Introduction
Traumatic brain injury (TBI) has deleterious effects
on public health and is associated with high mortality and
morbidity rates worldwide, with an incidence of 69 million
individuals suffering from TBI each year (1,2).
Global mortality rate has been recorded to be 30-40% in severely
affected patients with TBI (2).
Patients who survive TBI commonly suffer from physical and
cognitive disabilities that increase their susceptibility to other
neurological disorders (3). In
addition to primary mechanical damage, TBI activates a cascade of
pathophysiological mechanisms leading to secondary brain injury
(4,5). The secondary effects include glutamate
excitotoxicity, free radical production, oxidative stress,
mitochondrial dysfunction, loss of ATP, inflammation and ultimately
neuronal death (4-7).
TBI evolves within weeks to months, and can induce behavioural
perturbations (8). It is therefore
crucial to develop novel strategies to treat multifaceted and
complex pathophysiological mechanisms associated with TBI.
TBI induces synaptic damage that results in neuronal
dysfunction and subsequent neuronal apoptosis (9,10).
Synaptic structure and function serve a crucial role in brain
development and cognitive functions. Under normal conditions, the
synaptic vesicle in neurons fuses with the plasma membrane and
releases neurotransmitters into the synaptic cleft (11). It is therefore essential to
determine the neurological synaptic molecules that may be released
following TBI. At present, >5,000 synaptic proteins have been
identified; however, only a few are associated with synaptic
dysfunction post-TBI (10). The
postsynaptic compartment of excitatory synapses contains an
electron-dense region known as the postsynaptic density (PSD). The
postsynaptic density protein-95 (PSD95) of the PSD family acts as a
scaffolding protein during synaptogenesis and regulates synaptic
maturation (12). In addition,
PSD95 serves a vital role in repairing injuries affecting the PSD
region, since it is essentiel for synaptic integration and
functional recovery following neuronal damage (13,14).
PSD95 interacts with the N-methyl-d-aspartic acid (NMDA) receptor
subunit (NR2B) and neuronal nitric oxide synthase (nNOS) to
modulate glutamate transmission and maintain excitatory synapse
balance (10).
The present study hypothesized that
pharmacologically targeting the PSD95-NMDA interaction may provide
novel insight into neuroprotective strategies post-TBI. Numerous
neuroprotective agents against TBI have been identified; however
these agents have rarely been successful during clinical trials.
Dexmedetomidine (Dex), which is an alpha-2 adrenergic receptor
agonist drug, has been approved by the Food and Drug Administration
(FDA) and is known for its anaesthetic, analgesic and
neuroprotective effects (15). Dex
also exerts a positive impact on neuronal development by regulating
PSD95expression (16); however, the
role of Dex in post-TBI neuroprotection remains unknown. The
present study investigated the effect of Dex on the PSD95-NMDA
interaction and subsequent functional recovery post-TBI.
Materials and methods
Animals and TBI induction
Male C57 BL/6 mice (8 weeks old; n=72) were obtained
from the Shanghai Laboratory Animal Center. All procedures were
approved by the Research Review and Ethics Board (RREB) of the
Shanghai Ninth People's Hospital and was performed according to the
guidelines from the National Research Council Guide. Mice (n=72)
were subjected to controlled cortical impact injury (CCI) (that is
representative of TBI induction) as previously described (17). Prior to surgery, anesthetized mice
(3% isoflurane) were placed in a stereotaxic frame. The skin was
removed to expose the skull, and a 4-mm craniotomy was performed
between the lambda and bregma sutures under sterile procedure. The
skullcap was removed carefully without damaging the dura
underneath. A pneumatic impactor was used to control the contact
velocity and the level of cortical deformation, which determined
the severity of the injury. The contact velocity and degree of
deformation were set at 3.5 m/sec and 0.5 mm, respectively. These
settings provided an injury of moderate severity. Immediately after
the injury, the skin incision was sutured. Sham animals (n=18) were
not subjected to CCI injury; however craniotomy was performed on
them.
Drug administration
Following surgery, mice were divided into different
groups: TBI, TBI+vehicle, TBI+Dex and sham (n=18 in each group).
Mice in TBI+vehicle and TBI+Dex groups received intraperitoneal
injections of saline (n=18) and Dex 100 µg/kg (18) (n=18; Sigma Aldrich; Merck KGaA),
respectively, at 1 h and 12 h following surgery. At 24 h
post-injury, 10 animals out of 18 in each group were sacrificed
[according to ARRIVE and 2013 AVMA euthanasia guidelines (19,20)]
to isolate brain tissue for Fluoro-Jade B (FJB) staining and RNA
and protein extraction.
For the neurobehavioral tests involving motor and
cognitive function, TBI mice were placed in two groups (the
remaining n=8 in each group) and were injected with saline or Dex
intraperitoneally as aforementioned. The motor function was
assessed over the course of five days post-TBI and cognitive
function was evaluated from days 14 to 19. The sham surgery mice
(n=8) served as the control.
Histological analysis
Mice were sacrificed using the anesthetic agent
Avertin (400 mg/kg of body weight) administered intraperitoneally
and were transcardially perfused with cold saline and 4%
paraformaldehyde (PFA). Subsequently, brains were removed, fixed
with 4% PFA overnight at 4˚C and kept in 30% sucrose for 48 h at
4˚C. Brain sections (30 µm) were cut using a cryostat for
histological analysis and stored at -80˚C, whereas brain samples
were cut and dissected using a brain chisel, and mechanically lysed
in the ice-cold lysis buffer containing phenylmethylsulfonyl
fluoride (Beyotime Institute of Biotechnology) for western blot
analysis.
FJB staining
FJB stain is a fluorochrome that is commonly used to
label degenerating neurons. The isolated frozen sections that were
obtained after histological processing were mounted on Superfrost
plus slides (Thermo Fisher Scientific, Inc.). Slides were rinsed in
water and transferred into 0.06% potassium permanganate solution
for 20 min at room temperature (RT). Sections were washed with
double-distilled water and incubated with 0.0004% FJB solution
(Merck KGaA) containing 0.1% DAPI for 20 min at RT. Slides were
washed with dd water, air-dried thoroughly until completely dry and
visualised under a fluorescence microscope (490/525 wavelength;
magnification, x20).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from the excised tissue using
RNA isolation kit (Thermo Fisher Scientific, Inc.) and the purity
was tested using Nanodrop1000 system (Thermo Fisher Scientific,
Inc.). RNA was transcribed into cDNA using the First Strand cDNA
Synthesis Kit (Thermo Fisher Scientific, Inc.). According to the
manufacturer's instructions, RT-qPCR was performed to detect the
mRNA expression level using SyBr Green PCR Master Mix (Thermo
Fisher Scientific, Inc.). qPCR (Applied Biosystem) was performed
(95˚C initial template denaturation, and 40 cycles of 95˚C
denaturation and 60˚C anneal/extension) to assess the relative mRNA
expression level of PSD95 following TBI. PSD95 relative expressions
level was normalized to the endogenous control GAPDH and was
expressed as 2-ΔΔCq (21). The sequences of primers used are
presented in Table I.
 | Table IPrimer sequences for RT-PCR
reaction. |
Table I
Primer sequences for RT-PCR
reaction.
| Gene | Forward primer | Reverse primer |
|---|
| PSD95 |
5'-TCTGTGCGAGAGGTAGCAGA-3' |
5'-AAGCACTCCGTGAACTCCTG-3' |
| GAPDH |
5'-TGCACCACCAACTGCTTAGC-3' |
5'-GGCATGGACTGTGGTCATGAG-3' |
Western blotting
Brain samples were cut and dissected using a brain
chisel, and mechanically lysed in the ice-cold lysis buffer
containing phenylmethylsulfonyl fluoride (Beyotime Institute of
Biotechnology). The protein concentration was measured using the
bicinchoninic acid assay kit (Thermo Fisher Scientific, Inc.).
Proteins (20 µg) were separated by 12% SDS-PAGE and transferred
onto nitrocellulose membranes. Membranes were blocked using 5%
skimmed milk dissolved in PBS for 1 h at RT and incubated with the
primary antibodies. Primary antibodies used were as follows: Rabbit
polyclonal anti-PSD95 (Abcam; cat. no. ab18258, 1:1,000), rabbit
monoclonal anti-nNOS (Abcam; cat. no. ab76067; 1:1,000), rabbit
polyclonal anti-NR2B (Abcam; cat. no. ab65783; 1:1,000), rabbit
polyclonal anti-MMP2 (Abcam; cat. no. ab97779; 1:1,000), rabbit
polyclonal anti-MMP9 (Abcam; cat. no. ab38898; 1:1,000), rabbit
polyclonal caspase-3 (Abcam; cat. no. ab13847; 1:1,000) and mouse
monoclonal anti-GAPDH (Abcam; cat. no. ab8245; 1:500) overnight at
4˚C. Membranes were washed three times with PBS containing 0.1%
Tween and incubated with horeradish peroxidase goat anti-rabbit
(Abcam; cat. no. ab7090; 1:1,000) or anti-mouse immunoglubulin G
secondary antibodies (Abcam; cat. no. ab150117. 1:1,000) for 2 h at
room temperature. Bands were detected using enhanced
chemiluminescence kit (Thermo Fisher Scientific, Inc.). Relative
expression of proteins was normalized to GAPDH endogenous control
using Image J software version 1.50 (National Institute of
Health).
Immunoprecipitation analysis
Brain samples were lysed using ice-cold RIPA lysis
buffer (Thermo Fisher Scientific, Inc.). The lysate was incubated
overnight at 4˚C with protein-specific antibodies for proteins
PSD95, nNOS, NR2B or rabbit IgG (Abcam; cat. no. ab7090), which
served as the negative control. Protein A/G Sepharose beads (Abcam;
cat. no. ab193262; 2 µl/µg of total protein) were added to each
immune complex. The mixture was kept for 4 h at 4˚C with rotational
shaking. The beads were washed three times with RIPA lysis buffer.
Furthermore, the lysate bead mixture was eluted by heating the
samples in 4X SDS loading buffer for 10 min at 50˚C. Protein bands
were detected using western blot analysis following the
aforementioned protocol.
Motor function
Motor performance was evaluated using beam-balance
and beam-walk tests (22,23). For the beam balance test, mice were
placed on 1.5 cm wide elevated narrow beam-balance and the time
each mouse remained on beam was recorded. A maximum of 60 sec was
allowed (22). For the beam-walk
test, the time needed to traverse the 2.5 cm width and 100 cm
length beam was recorded (23). A
pre-assessment test was performed prior to the surgery to obtain a
baseline. Each experiment consisted of three trials per day with 60
sec of maximum allotted time for each task. The average daily
scores were analyzed.
Cognitive function
The cognitive function was assessed using the Morris
Water Maze test (24). A plastic
pool of 180 cm diameter and 60 cm height constituted the maze. The
pool was filled with water (temperature 26±1˚C, 28 cm depth) and
was placed in a space with prominent visual cues. A clear Plexiglas
stand of 10 cm diameter and 26 cm height was kept in the southwest
quadrant of the maze 26 cm was away from the wall of the maze. This
position of the platform was held constant for each mouse. To
evaluate the spatial learning, the mice were given four trials with
4 min inter-trial interval for 5 days from days 14 to 19 following
surgery. A maximum of 120 sec was permitted for the mice to find
the platform that was hidden at 2 cm under the water surface. At
day 19 post-surgery, the platform was kept 2 cm over the water
surface so it was visible to mice. This test provided information
on the effect of non-spatial factors, including sensorimotor
performance, visual acuity and motivation on cognitive function.
Every trial continued until the mice successfully climbed onto the
platform or the allowed threshold time of 120 sec had passed. If
the platform remained unfound in the given time, mice were manually
directed towards it. Mice were returned to a heated incubator
between each trial after 30 sec spent on the platform. The time
recorder for each trial during one day was averaged and
statistically analyzed. Spontaneous motor activity recording and
tracking system (SMART 2.0 tracking software; Panlab) was used to
record the time needed for the mice to locate the platform.
Statistical analysis
Data were represented as the means ± standard error
of the mean. One way analysis of variance followed by Bonferroni
post-hoc test was used for the comparison of variables using
GraphPad Prism 5.0 (GraphPad Software, Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
PSD95 expression is reduced in mice
following TBI
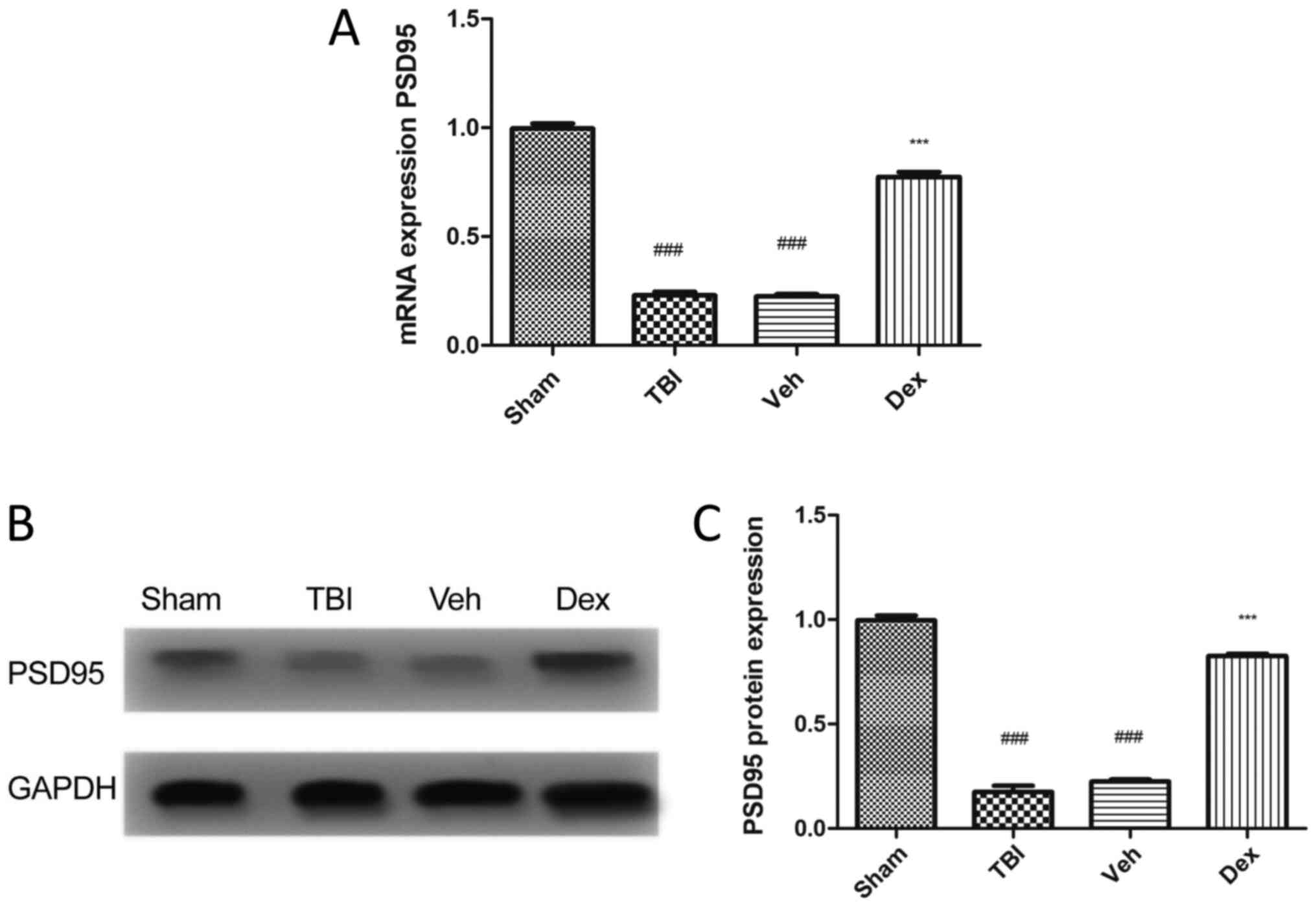
To evaluate the expression of PSD95 following TBI,
RT-qPCR and western blotting were performed. The results
demonstrated that PSD95 expression level was significantly
decreased in mice following TBI compared with mice in the sham
group (0.7705 fold decrease; P<0.001; Fig. 1A). This result was supported by the
reduced protein expression of PSD95 in mice following TBI compared
with mice in the sham group (0.8205 fold decrease; P<0.001;
Fig. 1B and C). Conversely, treatment with Dex
following TBI induced an increased PSD95 expression at mRNA
(0.5505-fold increase; P<0.001 vs. vehicle; Fig. 1A) and protein (0.6005 fold increase;
P<0.001 vs. vehicle; Fig. 1C)
levels.
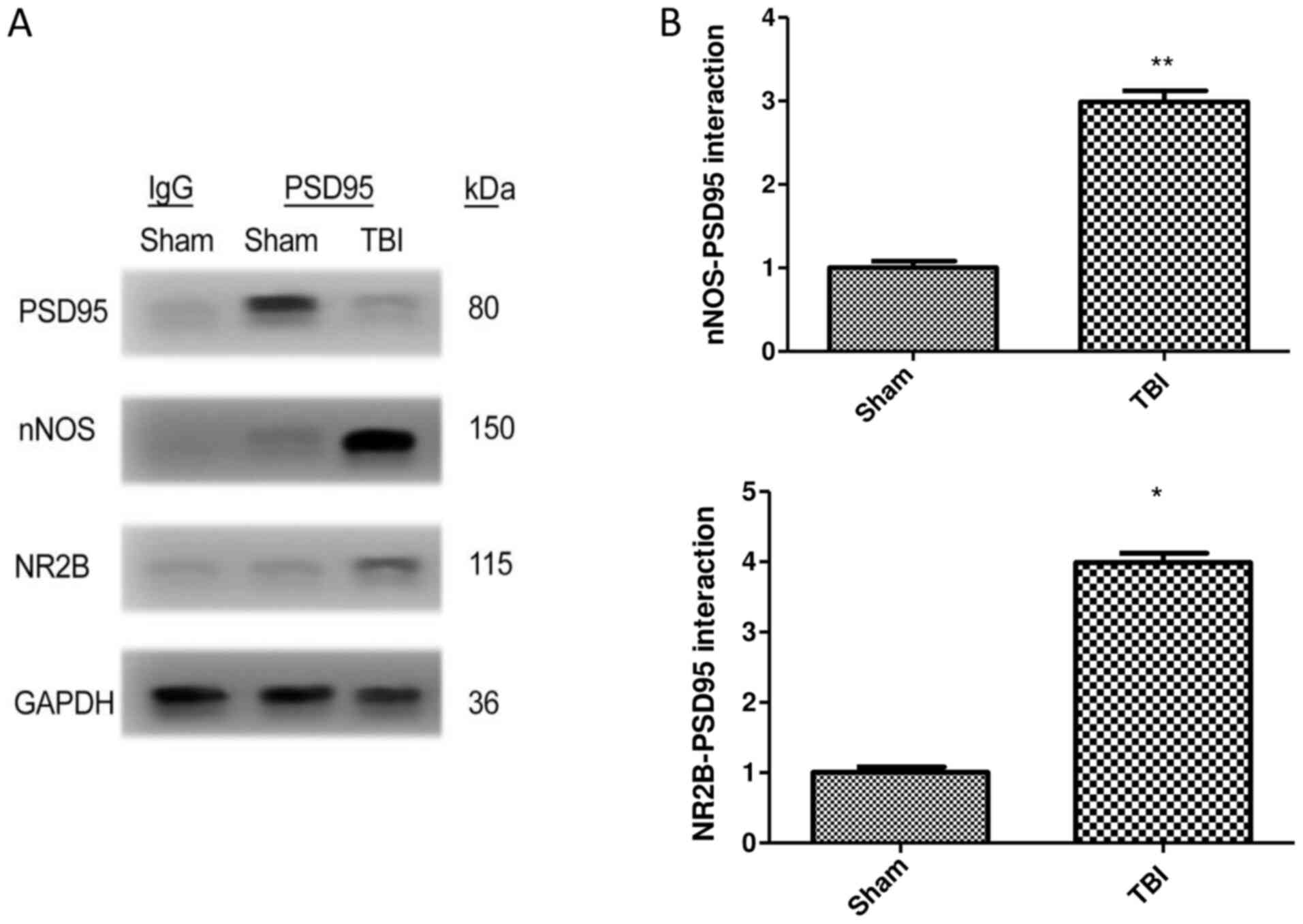
TBI increases the interaction of PSD95
with NR2B and nNOS
The interaction of PSD95 with NR2B and nNOS was
examined by co-immunoprecipitation in brain samples of mice
following TBI. The results demonstrated that the generation of
PSD95-NR2B-nNOS complex was significantly increased in the TBI
group compared with the sham group. Furthermore, there was a 1.99
and 2.98 fold increase in nNOS and NR2B interaction, respectively,
with PSD95 in the TBI group compared with the sham group (P<0.01
vs. sham). TBI therefore increased the expression of NR2B and nNOS
when co-immunoprecipitated with PSD95 (Fig. 2A and B).
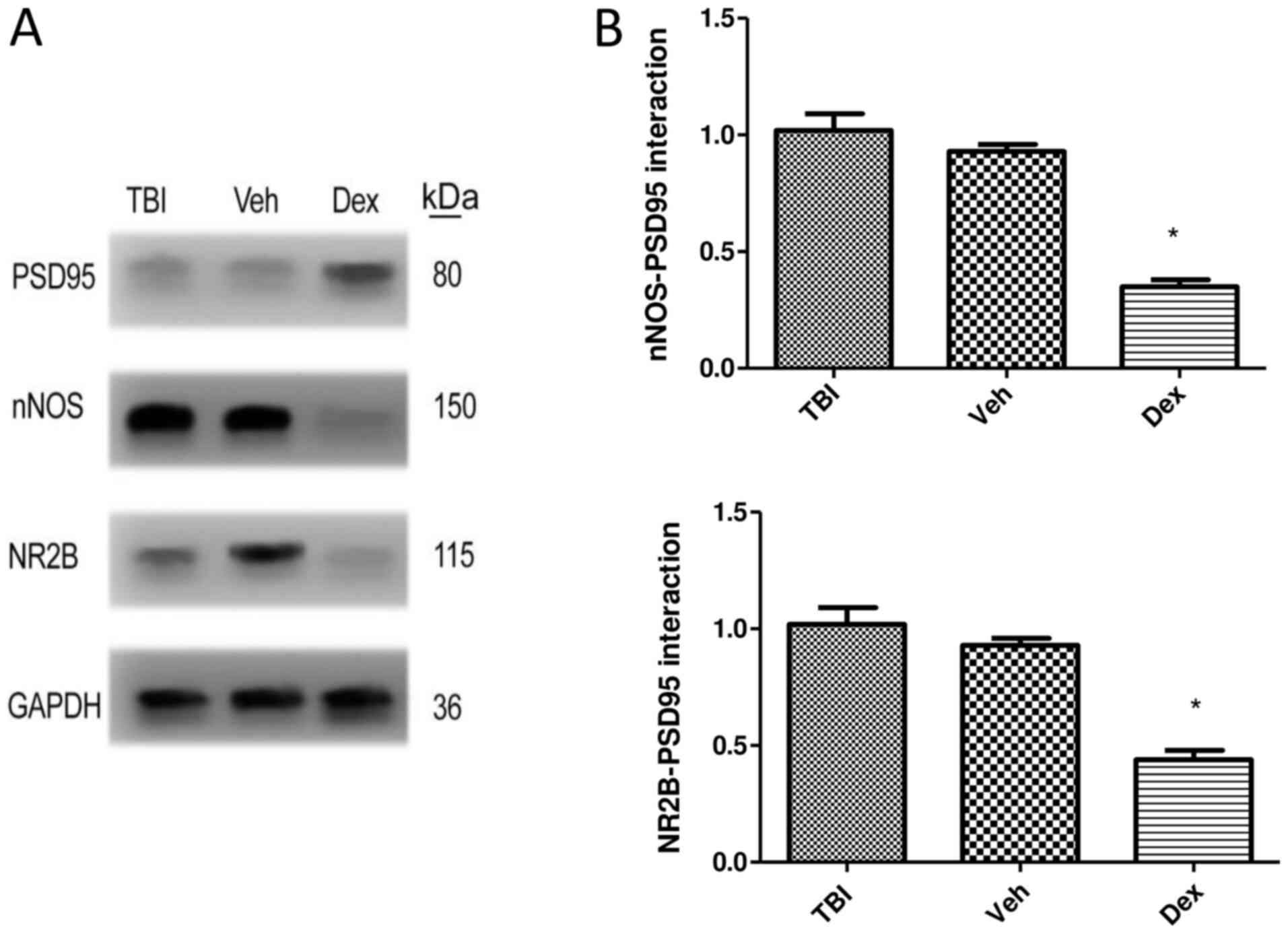
Dex treatment alleviates
PSD95-NR2B-nNOS complex generation following TBI
The formation of the PSD95-NR2B-nNOS complex
following Dex treatment was assessed by co-immunoprecipitation. The
results demonstrated that Dex treatment prevented the interaction
of PSD95 with NR2B-nNOS following TBI. The expression of NR2B
(0.58-fold decrease; P<0.05 vs. vehicle) and nNOS (0.67-fold
decrease; P<0.05 vs. vehicle) was significantly reduced when
co-immunoprecipitated with PSD95 following Dex treatment compared
with vehicle group (Fig. 3A and
B).
Effect of Dex treatment on TBI-induced
secondary brain injury
To study the effect of Dex administration on
secondary brain injury following TBI, FJB staining was performed at
24 h post-TBI. FJB staining was conducted to evaluate the effect of
Dex on neuronal degeneration post-TBI. The results demonstrated
that number of cells stained with FJB was increased in the TBI
group (65.75% increase; P<0.001 vs. sham) implying increased
neuronal degeneration, whereas the number of FJB-positive cells was
reduced following Dex treatment compared with the vehicle group
(75.7% decrease; P<0.001 vs. vehicle; Fig. 4A and B). To further study the effect of Dex on
neuronal death following TBI, the expression of caspase-3 in the
brain tissue near the injury was detected by western blotting. The
results indicated that Dex treatment inhibited neuronal apoptosis
post-TBI (0.594 fold decrease; P<0.05 vs. vehicle; Fig. 4C and D).
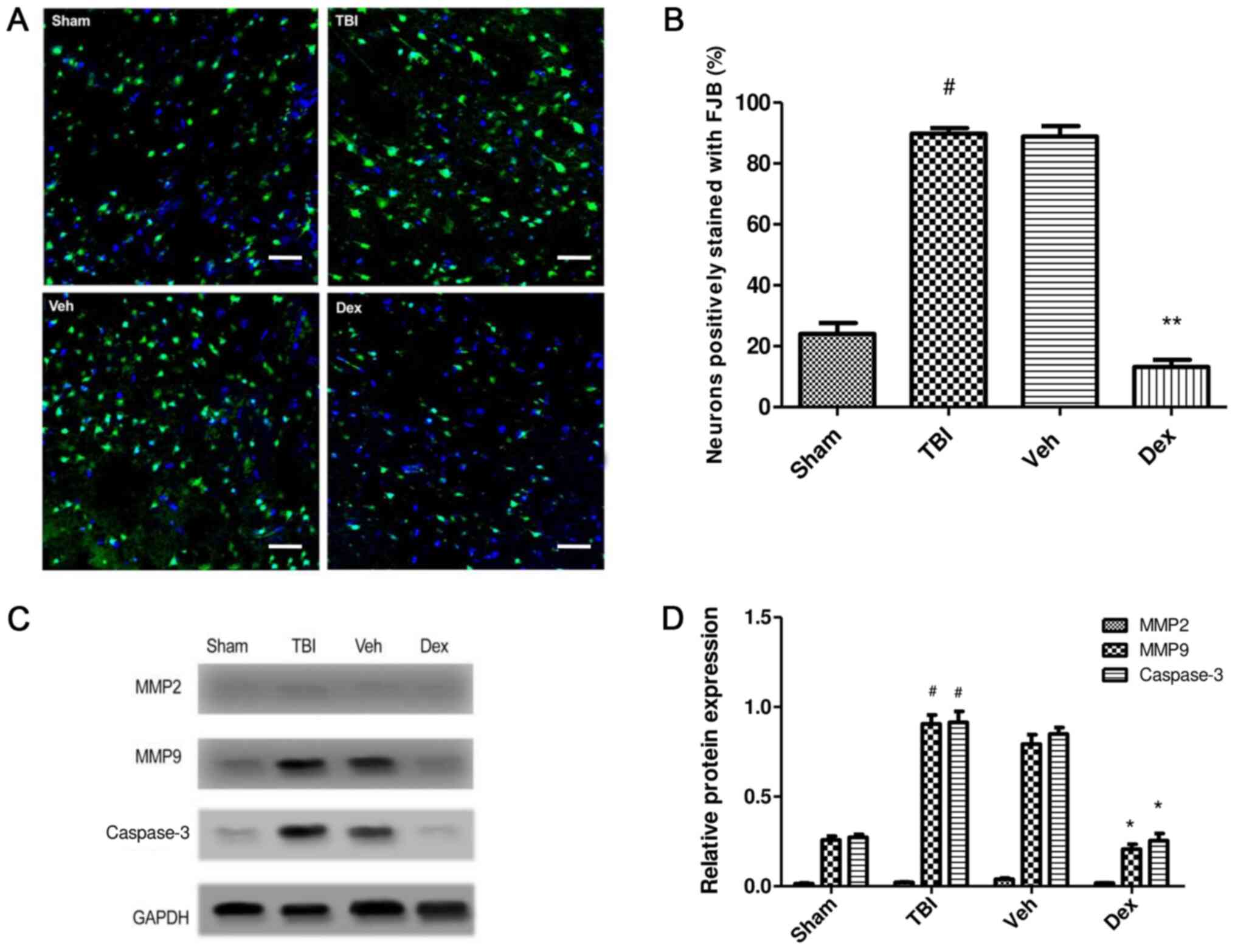 | Figure 4Effect of Dex on secondary brain
injury leading to neuronal death. (A) FJB staining in TBI, sham,
Veh and Dex groups. Magnification, x20, Scale bar= 20 µm. (B)
Quantification of FJB staining. The positively stained cells in
each group were counted, normalized to the total number of cells
(stained by DAPI) and expressed as percentage in the examined area.
(C) MMP2, MMP9 and Casp3 expression assessed by western blotting in
TBI, sham, Veh and Dex groups. (D) Quantification of protein
expression. Data were represented as the means ± standard error of
the mean. #P<0.05 vs. sham. *P<0.05 and
**P<0.01 vs. Veh. Casp3, caspase 3; Dex,
dexmedetomidine; FJB, Fluoro-Jade B; MMP2, matrix metalloproteinase
2; MMP9, matrix metalloproteinase 9; TBI, traumatic brain injury;
Veh, vehicle. |
The effect of Dex treatment on protein expression of
matrix metalloproteinase (MMP)2 and MMP9 following TBI was also
evaluated. The results demonstrated that MMP9 expression was
significantly increased at 24 h post-TBI (0.672-fold increase;
P<0.05 vs. sham) but was significantly decreased following Dex
treatment (0.586-fold decrease; P<0.05 vs. vehicle). No change
in MMP2 expression was observed following TBI and Dex treatment
(P>0.05). These findings demonstrated that Dex administration
efficiently reduced the TBI-induced activation of MMP9, which may
be consecutive to the inhibition of PSD95-NMDA complex formation
(Fig. 4C and D).
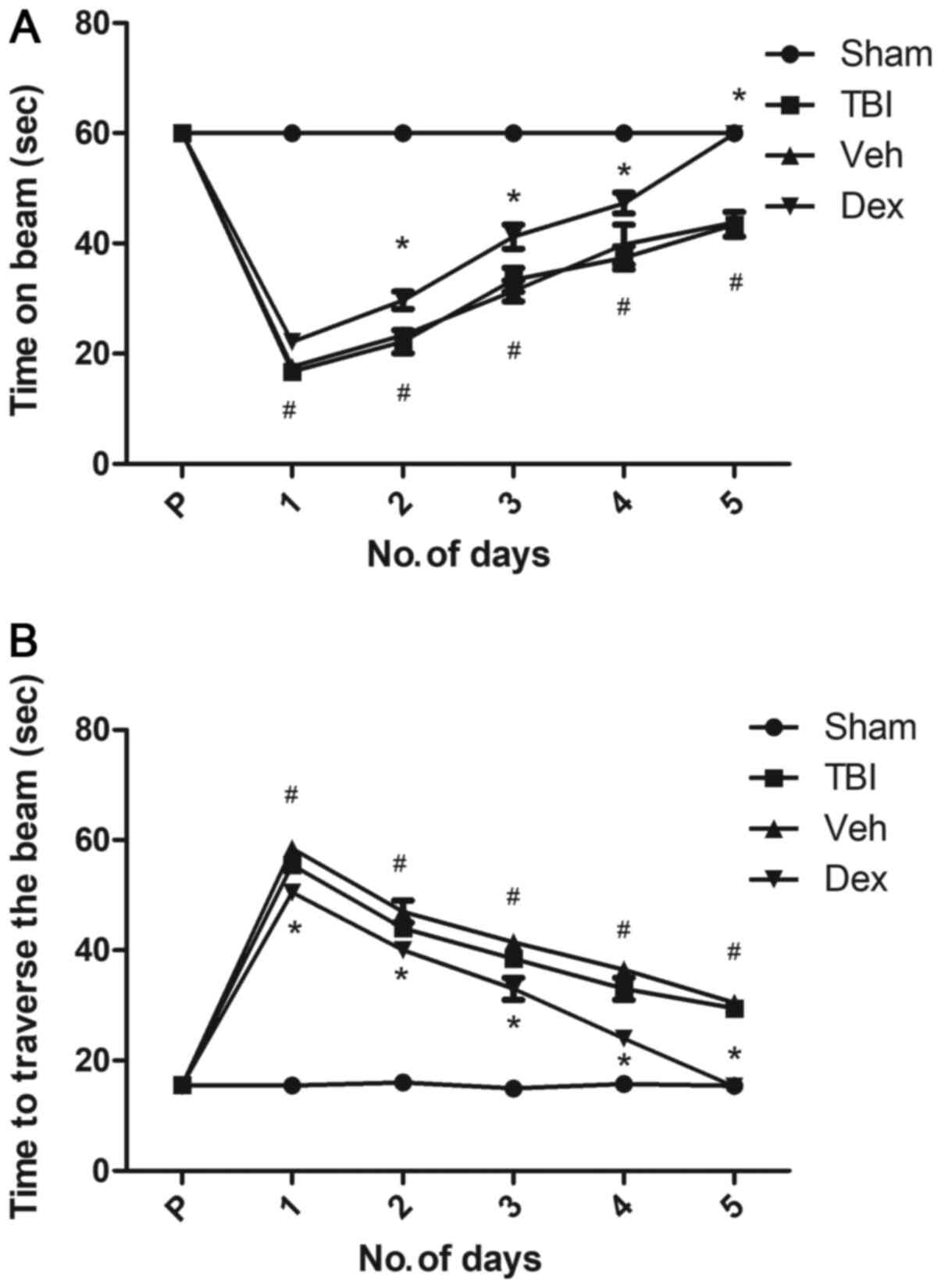
Dex treatment improves motor
function
The motor function of mice following TBI was
evaluated using the beam balance and beam walk tests. Following
surgery, mice were tested twice daily for 5 days. Prior to surgery,
mice motor performance, which corresponds to the duration spent on
the beam and the distance traversed was recorded and served as a
baseline. The results demonstrated that mice in each group were
capable of balancing on the beam for 60 sec for three trials and
presented no pre-surgical variance among groups. Following TBI, all
injured mice exhibited significantly impaired balance compared with
sham mice (P<0.05 vs. sham; Fig.
5A), whereas Dex-treated mice presented improved balance
(P<0.05 vs. vehicle; Fig.
5A).
Following TBI, there was a significant increase in
the time needed for mice to traverse the beam compared with the
sham group (55.5 sec on day 1 post-TBI; P<0.05 vs. sham;
Fig. 5B). Furthermore, a dramatic
decrease in the time needed for mice to traverse the beam was
observed in the vehicle group at five days following TBI (29.5 sec
on day 5 post-TBI; P<0.05 vs. sham; Fig. 5B). This result could be due to to
the skill acquired by the mice during the test. However, Dex
treatment facilitated a rapid recovery, and on day 5, Dex-treated
mice needed a minimum time to traverse the beam (15.3 sec;
P<0.05 vs. vehicle; Fig. 5B).
This result suggested that Dex treatment may improve motor function
recovery in mice following TBI.
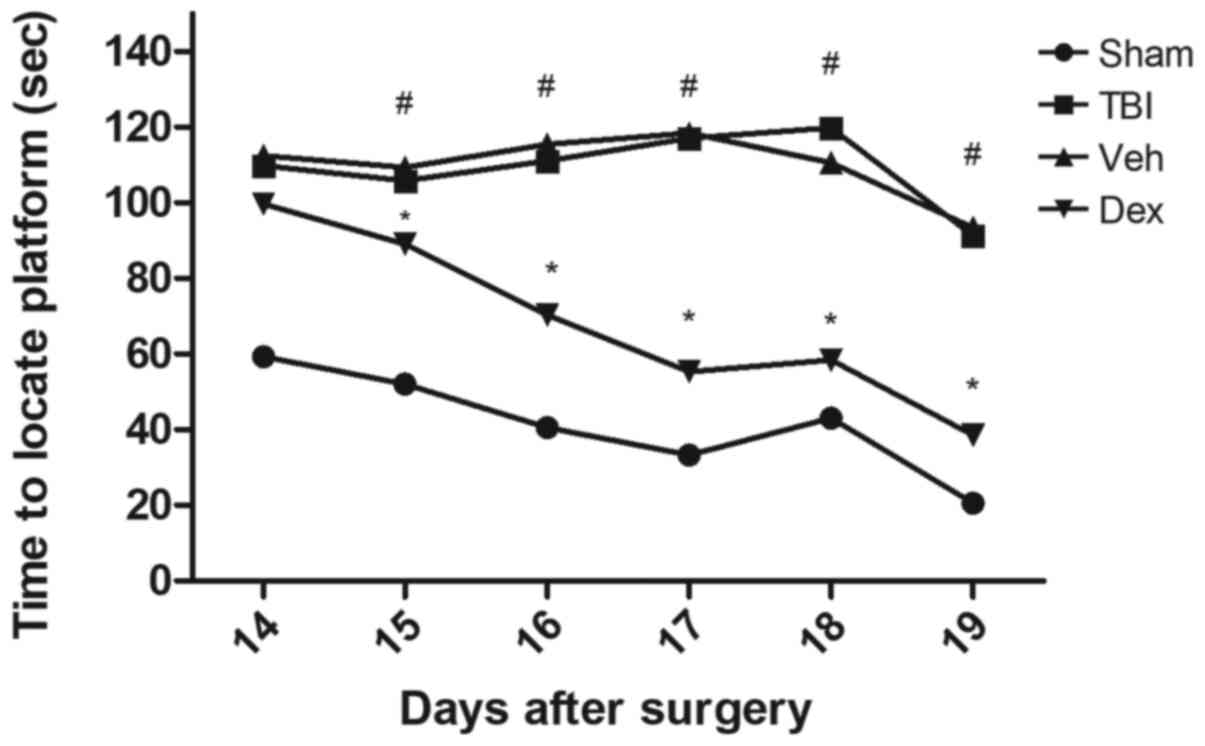
Dex treatment rescues cognitive
impairment caused by TBI
The Morris Water Maze was used to assess cognitive
impairment of mice following TBI and to determine the effect of Dex
treatment on mice 14-19 days after TBI (Fig. 6). The results demonstrated that TBI
group requested longer time to find the hidden platform (110.1 sec
on day 14 post-TBI; P<0.05 vs. sham), which was close to the
threshold of 120 sec allowed for each animal. However, Dex
treatment significantly reduced the amount of time needed to mice
to find the underwater platform compared with the vehicle group
(98.3 sec on day 14 post-TBI; P<0.05 vs. vehicle; Fig. 6). In addition, when the platform was
elevated on day 19 so that it was visible to mice, the time needed
to find the platform was reduced in all groups. However, mice that
were treated with Dex exhibited a greater cognitive recovery
compared with mice in the vehicle group (38.5 sec on day 19
post-TBI; P<0.05 vs. vehicle; Fig.
6). These results suggested that Dex treatment may serve a
crucial role in the cognitive recovery following TBI.
Discussion
The present study examined the underlying mechanism
of Dex on the PSD95-NMDA receptor interaction to promote functional
recovery in mice following TBI. The results demonstrated that Dex
treatment reduced the PSD95-NR2B-nNOS complex formation, which
subsequently improved the motor and cognitive function in mice
following TBI.
TBI can induce secondary brain damage, initiating a
cascade of pathophysiological events leading to motor dysfunction
and cognitive decline (1,2); however, no effective treatment for the
multifaceted and complex disorders associated with TBI is
available. One of the most preferred clinical approaches to treat
severe TBI is to control intracranial pressure through
pharmacologic sedation (25). The
sedative agents used on patients following TBI should have a rapid
effect with a short elimination half-time and no adverse effects on
other organ systems (25,26). The commonly used sedative agents,
including propofol and benzodiazepines, are associated with
respiratory depression and hypotension, which interfere with
neurological evaluations (26).
Conversely, Dex is an alpha-2 receptor adrenergic agonist FDA
approved, with an elimination half-time of ~2 h, which is
acceptable in humans (27);
however, the underlying mechanisms of Dex treatment following TBI
has not been thoroughly investigated. The present study
investigated the molecular mechanism by which Dex may allow
cognitive and motor recovery following TBI.
Functional recovery following TBI largely depends on
brain plasticity, which is determined by the synapse number and the
enhanced function of synapses in the neurons. Improved synaptic
function inhibits neuronal apoptosis, which enhances the action of
peripheral neurons following TBI (10). Numerous studies have targeted the
postsynaptic membrane-associated proteins for the treatment of
neurological disorders. For example, PSD95 is abundantly expressed
in excitatory neurons and is connected with the activation of the
NMDA receptor with nitric oxide (NO)-mediated neurotoxicity
(11-13).
Following TBI, NMDA excitatory potential in the neurons is reduced
and attenuates glutamate-induced excitatory currents (10).
The results from the present study demonstrated that
PSD95 expression was reduced following TBI, and that the formation
of PSD95-NMDA complex was increased post-TBI. It has been
demonstrated that excessive generation of the PSD95-NMDA complex
can stimulate NO production and activate MMP9, which can induce
neuronal apoptosis (28). The
present study reported that Dex treatment could decrease the
PSD95-NMDA interaction and consequently reduce neuronal death
caused by TBI. It was also demonstrated that Dex treatment
contributed to the cognitive and motor recovery enhancement
following TBI.
Previous findings have shown Dex to be a safe and
effective treatment in neurosurgical patients. For example,
post-surgical treatment with Dex can improve neurological scores
and reduces brain oedema following sub-arachnoid haemorrhage
(29). Furthermore, Dex exerts a
neuroprotective effect against TBI through the activation of the
PI3K/Akt/mTOR signalling pathway (30). In addition, Dex was demonstrated to
be neuroprotective in hippocampal slice cultures (31,32),
and a recent report substantiated neuroprotective property of Dex
in an in vivo model of TBI (18). An extensive review has discussed
that alpha 2-adrenergic agonists are neuroprotective agents
(33). These agents can reduce the
release of excitatory neurotransmitters at the supracellular level
and inhibit adenylate and guanylate cyclases at the cellular level
(33). The modulation of NMDA
receptor function via alpha 2-adrenergic agonists is a vital
mechanism in neuroprotection (34).
In the present study, the regulation of PSD95-NMDA interaction by
Dex may be attributed to its agonistic effect on the alpha-2
adrenergic receptor.
Following TBI, post-synaptic glutamate receptors are
activated, inducing an increased release of glutamate and reduced
glutamate intake. Following glutamate stimulation, NMDA receptors
impose a toxic effect through PSD95. PSD95 anchors the NMDA
receptor and stimulates the migration of downstream signalling
molecules towards the calcium channel of the NMDA receptor.
Intracellular calcium excess can lead to oxidative stress by
generating large amounts of reactive oxygen and nitrogen species.
The subsequent release of inflammatory cytokines and caspase-3
cascade activation lead therefore to neuronal apoptosis (35). Dex treatment could inhibit the
activation of MMP9 and caspase-3 and therefore reduce the neuronal
excitotoxicity.
The present study demonstrated that Dex
administration following TBI reduced cognitive impairment. Previous
studies reported that cognitive recovery depends on the interaction
between synapses (36-38).
Following the initial trauma, the loss of PSD95 is directly
associated with cognitive decline that is observed within weeks to
months (39). Another study
reported that activation of the protein kinase R-like endoplasmic
reticulum kinase following TBI causes memory impairment through
PDS95 and cAMP response element binding protein downregulation
(40). Furthermore, post-traumatic
hypothermia increases PSD95 expression to restore learning and
memory function (41). PSD95 is
therefore considered as an important therapeutic target that
promotes cognitive and motor recovery after TBI-induced secondary
brain damage.
In conclusion, the present study described a
potential mechanism of action of Dex treatment in mice following
TBI. Dex treatment reduced neuronal death as well as promoted motor
and cognitive recovery. Furthermore, improvement of cognitive and
motor function post-TBI in mice treated with Dex may be attributed
to the inhibition of PSD95-NMDA receptor activation. The regulation
of PSD95-NMDA receptor complex may largely contribute to synaptic
plasticity and learning abilities following brain injury. In
addition, the present study demonstrated that Dex treatment
inhibited PSD95 interaction with NR2B and nNOS, which resulted in
cognitive and motor recovery following TBI. The long-term effect of
Dex treatment and other associated molecular targets on functional
recovery following TBI will be further investigated.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
ZZ and YH conceptualised and designed the
experiments, ZZ performed the experiments, YR and HJ performed the
statistical analysis and provided assistance for the current study.
ZZ and YH drafted the manuscript. YH revised the manuscript
critically and approved the final version to be submitted. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All the experiments were ethically approved and
performed according to the National Institutes of Health guide for
the care and use of laboratory animals, ARRIVE guidelines
(http://www.nc3rs.org.uk/arrive-guidelines) and the
AVMA euthanasia guidelines 2013.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dewan MC, Rattani A, Gupta S, Baticulon
RE, Hung YC, Punchak M, Agrawal A, Adeleye AO, Shrime MG, Rubiano
AM, et al: Estimating the global incidence of traumatic brain
injury. J Neurosurg. 1:1–18. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Blennow K, Brody DL, Kochanek PM, Levin H,
McKee A, Ribbers GM, Yaffe K and Zetterberg H: Traumatic brain
injuries. Nat Rev Dis Primers. 2(16084)2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Stocchetti N and Zanier ER: Chronic impact
of traumatic brain injury on outcome and quality of life: A
narrative review. Crit Care. 20(148)2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Sheriff FG and Hinson HE: Pathophysiology
and clinical management of moderate and severe traumatic brain
injury in the ICU. Semin Neurol. 35:42–49. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Joseph B, Haider A and Rhee P: Traumatic
brain injury advancements. Curr Opin Crit Care. 21:506–511.
2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Corps KN, Roth TL and McGavern DB:
Inflammation and neuroprotection in traumatic brain injury. JAMA
Neurol. 72:355–362. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Rodríguez-Rodríguez A, Egea-Guerrero JJ,
Murillo-Cabezas F and Carrillo-Vico A: Oxidative stress in
traumatic brain injury. Curr Med Chem. 21:1201–1211.
2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Mckee AC and Daneshvar DH: The
neuropathology of traumatic brain injury. Handb Clin Neurol.
127:45–66. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Nakayama K, Kiyosue K and Taguchi T:
Diminished neuronal activity increases neuron-neuron connectivity
underlying silent synapse formation and the rapid conversion of
silent to functional synapses. J Neurosci. 25:4040–4051.
2005.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Merlo L, Cimino F, Angileri FF, La Torre
D, Conti A, Cardali SM, Saija A and Germanò A: Alteration in
synaptic junction proteins following traumatic brain injury. J
Neurotrauma. 31:1375–1385. 2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Travaglia A, Bisaz R, Cruz E and Alberini
CM: Developmental changes in plasticity, synaptic, glia and
connectivity protein levels in rat dorsal hippocampus. Neurobiol
Learn Mem. 135:125–138. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Keith D and El-Husseini A: Excitation
control: Balancing PSD-95 function at the synapse. Front Mol
Neurosci. 1(4)2008.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Christopherson KS, Hillier BJ, Lim WA and
Bredt DS: PSD-95 assembles a ternary complex with the
N-methyl-D-aspartic acid receptor and a bivalent neuronal NO
synthase PDZ domain. J Biol Chem. 274:27467–27473. 1999.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Mo SF, Liao GY, Yang J, Wang MY, Hu Y,
Lian GN, Kong LD and Zhao Y: Protection of neuronal cells from
excitotoxicity by disrupting nNOS-PSD95 interaction with a small
molecule SCR-4026. Brain Res. 1648:250–256. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Dahmani S, Rouelle D, Gressens P and Mantz
J: Characterization of the postconditioning effect of
dexmedetomidine in mouse organotypic hippocampal slice cultures
exposed to oxygen and glucose deprivation. Anesthesiology.
112:373–383. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Lv J, Ou W, Zou XH, Yao Y and Wu JL:
Effect of dexmedetomidine on hippocampal neuron development and
BDNF-TrkB signal expression in neonatal rats. Neuropsychiatr Dis
Treat. 12:3153–3159. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Gao X, Deng-Bryant Y, Cho W, Carrico KM,
Hall ED and Chen J: Selective death of newborn neurons in
hippocampal dentate gyrus following moderate experimental traumatic
brain injury. J Neurosci Res. 86:2258–2270. 2008.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Wu J, Vogel T, Gao X, Lin B, Kulwin C and
Chen J: Neuroprotective effect of dexmedetomidine in a murine model
of traumatic brain injury. Sci Rep. 8(4935)2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kilkenny C, Browne W, Cuthill IC, Emerson
M and Altman DG: National Centre for the Replacement, Refinement
and Reduction of Animals in Research. Animal research: Reporting
in vivo experiments-the ARRIVE guidelines. J Cereb Blood
Flow Metab. 31:991–993. 2011.PubMed/NCBI View Article : Google Scholar
|
|
20
|
American Veterinary Medical Association:
AVMA Guidelines for the Euthanasia of Animals: 2013 Edition.
American Veterinary Medical Association, Schaumburg, IL, 2013.
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Luong TN, Carlisle HJ, Southwell A and
Patterson PH: Assessment of motor balance and coordination in mice
using the balance beam. J Vis Exp. 49(pii: 2376)2011.PubMed/NCBI View
Article : Google Scholar
|
|
23
|
Hausser N, Johnson K, Parsley MA, Guptarak
J, Spratt H and Sell SL: Detecting behavioral deficits in rats
after traumatic brain injury. J Vis Exp. 131(56044)2018.PubMed/NCBI View
Article : Google Scholar
|
|
24
|
Guidi M and Foster TC: Behavioral model
for assessing cognitive decline. Methods Mol Biol. 829:145–153.
2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Oddo M, Crippa IA, Mehta S, Menon D, Payen
JF, Taccone FS and Citerio G: Optimizing sedation in patients with
acute brain injury. Crit Care. 20(128)2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Flower O and Hellings S: Sedation in
traumatic brain injury. Emerg Med Int. 2012(637171)2012.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Bejian S, Valasek C, Nigro JJ, Cleveland
DC and Willis BC: Prolonged use of dexmedetomidine in the
paediatric cardiothoracic intensive care unit. Cardiol Young.
19:98–104. 2009.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Gu Z, Kaul M, Yan B, Kridel SJ, Cui J,
Strongin A, Smith JW, Liddington RC and Lipton SA: S-nitrosylation
of matrix metalloproteinases: Signaling pathway to neuronal cell
death. Science. 297:1186–1190. 2002.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wang Y, Han R and Zuo Z: Dexmedetomidine
post-treatment induces neuroprotection via activation of
extracellular signal-regulated kinase in rats with subarachnoid
haemorrhage. Br J Anaesth. 116:384–392. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Shen M, Wang S, Wen X, Han XR, Wang YJ,
Zhou XM, Zhang MH, Wu DM, Lu J and Zheng YL: Dexmedetomidine exerts
neuroprotective effect via the activation of the PI3K/Akt/mTOR
signaling pathway in rats with traumatic brain injury. Biomed
Pharmacother. 95:885–893. 2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Schoeler M, Loetscher PD, Rossaint R,
Fahlenkamp AV, Eberhardt G, Rex S, Weis J and Coburn M:
Dexmedetomidine is neuroprotective in an in vitro model for
traumatic brain injury. BMC Neurol. 12(20)2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zhang MH, Zhou XM, Cui JZ, Wang KJ, Feng Y
and Zhang HA: Neuroprotective effects of dexmedetomidine on
traumatic brain injury: Involvement of neuronal apoptosis and HSP70
expression. Mol Med Rep. 17:8079–8086. 2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhang Y and Kimelberg HK: Neuroprotection
by alpha 2-adrenergic agonists in cerebral ischemia. Curr
Neuropharmacol. 3:317–323. 2005.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Mori-Okamoto J, Namii Y and Tatsuno J:
Subtypes of adrenergic receptors and intracellular mechanisms
involved in modulatory effects of noradrenaline on glutamate. Brain
Res. 539:67–75. 1991.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Luo P, Fei F, Zhang L, Qu Y and Fei Z: The
role of glutamate receptors in traumatic brain injury: Implications
for postsynaptic density in pathophysiology. Brain Res Bull.
85:313–320. 2011.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Cheung ZH and Ip NY: From understanding
synaptic plasticity to the development of cognitive enhancers. Int
J Neuropsychopharmacol. 14:1247–1256. 2011.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Sultana R, Banks WA and Butterfield DA:
Decreased levels of PSD95 and two associated proteins and increased
levels of BCl2 and caspase 3 in hippocampus from subjects with
amnestic mild cognitive impairment: Insights into their potential
roles for loss of synapses and memory, accumulation of Abeta, and
neurodegeneration in a prodromal stage of alzheimer's disease. J
Neurosci Res. 88:469–477. 2010.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Shao CY, Mirra SS, Sait HB, Sacktor TC and
Sigurdsson EM: Postsynaptic degeneration as revealed by PSD-95
reduction occurs after advanced Aβ and tau pathology in transgenic
mouse models of Alzheimer's disease. Acta Neuropathol. 122:285–292.
2011.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Wakade C, Sukumari-Ramesh S, Laird MD,
Dhandapani KM and Vender JR: Delayed reduction in hippocampal
post-synaptic density protein-95 expression temporally correlates
with cognitive dysfunction following controlled cortical impact in
mice. J Neurosurg. 113:1195–1201. 2010.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Sen T, Gupta R, Kaiser H and Sen N:
Activation of PERK elicits memory impairment through inactivation
of CREB and downregulation of PSD95 after traumatic brain injury. J
Neurosci. 37:5900–5911. 2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Wang CF, Zhao CC, Jiang G, Gu X, Feng JF
and Jiang JY: The role of posttraumatic hypothermia in preventing
dendrite degeneration and spine loss after severe traumatic brain
injury. Sci Rep. 6(37063)2016.PubMed/NCBI View Article : Google Scholar
|