 16:1161–1174.
2012.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Joana DA, Joana MX, Clifford JS and
Cecília MP: Targeting the p53 pathway of apoptosis. Curr Pharm
Design. 16:2493–2503. 2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Belliotti TR, Brink WA, Kesten SR, Rubin
JR, Wustrow DJ, Zoski KT, Whetzel SZ, Corbin AE, Pugsley TA,
Heffner TG and Wise LD: Isoindolinone enantiomers having affinity
for the dopamine D4 receptor. Bioorg Med Chem Lett. 8:1499–1502.
1998.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhuang ZP, Kung MP, Mu M and Kung HF:
Isoindol-1-one Analogues of
4-(2-methoxyphenyl)-1-[2-[N-(2-pyridyl)-p-iodobenzamido] ethyl]
piperazine (p-MPPI) as 5-HT1A receptor ligands. J Med Chem.
41:157–166. 1998.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kanamitsu N, Osaki T, Itsuji Y, Yoshimura
M, Tsujimoto H and Soga M: Novel water-soluble sedative-hypnotic
agents: Isoindolin-1-one derivatives. Chem Pharm Bull.
55:1682–1688. 2007.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Bernstein PR, Aharony D, Albert JS,
Andisik D, Barthlow HG, Bialecki R, Davenport T, Dedinas RF,
Dembofsky BT, Koether G, et al: Discovery of novel, orally active
dual NK1/NK2 antagonists. Bioorg Med Chem Lett. 11:2769–2773.
2001.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wacker DA, Varnes JG, Malmstrom SE, Cao X,
Hung CP, Ung T, Wu G, Zhang G, Zuvich E, Thomas MA, et al:
Discovery of
(R)-9-Ethyl-1,3,4,10b-tetrahydro-7-trifluoromethylpyrazino[2,1-a]isoindol-6(2H)-one,
a selective, orally active agonist of the 5-HT(2C) receptor. J Med
Chem. 50:1365–1379. 2007.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Pendrak I, Barney S, Wittrock R, Lambert
DM and Kingsbury WD: Synthesis and anti-HSV activity of
a-ring-deleted mappicine ketone analog. J Org Chem. 59:2623–2625.
1994.
|
|
27
|
Taylor EC, Zhou P, Jennings LD, Mao Hu ZB
and Jun JG: Novel synthesis of a conformationally-constrained
analog of DDATHF. Tetrahedron Lett. 38:521–524. 1997.
|
|
28
|
Hu CM, Zheng LY, Pei YZ and Bai X:
Synthesis of novel 3-aryl isoindolinone derivatives. Chem Res Chin
Univ. 29:487–494. 2013.
|
|
29
|
Hu CM: Design, synthesis and anticancer
activity of novel 3-aryl isoindolinone derivatives. Changchun Jilin
University, 2012.
|
|
30
|
Valente LJ, Aubrey BJ, Herold MJ, Kelly
GL, Happo L, Scott CL, Newbold A, Johnstone RW, Huang DC, Vassilev
LT and Strasser A: Therapeutic response to non-genotoxic activation
of p53 by Nutlin3a Is driven by PUMA-mediated apoptosis in lymphoma
cells. Cell Rep. 14:1858–1866. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Shah JH, Swartz GM, Papathanassiu AE,
Treston AM, Fogler WE, Madsen JW and Green SJ: Synthesis and
enantiomeric separation of 2-phthalimidino-glutaric acid analogues:
Potent inhibitors of tumor metastasis. J Med Chem. 42:3014–3017.
1999.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hardcastle IR, Liu J, Valeur E, Watson A,
Ahmed SU, Blackburn TJ, Bennaceur K, Clegg W, Drummond C, Endicott
JA, et al: Isoindolinone inhibitors of the murine double minute 2
(MDM2)-p53 protein-protein interaction: Structure-activity studies
leading to improved potency. J Med Chem. 54:1233–1243.
2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Yin C, Knudson CM, Korsmeyer SJ and Van
Dyke T: Bax suppresses tumorigenesis and stimulates apoptosis in
vivo. Nature. 385:637–640. 1997.PubMed/NCBI View
Article : Google Scholar
|
|
34
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
35
|
DiPaola RS: To arrest or not to G2-M
cell-cycle arrest. Clin Cancer Res. 8:3311–3314. 2002.PubMed/NCBI
|
|
36
|
Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan
KH and Brody C: BRCA1 regulates the G2/M checkpoint by activating
Chk1 kinase upon DNA damage. Nat Genet. 30:285–289. 2002.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wang WS and Hu YZ: Small molecule agents
targeting the p53-MDM2 pathway for cancer therapy. Med Res Rev.
32:1159–1196. 2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zhao DK, Tahaney WM, Mazumdar A, Savage MI
and Brown PH: Molecularly targeted therapies for p53-mutant
cancers. Cell Mol Life Sci. 74:4171–4187. 2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Deng CX, Zhang PM, Harper JW, Elledge SJ
and Leder P: Mice lacking p21CIP1/WAF1 undergo normal development,
but are defective in G1 checkpoint control. Cell. 82:675–684.
1995.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Taylor RC, Cullen SP and Martin SJ:
Apoptosis: Controlled demolition at the cellular level. Nat Rev Mol
Cell Biol. 9:231–241. 2008.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Wang CX and Youle RJ: The role of
mitochondria in apoptosis. Annu Rev Genet. 43:95–118.
2009.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Su TT: Cellular responses to DNA damage:
One signal, multiple choices. Annu Rev Genet. 40:187–208.
2006.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Bucher N and Britten CD: G2 checkpoint
abrogation and checkpoint kinase-1 targeting in the treatment of
cancer. Br J Cancer. 98:523–528. 2008.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Smith J, Tho LM, Xu N and Gillespie DA:
The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and
cancer. Adv Cancer Res. 108:73–112. 2010.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Sur S and Agrawal DK: Phosphatases and
kinases regulating CDC25 activity in the cell cycle: Clinical
implications of CDC25 overexpression and potential treatment
strategies. Mol Cell Biochem. 416:33–46. 2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Mah LJ, El-Osta A and Karagiannis TC:
gammaH2AX: A sensitive molecular marker of DNA damage and repair.
Leukemia. 24:679–686. 2010.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Ola MS, Nawaz M and Ahsan H: Role of Bcl-2
family proteins and caspases in the regulation of apoptosis. Mol
Cell Biochem. 351:41–58. 2011.PubMed/NCBI View Article : Google Scholar
|