Introduction
X-linked juvenile retinoschisis (XLRS) is a
condition featuring the degeneration of the macula that commonly
onsets early in males; major symptoms are vision loss and splitting
or schisis of the retinal layer (1). Certain patients with this condition
may also suffer from complications associated with vitreous
hemorrhage and retinal detachment (1). XLRS is a monogenic, X
chromosome-linked recessive disease that is caused by mutations in
the retinoschisin (RS1) gene and affects between 1 in 5,000 and 1
in 25,000 males. By contrast, females who carry this trait do not
suffer from loss of vision with the same frequency (2).
The RS1 gene is located at Xp22.1 and encodes that
the 24-kDa discoidin domain-containing protein retinoschisin, which
is secreted as a homo-oligomeric complex. RS1 is typically
expressed in the retina and pineal gland (3), where it is predicted to serve as an
adhesive protein in maintaining the structural and functional
integrity of the retina (4).
Clinical retinoschisis is characterized by splitting
that may occur in both the nerve fiber layer on the retinal surface
and the deeper layers of the retina. Peripheral retinoschisis
occurs in <50% of affected individuals, whilst foveal
involvement is present in all affected patients. Peripheral
retinoschisis is commonly observed in the inferotemporal retina.
With the use of optic coherence tomography (OCT), the diagnostic
approach for XLRS has changed (5).
The purpose of the present study was to examine the
clinical features of XLRS in a Chinese family over a 7-year
monitoring period and to further identify the possible genetic
mutations that are associated with this disease.
Materials and methods
Patients
A total of two patients from the same family were
recruited at The Department of Ophthalmology, Second Affiliated
Hospital of Harbin Medical University (Harbin, China) between May
and June 2011. They were healthy apart from having XLRS. The
diagnosis of XLRS was made based on the presence of macular schisis
with OCT examination. The present study was performed with approval
from the Ethics Committee of The Second Affiliated Hospital of
Harbin Medical University (Harbin, China). Informed consent was
obtained from all participants.
Ophthalmic examinations
All patients were assessed for uncorrected visual
acuity and best-corrected visual acuity, by fundus photography and
spectral-domain OCT. The younger patient was examined by fundus
autofluorescence and fundus fluorescence (FFA) and multifocal
electroretinograms (ERG) during the first visit.
Mutation analysis
Blood samples were collected from all participants
after obtaining informed consent according to the Declaration of
Helsinki, which included the two patients, their parents and 100
healthy subjects (age range, 20-40 years; 50 males and 50 females)
that were recruited at The Department of Ophthalmology, Second
Affiliated Hospital of Harbin Medical University (Harbin, China)
between May and December 2011, who were used as normal controls and
were unrelated to the family. Individuals with ocular and systemic
diseases were excluded. Genomic DNA was extracted from the
peripheral blood using the Relaxgene Blood DNA System (Tiangen
Biotech Co., Ltd.). RS1 gene coding regions and the flanking intron
sequences were amplified by PCR (6)
using a DNA polymerase from Takara Biotechnology Co., Ltd. The
thermocycling conditions were as follows: Initial denaturation at
95˚C for 10 min, followed by 35 cycles of 95˚C for 30 sec, 55˚C for
30 sec and 72˚C for 1 min, and final extension at 72˚C for 7 min.
The coding regions of the RS1 gene that encode retinoschisin were
directly sequenced on an automated sequencer (ABI 3730xl Genetic
Analyzer; Applied Biosystems; Thermo Fisher Scientific, Inc.) to
perform mutation analysis. To identify the sequence variations,
reference sequences of RS1 (NM_000330.3) were used.
Results
Clinical manifestations
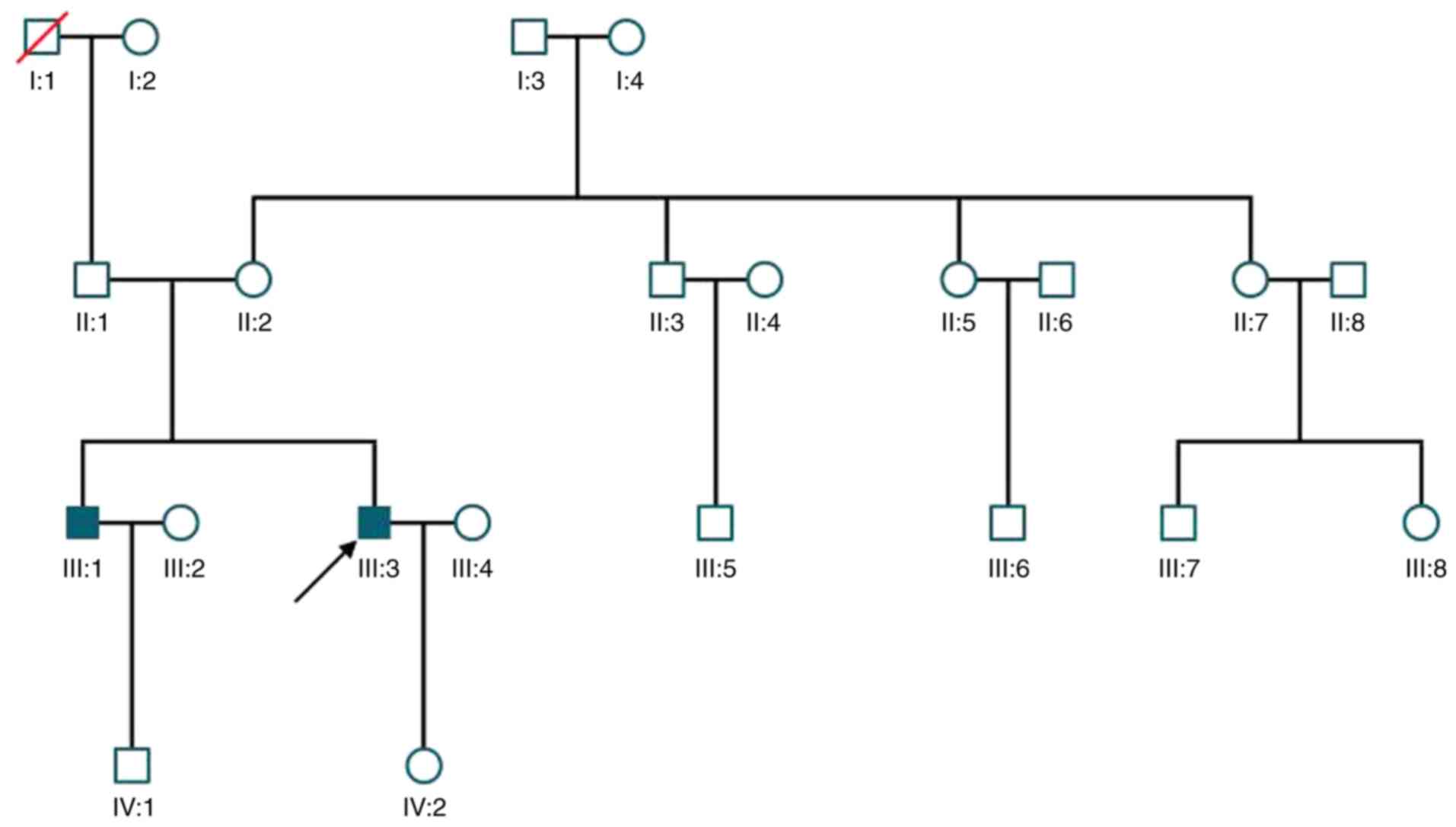
A total of two male siblings were diagnosed with
XLRS in the family (Fig. 1); they
were both indicated to have macular abnormalities. The younger
sibling was the proband, who presented with blurred vision at the
age of 12 years, which could not be corrected. The patient was
diagnosed at 30 years old with XLRS at The Second Affiliated
Hospital of Harbin Medical University (Harbin, China) in May 2011
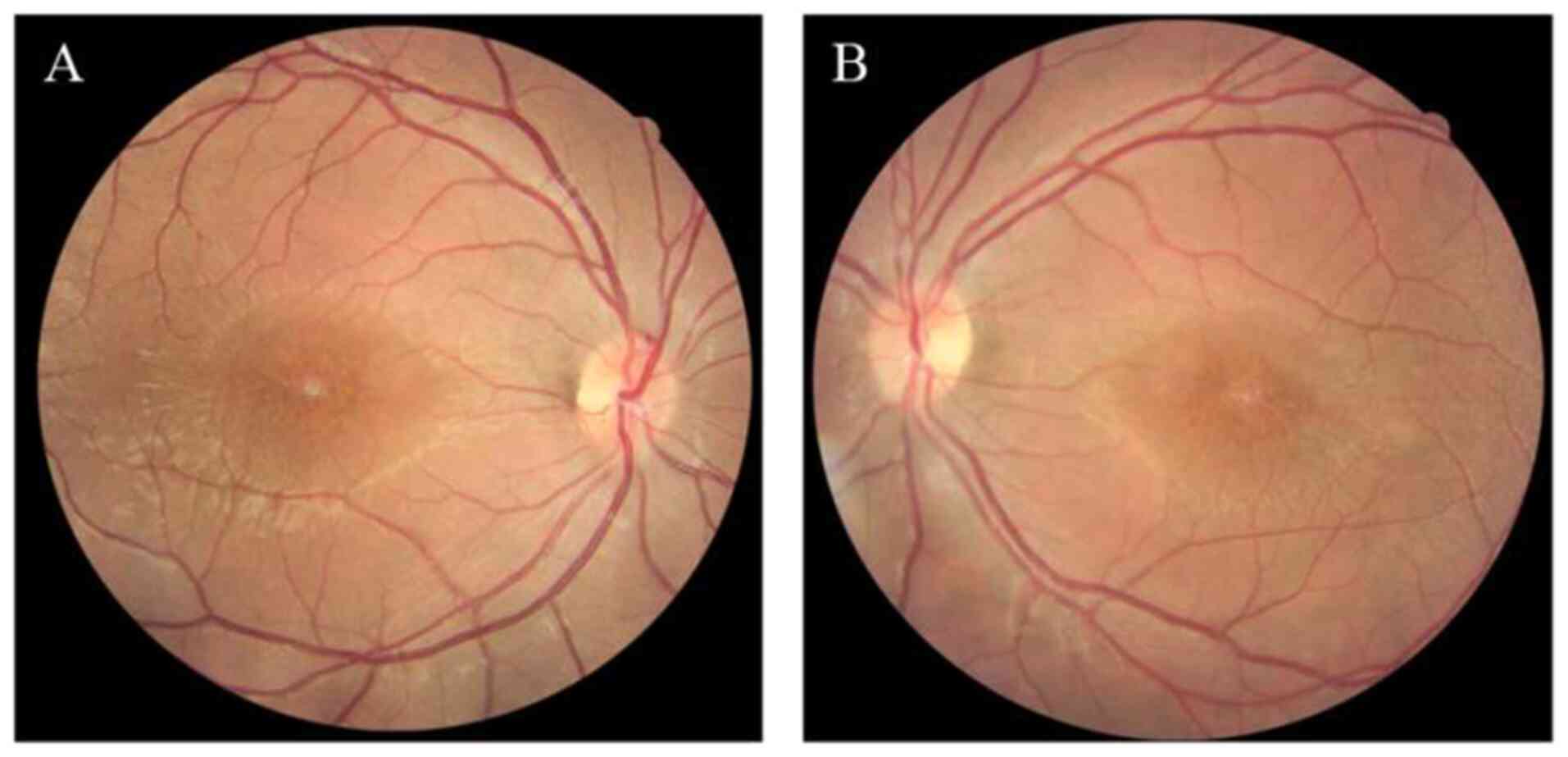
and was followed up for 7 years. At the first visit, a ‘spoke
wheel’ pattern and macular retinoschisis were observed in the
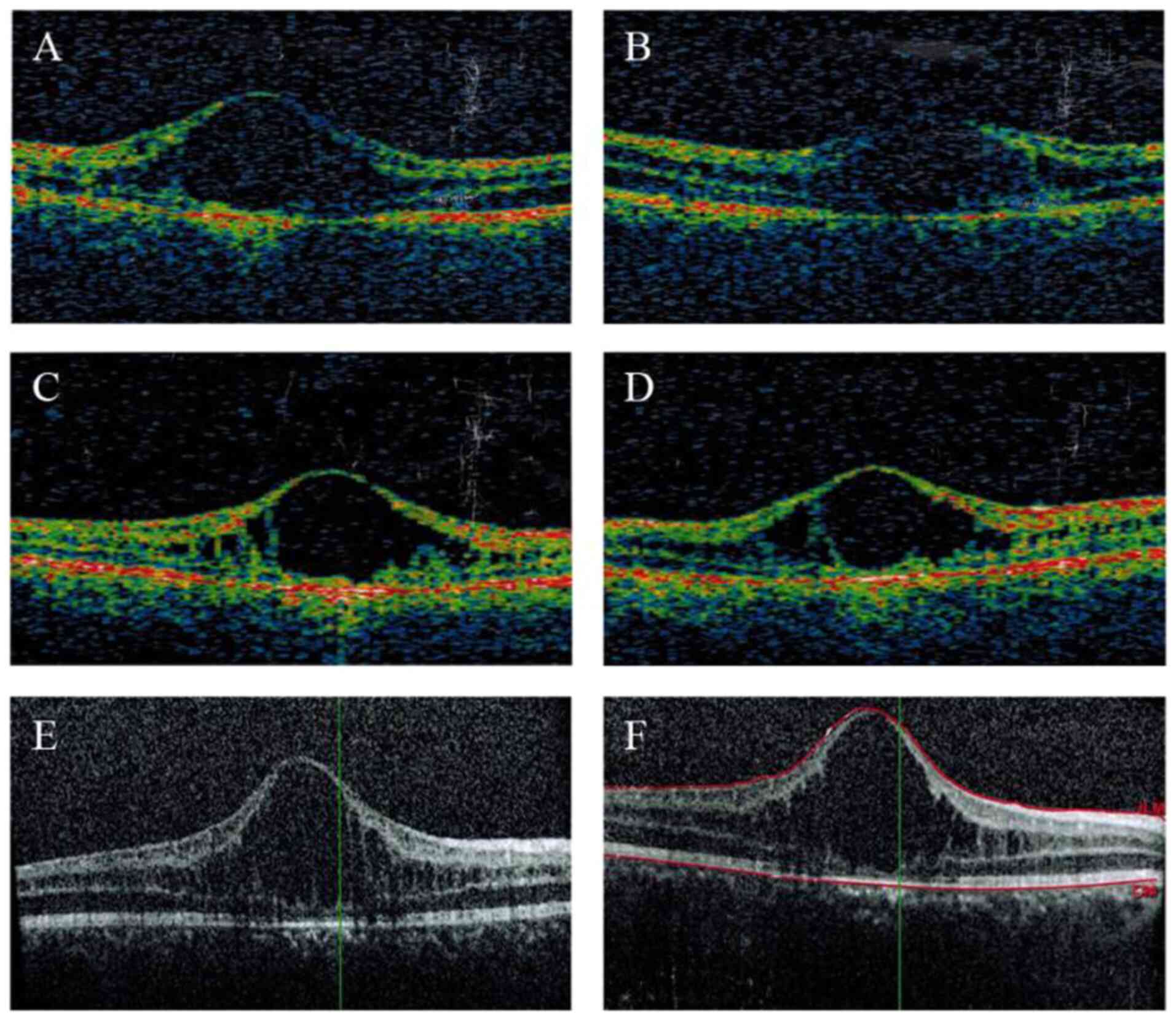
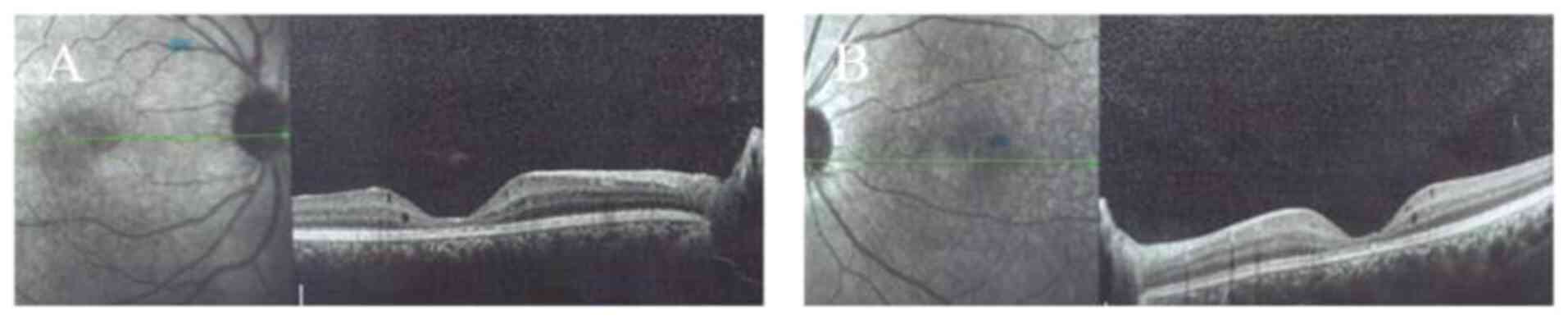
fundus (Fig. 2). Examination by OCT
indicated macular schisis and there were increases in the thickness
of the macula in both eyes compared with that in normal eyes. The
older male sibling of the proband visited 1 month later, he started
exhibiting blurred vision at 11 years old, was diagnosed at 33
years old, and bilateral macular schisis, intraretinal cysts and
macular atrophy were observed in this patient (Table I). Over 7 years, notable increases
in the thickness of the macula were detected in both eyes of the
proband, whilst macular atrophy persisted in the older sibling.
There were no changes in visual acuity in both patients (Figs. 3 and 4).
 | Table IClinical manifestations. |
Table I
Clinical manifestations.
| Patient no. | Age (years) | Eye | 1st BCVA | 2nd BCVA | 3rd BCVA | Macular
abnormalities |
|---|
| III:3 (proband) | 30 | OD | 20/100 | 20/100 | 20/100 | Schisis |
| | | OS | 20/80 | 20/80 | 20/100 | Schisis |
| III:1 (elder
brother) | 33 | OD | 20/50 | N/A | 20/50 | Schisis, atrophy |
| | | OS | 20/50 | N/A | 20/50 | Schisis, atrophy |
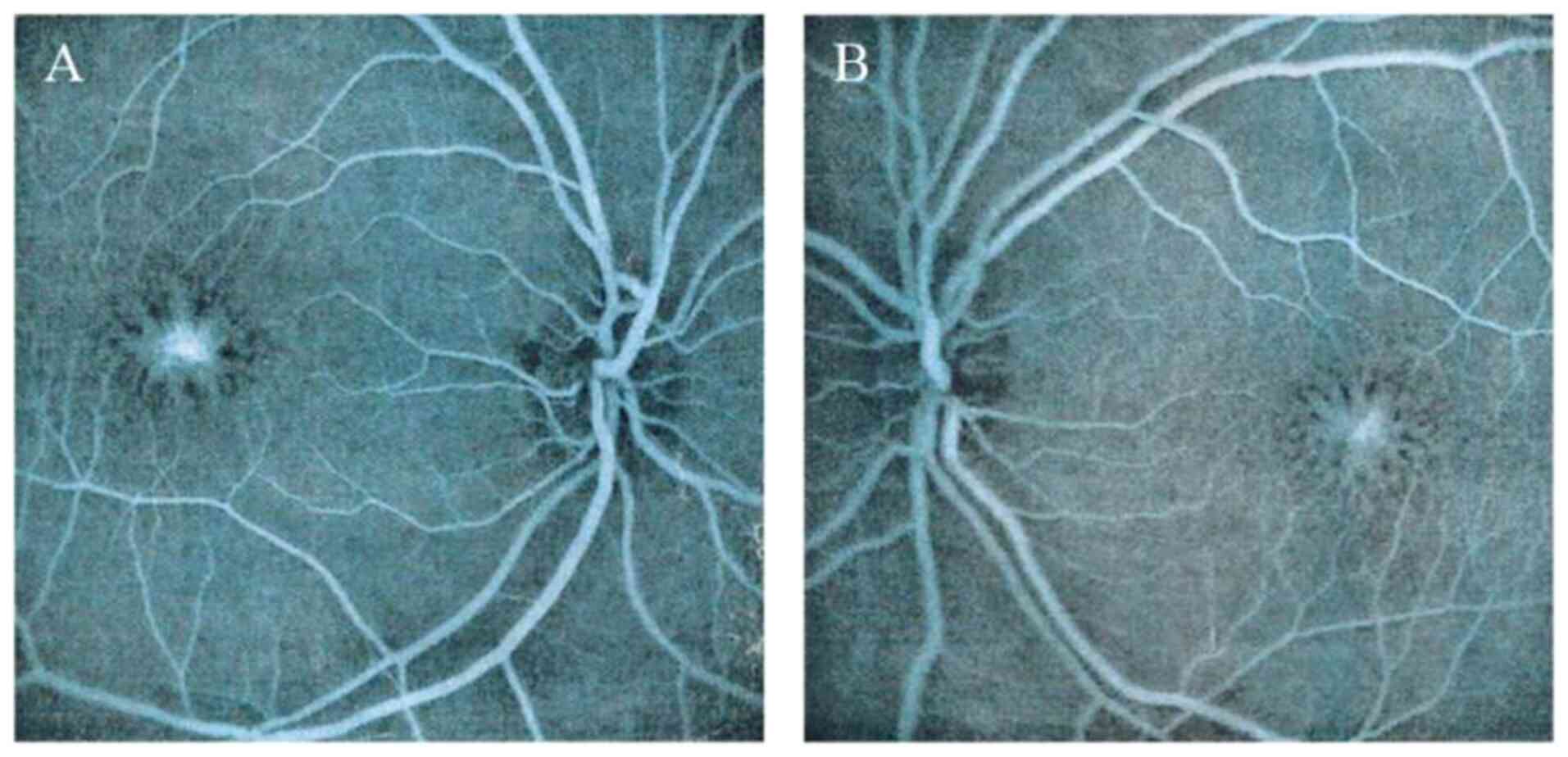
Fundus fluorescence angiography
FFA images acquired at the first visit revealed a
‘spoke wheel’ pattern of hyperfluorescence in the central macular
area of the proband (Fig. 5).
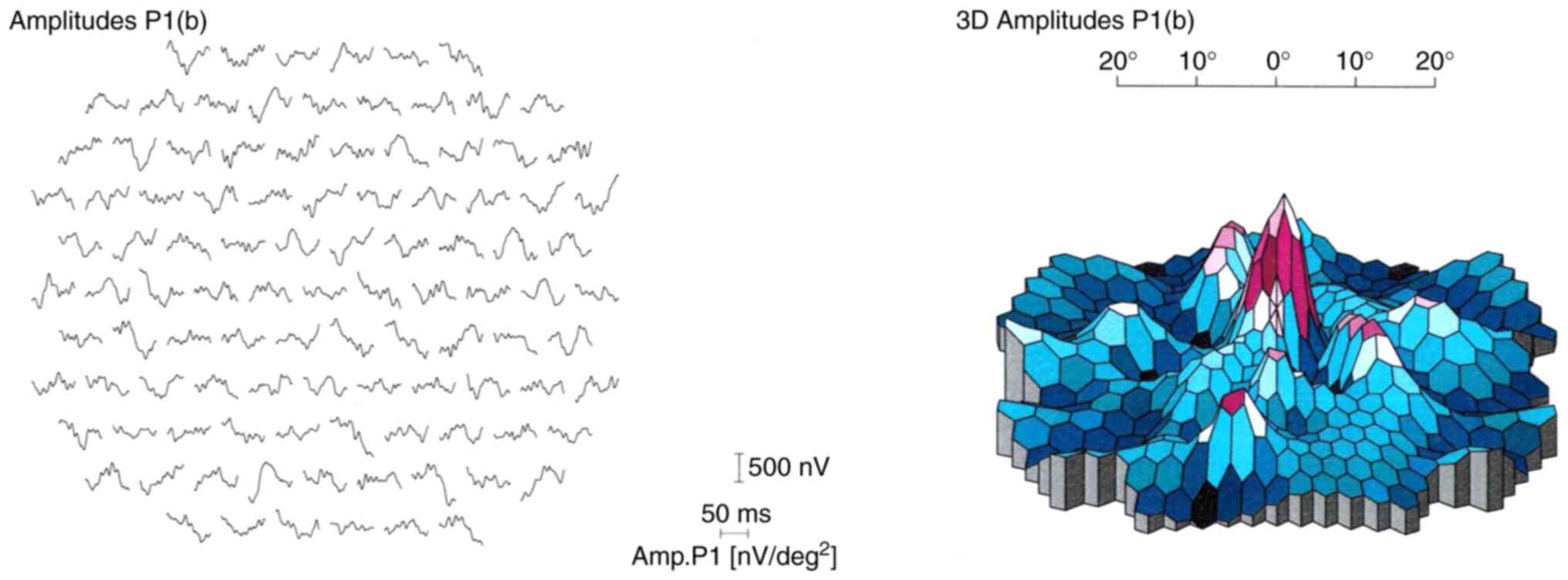
ERG
Multifocal ERG (mfERG) was recorded from the central
30˚ of the visual field of the proband, where responses were
revealed to be reduced bilaterally in the central and outer rings
at the first visit. The mfERG results suggested central cone
dysfunction affecting not only the macular area but across the
central 30˚ of the visual field tested in each eye (Fig. 6).
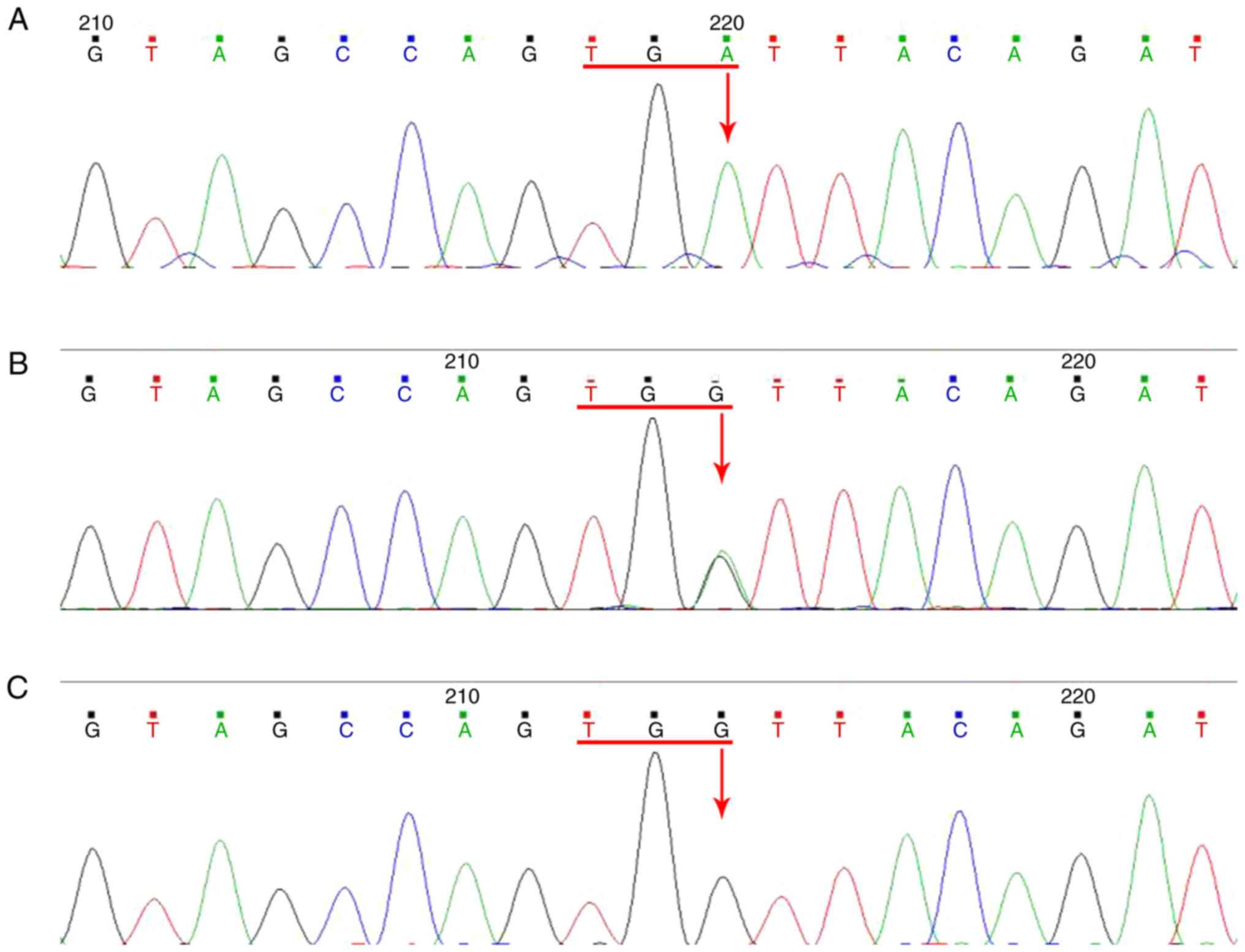
Genetic analysis
The two patients were revealed to carry a genetic
mutation in the RS1 gene; the mutation was determined to be
localized in exon 5 (c.366G>A). This G>A substitution changed
the amino acid from tryptophan to a stop codon, which produced a
nonfunctional and truncated protein that was 22 amino acids in
length.
None of the healthy family members or the 100
control subjects examined tested positive for this mutation
(Fig. 7). The mother of the proband
was demonstrated to be a heterozygous carrier of the mutation
without the manifestation of any symptoms. Base changes were
neither identified in the sequences of other normal members of the
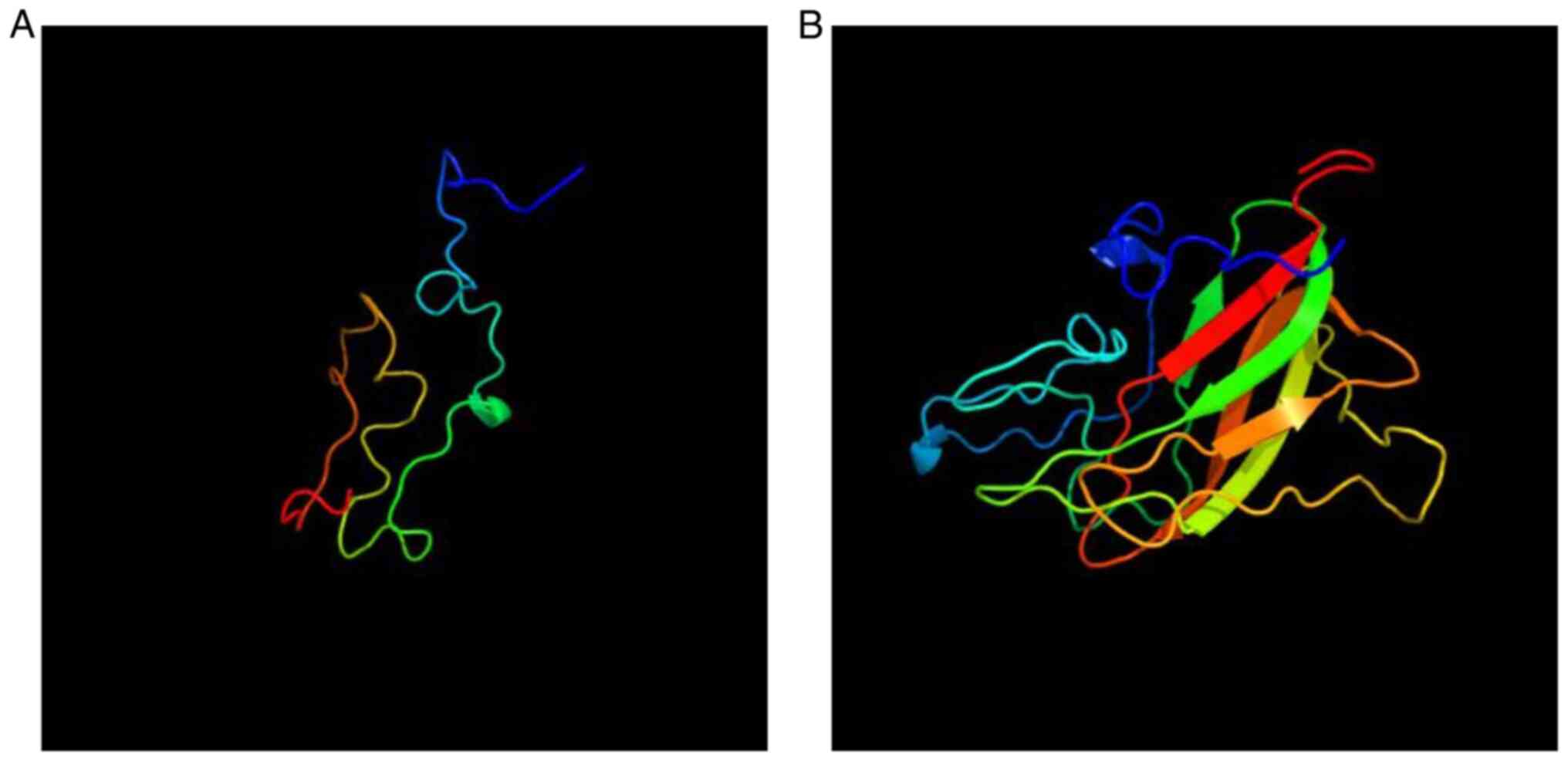
family nor in the normal control group. The 3D protein structures
of RS1 were analyzed using the Phyre2 software. Molecular modeling
indicated that the p.W122* substitution significantly
changed the secondary structure of the RS1 protein and made the
secondary structure shorter, which was caused by the mutation of
c.366G> A to generate a stop codon. This observation suggested
that this mutation is likely to markedly impact the protein
function (Fig. 8).
There are currently no effective treatments for
XLRS. The two patients did not receive any treatment but were only
clinically followed. It has been reported that carbonic anhydrase
inhibitors (CAIs), which belong to sulfanilamide drugs, were
effective in improving cystoid macular edema in patients with XLRS
(7). The elder brother's macula was
already atrophic at presentation, and although the younger
brother's macular thickness increased, he had a history of
sulfanilamide allergy, therefore he was not treated with CAIs.
Discussion
XLRS is a congenital disease that was first
described by Haas (8) in 1898. It
causes changes to the retinal structure early in life. A number of
studies have previously demonstrated that XLRS is a
neurodevelopmental abnormality caused by gene mutations, leading to
retinoschisis. In total, ~251 different mutations in the RS1 gene
have been reported to cause XLRS, the clinical characteristics of
which include splitting of the retina and visual impairment.
Kjellström et al (9)
previously followed up 10 patients for 12 years and determined no
changes in visual acuity after childhood. Similar results were also
reported by another study (10),
suggesting XLRS to be a progressive retinal degenerative condition
with a slow onset. Although a ‘spoke wheel’-like appearance in the
macula is the most typical characteristic feature of XLRS,
subretinal fibrosis, macular atrophy, mimicking maculopathy,
vitreous hemorrhage, peripheral retinoschisis and peripheral
pigmentary changes are also occasionally present in patients with
XLRS (11).
XLRS is a clinically heterogeneous disease with
>100 different mutations in the RS1 gene now known to cause this
disease, where two patients in a family with the same mutation may
present with different phenotypes. In a previous study, Kondo et
al (12) reported that foveal
schisis was more frequently associated with missense mutations in
67 Japanese patients from 56 families affected with XLRS, where
peripheral schisis was detected in 50 and 67% of the eyes of
patients harboring truncation and missense mutations,
respectively.
Hu et al (13) examined 30 patients suspected to
exhibit XLRS and 51 patients with confirmed XLRS in China and
identified 28 mutations associated with this disease, 8 of which
were novel. Tian et al (14)
reported on 6 families with XLRS in China, whilst certain sporadic
cases of XLRS and novel mutations were also reported in China
(15).
The present study reported on the clinical
characteristics and data obtained by OCT, FFA, ERG and RS1 gene
mutation analysis of 2 patients from a Chinese family who were
followed up for 7 years. The eyes of the younger sibling had a
‘spoke wheel’-like appearance, whereas those of the older sibling
exhibited intraretinal cysts and macular atrophy. The younger
sibling was followed up for 7 years, who was suffering from blurred
vision at 12 years of age but no change in visual acuity was
observed over the 7 years after presentation in the hospital. OCT
analysis revealed a minor change in the central macula; however,
the macular thickness was determined to be increased during 7 years
of follow-up. The older sibling was diagnosed with XLRS at the age
of 33 years and atrophy persisted in the central macula after 7
years.
In addition, the present study also identified a
novel genetic mutation in the RS1 gene of the two patients
examined, which was localized in exon 5 (c.366 G>A), where this
G>A substitution changed the amino acid from tryptophan to a
stop codon, producing a truncated nonfunctional 22-amino acid
protein. Subsequent molecular modeling indicated that
p.W122* substitution significantly shortened the
secondary structure of the RS1 protein. This mutation was indicated
to be clustered in the discoidin domain and occurred within the
conserved residues, where several studies have previously reported
that mutations may cause protein misfolding and retention in the
endothelial reticulum (16). This
observation suggested that this mutation may impact RS1
function.
In 1997, Sauer et al (17) discovered that the causative gene of
XLRS was RS1 located at Xp22. RS1 is expressed in photoreceptor
cells and bipolar cells, it contains 6 exons and encodes a secreted
protein called the retinal splitting protein (18). The retinal splitting protein is
comprised of 224 amino acids and contains a highly hydrophobic
signal peptide. This signal peptide is cleaved by peptidases to
form a mature protein serving as a connection between cells in the
inner nuclear layer, which is closely associated with synaptic
connections of photoreceptors, where the bipolar cells are located
(19). The retinal splitting
protein contains a disc-like domain that consists of 157 amino
acids and 10 cysteines, which is encoded by exons 4, 5 and 6. This
domain is highly conserved and has been previously associated with
cell adhesion and signal transmission throughout evolution
(20). The discoid domain receptor
is a transmembrane receptor that is able to interact with collagen,
mediate adhesion between cells and regulate the extracellular
matrix (21).
RS1 is mainly expressed in photoreceptor and bipolar
cells, potentially providing a reason for the lack of systemic
tissue abnormalities in male patients (22). Examination of female carriers may
prove difficult due to the absence of clinical symptoms (22). However, a previous study has
suggested that female carriers may also exhibit an abnormal ERG
performance, with observed splitting in the periphery or center of
the retina (22). The division of
the retina may be due to the inactivation of the X chromosome,
resulting in this abnormal clinical manifestation (22).
It has been indicated that mutations in the RS1 gene
cause dysfunction in the secretion of protein products and loss of
adhesion function, impaired intercellular communication, weakened
adhesion between the retinal layers and the formation of internal
retinal fission cavities (23). The
new mutation identified in the present cases whose mother was
determined to be a carrier leads to protein truncation. By applying
a software prediction of the changes in protein structure, it was
indicated that the structure of the protein was significantly
changed due to the mutation. The site was indicated to be located
in the highly conserved disc-like domain of the RS1 gene (24). It may be speculated that this
mutation affects the communication between cells and adhesion
between the retinal layers, resulting in the splitting of the inner
retina (25).
The biochemical mechanisms of XLRS remain poorly
understood. There are currently no effective treatments for XLRS.
Congenital retinal splitting has a slow onset and is difficult to
observe during stable phases of the disease. When complications
occur, they may be treated symptomatically (26). A number of studies on patients with
XLRS indicated that the vision of adult patients deteriorates,
whilst the vision of the majority of patients aged >70 years
falls <0.1(27). It is promising
that the retinal structure and function improved after gene
transfer therapy in a mouse RS1 knockout model of XLRS (28,29).
Bashar et al (30)
previously reported that extracellular delivery of RS1 rescued the
structural and functional deficits in the RS1h knockout mouse
model, where this ex vivo gene therapy approach was able to
inhibit disease progression. Zeng et al (28) injected RS1h complementary DNA into
the eyeballs of adult RS1h knockout mice, which reversed the
abnormal negative waveform of ERG, restored the positive b wave and
led to the expression of the retinal splitting protein in the
entire layer of the retina. Therefore, it is believed that gene
transfer may be an effective treatment strategy for XLRS, which
brings optimism for future interventions of this disease.
Zhao et al (26) previously studied 32 eyes with severe
complications that underwent vitrectomy and determined that
vitreoretinal surgery significantly improved visual acuity and
restored the anatomic structure of the retina. In addition,
carbonic anhydrase inhibitors may be helpful in reducing the
cavities and retinal thickness (31). Although significant progress has
been made in recent years, numerous questions, for example the
potential effects of gene therapy, should be further explored.
Since there is as yet no effective treatment for
XLRS, screening for gene mutations is vital for understanding the
pathogenesis of XLRS to explore novel effective treatment methods
for this disease.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YQ designed the study; NZhang and YP performed the
experiments; NZhou acquired clinical data; NZhang wrote and revised
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was performed with approval from
the Ethics Committee of The Second Affiliated Hospital of Harbin
Medical University (Harbin, China). Informed consent was obtained
from all participants.
Patient consent for publication
The proband and the elder brother provided consent
for publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
George ND, Yates JR and Moore AT: X linked
retinoschisis. Br J Ophthalmol. 79:697–702. 1995.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Wieacker P, Wienker J, Dallapiccola B,
Bender K, Davies KE and Ropers HH: Linkage relationships between
retionschisis, Xg, and a cloned DNA seqence from the distal short
arm of the X chromosome. Hum Genet. 64:143–145. 1983.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Takada Y, Fariss RN, Tanikawa A, Zeng Y,
Carper D, Bush R and Sieving PA: A retinal neuronal developmental
wave of retinoschisin expression begins in ganglion cells during
layer formation. Invest Ophthalmol Vis Sci. 45:3302–3312.
2004.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wu WW, Wong JP, Kast J and Molday RS: RS1,
a discoidin domain-containing retinal cell adhesion protein
associated with X-linked retinoschisis, exists as a novel
disulfide-linked octamer. J Biol Chem. 280:10721–10730.
2005.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Renner AB, Kellner U, Fiebig B, Cropp E,
Foerster MH and Weber BHF: ERG variability in X-linked congenital
retinoschisis patients with mutation in the RS1 gene and the
diagnostic importance of fundus autofluorescence and OCT. Doc
Ophthalmol. 116:97–109. 2008.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Wang NK, Liu LL, Chen HM, Tsai S, Chang
TC, Tsai TH, Yang CM, Chao AN, Chen KJ, Kao LY, et al: Clinical
presentations of X-linked retinoschisis in Taiwanese patients
confirmed with genetic sequencing. Mol Vis. 21:487–501.
2015.PubMed/NCBI
|
|
7
|
Thobani A and Fishman GA: The use of
carbonic anhydease inhibitors in the retreatment of cystic macular
lesions in retinitis pigmentosa and X-linked retinoschisis. Retina.
31:312–315. 2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Haas J: Über das zusammenvorkommen von
veraenderungen der retina und choroides. Arch Augenheilkd.
37:343–348. 1898.
|
|
9
|
Kjellström S, Vijayasarathy C, Ponjavic V,
Sieving PA and Andréasson S: Long-term 12 year follow-up of
X-linked congenital retinoschisis. Ophthalmic Genet. 31:114–125.
2010.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kellner U, Brummer S, Foerster MH and
Wessing A: X-linked congenital retinoschisis. Graefes Arch Clin Exp
Ophthalmol. 228:432–437. 1990.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Apushkin MA, Fishman GA and Janowica MJ:
Correlated of optical coherencce tomography findings with visual
acuity and macular lesions in patients with X-linked retinoschisis.
Ophthalmology. 112:495–501. 2005.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kondo H, Oku K, Katagiri S, Hayashi T,
Nakano T, Iwata A, Kuniyoshi K, Kusaka S, Hiyoshi A, Uchio E, et
al: Novel mutations in the RS1 gene in Japanese patients with
X-linked congenital retinoschisis. Hum Genome Var.
6(3)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hu QR, Huang LZ, Chen XL, Xia HK, Li TQ
and Li XX: Genetic analysis and clinical features of X-linked
retinoschisis in Chinese patients. Sci Rep. 7(44060)2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Tian R, Jiang RX and Chen YX: Genetic and
phenotypic characteristics of six Chinese families with X-linked
juvenile retinoschisis. Chin Med J (Engl). 126:4392–4394.
2013.PubMed/NCBI
|
|
15
|
Huang XF, Tu CS, Xing DJ, Gan DK, Xu GZ
and Jin ZB: R102W mutation in the RS1 gene responsible for
retinoschisis and recurrent glaucoma. Int J Ophthalmol. 7:169–172.
2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wu WW and Molday RS: Defective discoidin
domain structure subunit assembly, and endoplasmic reticulum
processing of retinoschisin are primary mechanisms responsible for
X-linked retinoschisis. J Biol Chem. 278:28139–28146.
2003.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Sauer CG, Gehrig A, Warneke-Wittstock R,
Marquardt A, Ewing CC, Gibson A, Lorenz B, Jurklies B and Weber BH:
Positional cloning of the gene associated with X-linked juvenile
retinoschisis. Nat Genet. 17:164–170. 1997.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Gehrig AE, Warneke-Witstock R, Sauer CG
and Weber BH: Isolation and characterization of the murine X-linked
juvenile retinoschisis (Rs 1 h) gene. Mamm Genome. 10:303–307.
1999.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Robert SM, Ulrich K and Bernhard HF:
X-linked juvenile retinoschisis: Clinical diagnosis, genetic
analysis, and molecular mechanisms. Prog Retin Eye Res. 31:195–212.
2012.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Baumgartner S, Hofmann K,
Chiquet-Ehrismann R and Bucher P: The discoidin domain family
revisited: New members from prokaryotes and a homology-based fold
prediction. Protein Sci. 7:1626–1631. 1998.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Raymond A, Ensslin MA and Shur BD:
SED1/MFG-E8: A bi-motif protein that orchestrated diverse cellular
interaction. J Cell Biochem. 106:957–966. 2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kim LS, Seiple W, Fishman GA and Szlyk JP:
Multifocal ERG findings in carriers of X-linked retinoschisis. Doc
Ophthalmol. 114:21–26. 2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Grayson C, Reid SN, Ellis JA, Rutherford
A, Sowden JC, Yates JR, Farber DB and Trump D: Retinoschisin, the
X-linked retinoschisis protein, is a secreted photoreceptor
protein, and is expressed and released by Weri-Rb1 cell. Hum Mol
Genet. 9:1873–1879. 2000.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Fraternali F, Cavallo L and Musco G:
Effect of pathological mutations on the stability of a conserved
amino acid triad in retinoschisin. FEBS Lett. 544:21–26.
2003.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Sergeev YV, Caruso RC, Meltzer MR, Smaoui
N, MacDonald IM and Sieving PA: Molecular modeling of retinoschisin
with functional analysis of pathogenic mutations from human
X-linked retinoschisis. Hum Mol Genet. 19:1302–1313.
2010.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhao C, Zhang Q, Jin HY and Zhao PQ:
Clinical observations of vitreoretinal surgery for four different
phenotypes of X-linked congenital retinoschisis. Int J Ophthalmol.
11:986–990. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Tantri A, Vrabec TR, Cu-Unjieng G, Frost
A, Annesley WH Jr and Donoso LA: X-linked retinoshisis: A clinical
and molecular genetic review. Surv Ophthalmol. 49:214–230.
2004.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zeng Y, Takada Y, Kjellstrom S, Hiriyanna
K, Tanikawa A, Wawrousedk E, Smaoui N, Caruso R, Bushu RA and
Sieving PA: RS-1 gene delivery to an adult Rs1h knockout mouse
model restores ERG b-wave with reversal of the electronegative
waveform of X-linked retinoschisis. Invest Ophthalmol Vis Sci.
45:3279–3285. 2004.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Min SH, Molday LL, Seeliger MW, Dinculescu
A, Timmers AM, Janssen A, Tonagel F, Tanimoto N, Weber BH, Molday
RS and Hauswirth WW: Prolonged recovery of retinal
stucture/function after gene therapy in an Rs1h-deficient mouse
model of X-linked juvenile retinoschisis. Mol Ther. 12:644–651.
2005.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Bashar AE, Metcalfe AL, Viringipurampeer
IA, Yanai A, Gregory-Evans CY and Gregory-Evans K: An ex vivo gene
therapy approach in X-linked retinoschisis. Mol Vis. 22:718–733.
2016.PubMed/NCBI
|
|
31
|
Apushkin MA and Fishman GA: Use of
dorzolamide for patients with X-linked retinoschisis. Retina.
26:741–745. 2006.PubMed/NCBI View Article : Google Scholar
|