Introduction
Myocardial ischemia-reperfusion injury (MIRI), which
usually occurs in clinical settings, leads to severe outcomes for
patients if no effective strategies are applied to inhibit the
downstream apoptotic cascades. However, numerous animal studies
that have revealed various protective mechanisms have also
confirmed the efficacy of cardioprotection in overcoming MIRI
(1-6).
However, the results achieved in the clinical application of these
strategies have not been consistent with those achieved in
experimental research (7-9).
In this context, the mechanism by which the apoptotic pathway and
specific key molecules are induced during MIRI, particularly
pathways and molecules associated with cell death and ischemia,
such as endoplasmic reticulum stress (ERS)-associated apoptosis
signaling pathways, may be important. However, these factors are
currently unclear.
MIRI induces severe damage to the endoplasmic
reticulum. In 2016, Wu et al (10) suggested that ERS should be
considered in the occurrence of MIRI. IRI has been indicated to be
a multifactorial process that can result in multiple organ damage
via ERS and the associated occurrence of apoptosis. The underlying
mechanism involves excessive oxidative damage, ATP depletion and
energy imbalance, calcium homeostasis and other factors. A number
of studies have elucidated the effects of ameliorating ERS on the
prognosis of MIRI in animal models and in vitro cell models
(11-13).
Furthermore, numerous signaling pathways, such as the
miR-34a/sirtuin 1/nuclear factor erythroid 2-related factor 2
(Nrf2) (14), AMP-activated protein
kinase/Nrf2(15), PI3K/AKT
(16) and Toll-like receptor
4/myeloid differentiation factor 88/NF-κB pathways (17), have been demonstrated to mediate
ERS. Furthermore, in a notable review in 2019, Davidson et
al (18) opined that
multitargeted strategies are necessary to reduce MIRI, because any
single approach has a limited capacity to overcome the complex
state of MIRI.
The highly selective α2 adrenergic
receptor agonist dexmedetomidine (DEX) is frequently used
clinically, especially to provide protection to the heart and other
organs during surgery (19-22).
Currently, most of the functions of DEX appear to be associated
with its anti-inflammatory activity and ability to inhibit IRI.
However, the effects of DEX on ERS and the resulting apoptosis have
not yet been thoroughly elucidated. In previous studies, DEX
exhibited a protective role in the hearts of diabetic mice by
interfering with ERS or autophagy, thereby suppressing IRI
(6,23); however, the results were partially
attributed to the diabetes context. Furthermore, researchers have
focused on the study of cells other than cardiomyocytes, such as
endothelial cells, under IRI or hypoxia/reoxygenation (H/R)
conditions (24,25), and have examined several crucial ERS
chaperones, proteins and apoptosis indicators that are produced by
organs other than the heart under IRI or H/R conditions (6,26-30).
These studies have shown that DEX effectively regulates the
function of non-cardiomyocytes and interferes with the ERS
signaling pathway under these conditions. In addition, a couple of
studies have explored the function of DEX in preventing the injury
of H9c2 cardiomyocytes under H/R conditions (31,32).
In both studies, DEX was used to precondition the H/R H9c2 cell
model, and a significant alleviation of H/R injury was achieved;
the study by Wang et al (31) indicated that this was achieved
through the increased expression of mediator of RNA polymerase II
transcription subunit 13, while that by Yuan et al (32) demonstrated the involvement of
increased FK506 binding protein 1B expression. However, the exact
regulatory effect of DEX on ERS and the appropriate experimental
conditions for the evaluation of its regulatory effects on H/R
remain unknown. Furthermore, if DEX protects against IRI through
the inhibition of ERS alone or whether other functions are also
involved is unclear.
In the present study, we hypothesized that DEX
protects cardiomyocytes against H/R injury by mechanisms in
addition to its interference with ERS. The aims of the study were
to verify the capacity of DEX to protect injured cardiomyocytes
under in vitro H/R conditions and to optimize suitable
experimental conditions for future research.
Materials and methods
Cell culture
H9c2 embryonic rat cardiomyocytes were obtained from
the cell bank of the Central Experimental Laboratory of the Second
Hospital of Jiaxing University. The cells were cultured in DMEM
(Corning Inc.) containing 4.5 g/l glucose and supplemented with 10%
fetal bovine serum (Invitrogen; Thermo Fisher Scientific, Inc.),
100 g/ml streptomycin and 100 U/ml penicillin (Beijing Solarbio
Science & Technology Co., Ltd.). All cells used for experiments
were cultured in a 37˚C incubator containing 95% air and 5%
CO2.
Toxicity testing
The original DEX solution was prepared by dissolving
DEX hydrochloride powder (SML0956; Sigma-Aldrich; Merck KGaA) in
DMSO and then diluting the solution >1,000-fold in DMEM to
achieve media with final DEX concentrations of 0, 1, 5 and 10 nM,
and 0.1, 1, 5 and 10 µM. The H9c2 cells were incubated in 96-well
plates under normal conditions, and the medium was replaced with
fresh medium containing DEX 24 h prior to the cell viability
assay.
Establishment of the H/R injury
model
To establish an optimal in vitro H/R model,
methods similar to those used by Wang et al (31) and Yuan et al (32) were used. A single-cell suspension of
H9c2 cells (~5x105 cells/ml) was prepared and
5x103 cells/well were seeded in 96-well plates. The
cells were cultured in two plates, namely a control plate and an
experimental plate, and the cells in each plate were randomly
divided into four groups: The H3/R3, H3/R6, H3/R12 and H3/R24
groups. The control plate was incubated in a cell incubator at 37˚C
in 5% CO2. The experimental plates underwent a 3-h
exposure to anoxia after replacement of the medium with serum-free
DMEM. The medium in the control plate was not replaced with
serum-free DMEM at the same time point. At the end of the anoxic
culture step, the cell groups in the experimental plates were
cultured under normoxic conditions upon the initiation of
reoxygenation. The experimental H9c2 cells in the H3/R3, H3/R6,
H3/R12 and H3/R24 groups underwent 3, 6, 12 and 24 h of
reoxygenation, respectively, in a cell incubator at 37˚C and 5%
CO2. The parameters of the group in which the most
severe damages were observed, according to the results of cell
viability and injury assays were considered the optimal conditions
for H/R and were used for the H/R groups in subsequent
experiments.
To analyse the effect of DEX on the viability and
injury of H9c2 cells under H/R conditions, the cells were divided
into a control group, H/R group and H/R + DEX group. The H/R + DEX
group included 7 subgroups, to which 7 different concentrations of
DEX (1, 5 and 10 nM; 0.1, 1, 5 and 10 µM) were added 1 h prior to
the initiation of hypoxia. Cells in the H/R and H/R + DEX groups
were incubated for 3 h in a hypoxia chamber filled with 5%
CO2 and 95% N2 and were then subjected to 3 h
of reoxygenation under normoxic conditions. Correspondingly, the
control group was incubated under normoxic conditions for 6 h.
Experimental protocols
After the above procedures were complete, the
optimal concentration of DEX for intervention and experimental
conditions for H/R were identified and used in the following
experiments. To verify that the selected concentration of DEX (1
µM) was effective for attenuating H/R injury in H9c2 cells and
evaluate whether the experiment could be completed under the
selected experimental conditions (3 h hypoxia and 3 h
regeneration), cells cultured in 96- or 6-well plates were divided
into 7 groups, namely the control, DEX, H/R, H/R + DEX,
4-phenylbutyrate (4-PBA; P-21005; Sigma-Aldrich; Merck KGaA), H/R +
4-PBA and H/R + DEX + 4-PBA groups. A total of 1 mM 4-PBA, an
effective ERS inhibitor, was used to treat the cells 24 h prior to
H/R.
In addition to cell viability and injury assays,
flow cytometry assays were conducted to evaluate apoptosis in the
groups. The protein and mRNA expression levels of glucose-regulated
protein 78 (GRP78), C/EBP homologous protein (CHOP) and caspase-12
were also measured via reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) and western blot analysis, as
described below.
Cell viability and injury assays
According to the instructions of the Cell Counting
Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.), H9c2 cells
(~5x103 cells/well) were seeded in a 96-well plate. For
viability testing, the cells in each well were covered with 10 µl
CCK-8 solution for 1 h and incubated at 37˚C. Then, the optical
density was measured at 450 nm using a microplate reader (Molecular
Devices, LLC) and the cell viability (%) was calculated. In
addition, cell injury was assessed with a lactate dehydrogenase
(LDH) kit (cat. no. C0017; Beyotime Institute of Biotechnology)
according to the manufacturer's instructions, based on the amount
of LDH released into the supernatant.
Apoptosis assay
Apoptosis of the H9c2 cells in each group was
evaluated by flow cytometry with an Annexin V-PE/7-AAD kit (cat. no.
CT1030; Beijing Solarbio Science & Technology Co., Ltd.). In
brief, after digestion with 0.25% trypsin without EDTA and 3,000 x
g centrifugation for 5 min at room temperature, cells were washed
twice with PBS, resuspended in binding buffer, and incubated with 5
µl Annexin V-PE and 10 µl 7-AAD for 20 min at room temperature. The
apoptotic cells were detected using a flow cytometer (BD
Biosciences), and early and late apoptosis were presented in the
lower and upper right quadrants of the plots for each group,
respectively. The apoptosis rate was calculated with FlowJo X (Tree
Star, Inc.).
RT-qPCR
The primers used to amplify GRP78, caspase-12, CHOP
and β-actin were synthesized by Invitrogen (Thermo Fisher
Scientific, Inc.), and the sequences are presented in Table I. Following the aforementioned
treatments, total RNA was extracted from the H9c2 cells with
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), and the purity of the RNA from each group was determined.
RNA (500 ng/sample) was reverse transcribed to cDNA using a reverse
transcription kit (Takara Biotechnology Co., Ltd.) according to the
manufacturer's instructions. Subsequently, qPCR was conducted with
2 µl cDNA and other necessary reagents according to the
instructions of the SYBR Premix Ex Taq kit (Takara Biotechnology
Co., Ltd.), and the final amplification reaction (25 µl) was
conducted in an ABI Prism 7500 system (Thermo Fisher Scientific,
Inc.). The following thermal cycling conditions were used for
amplification: Initial denaturation at 95˚C for 3 min followed by
40 amplification cycles of denaturation at 95˚C for 30 sec,
annealing at 55˚C for 20 sec and elongation at 72˚C for 20 sec.
mRNA expression levels were calculated as the ratio of the target
gene expression level to that of β-actin using the
2-ΔΔCq method (33).
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Gene | Forward
(5'-3') | Reverse
(5'-3') |
|---|
| GRP78 |
ACTGGAATCCCTCCTGCTC |
CAAACTTCTCGGCGTCAT |
| CHOP |
TGCCTTTCGCCTTTGAGAC |
GCTTTGGGAGGTGCTTGTG |
| Caspase-12 |
GGGATAGCCACTGCTGATA |
GCCACTCTTGCCTACCTTC |
| β-actin |
TGAGAGGGAAATCGTGCGTG |
TTGCTGATCCACATCTGCTGG |
Western blot analysis
Following the aforementioned treatments, H9c2 cells
were washed thoroughly with ice-cold PBS solution, and RIPA buffer
(Beyotime Institute of Biotechnology) was then added to the wells
and incubated for 30 min on ice. Protein was quantified using a BCA
assay. A total of 30 µg proteins/lane were separated by 8% SDS-PAGE
and transferred to PVDF membranes at 4˚C and 200 mA for 2 h. The
membranes were blocked by 5% skimmed milk powder in TBS with
Tween-20 (TBS-T) solution for 2 h at room temperature and then
incubated at 4˚C overnight with the following primary antibodies:
Anti-CHOP (1:1,000; cat. no. DF6025; Affinity Biosciences),
anti-GRP78 (1:1,000; cat. no. AF5366; Affinity Biosciences),
anti-caspase-12 (1:1,000; cat. no. AF5199; Affinity Biosciences)
and mouse anti-β-actin (1:1,000; cat. no. T0022; Affinity
Biosciences). A horseradish peroxidase-conjugated secondary
antibody (1:2,000; cat. no. 111-095-003; Jackson ImmunoResearch
Laboratories, Inc.) was then added for 2 h at room temperature
after the membranes were washed five times with TBS-T buffer.
Finally, the membranes were washed with TBST, and signals were
visualized with an enhanced chemiluminescence detection kit
(Beyotime Institute of Biotechnology). The protein band densities
were quantified with ImageQuant TL software (version 7.0;
Cytiva).
Statistical analysis
All data are expressed as the mean ± standard
deviation (SD). The significance of differences among multiple
groups was evaluated by one-way analysis of variance followed by
Tukey's post hoc test in SPSS 19.0 software (IBM Corp.). GraphPad
Prism (GraphPad Software, Inc.) was used to generate the graphs.
P<0.05 was considered to indicate a statistically significant
difference.
Results
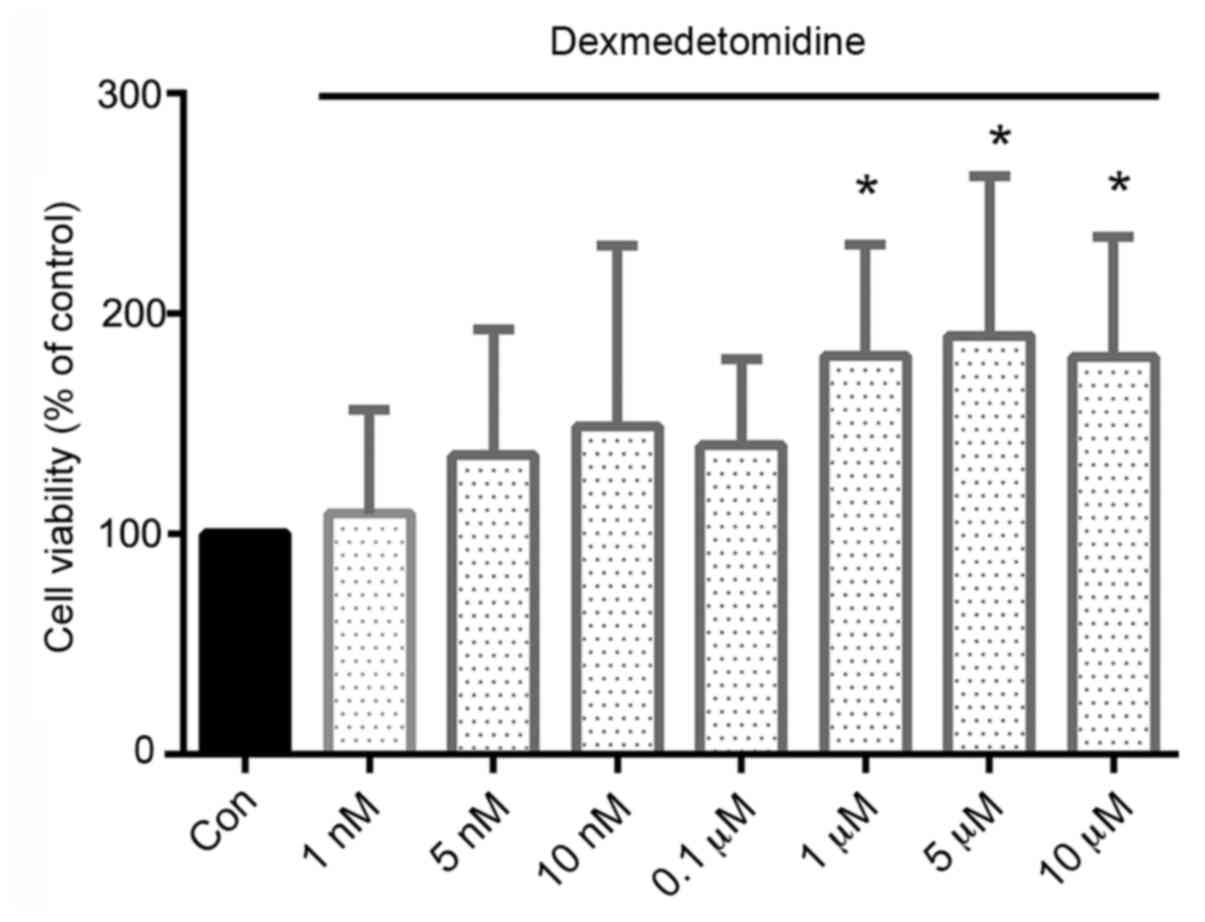
DEX promotes the proliferation of H9c2
cells
DEX caused no obvious cytotoxicity to H9c2 cells;
instead, the viability of the DEX-treated cells was increased
compared with that of the control cells. Moreover, DEX
concentrations of ≥1 µM resulted in significantly increased
viability, with increases of 81, 89 and 80% in cells treated with
1, 5 and 10 µM, respectively, compared with the viability of the
control cells (Fig. 1).
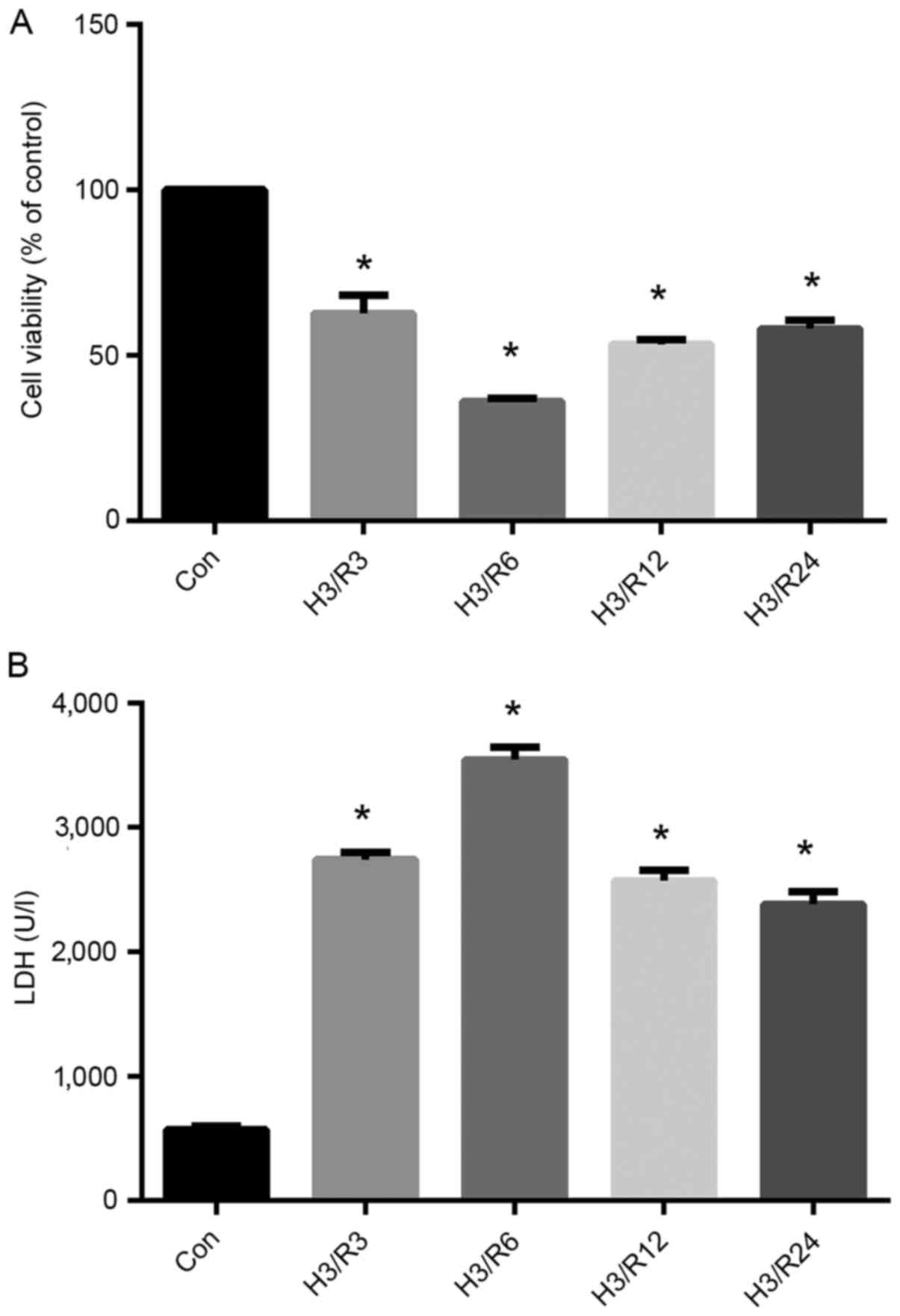
Optimal experimental level of hypoxia
is achieved by 3 h of hypoxia/3 h of reoxygenation
To establish the optimal H/R conditions that met
subsequent experimental requirements, several model conditions were
selected: 3 h hypoxia/3 h reoxygenation, 3 h hypoxia/6 h
reoxygenation, 3 h hypoxia/12 h reoxygenation and 3 h hypoxia/24 h
reoxygenation (Fig. 2). As shown in
Fig. 2A, the viability of the
hypoxic cells exhibited a decreasing trend after 3 h of
reoxygenation and remained relatively low until 24 h after
reoxygenation. The cell viability in the H3/R3, H3/R6, H3/R12 and
H3/R24 groups was decreased significantly to 62.67, 36, 53.33 and
58%, respectively, of that in the control group (P<0.05).
In addition, as shown in Fig. 2B, H/R significantly increased the
level of LDH in the supernatant. The significant increase in LDH
level started at 3 h of reoxygenation, at which time the mean LDH
level was 480% of the control (P<0.01), and maintained high
levels throughout the rest 24 h, as and peaked at 6 h of
reoxygenation.
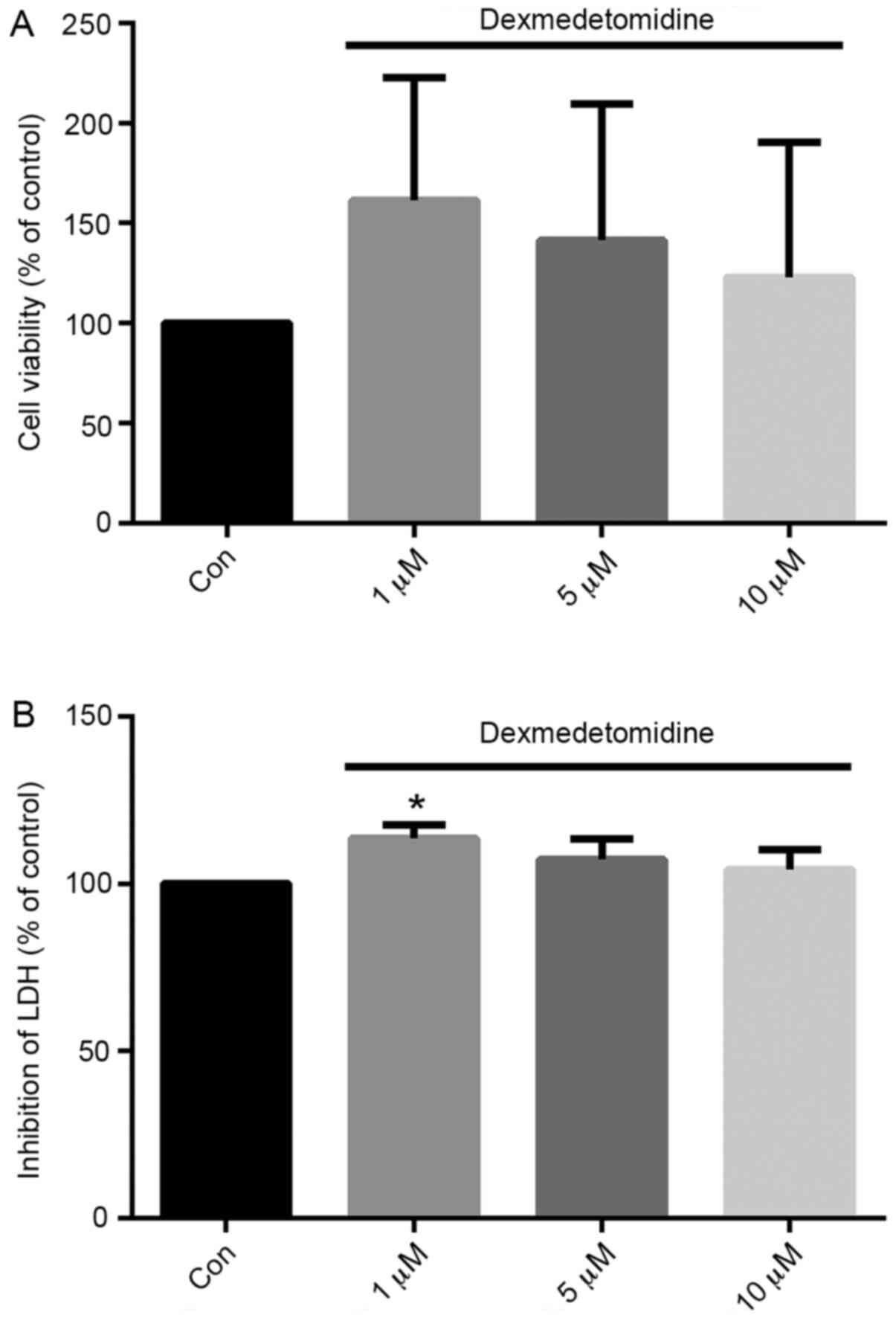
DEX (1 µM) effectively protects H9c2
cells from H/R injury
H9c2 cell viability and LDH release were evaluated
following H/R with DEX at the concentrations shown to most
effectively promote proliferation: 1, 5 and 10 µM. As shown in
Fig. 3, treatment with DEX at all
three concentrations increased the viability of H/R-exposed H9c2
cells compared with control group, although no significant
differences were found in 5 and 10 µM groups. Furthermore, the
effect appeared to gradually reduce from 1 to 10 µM. Concentrations
of 1 µM exhibited the greatest effect compared with control
treatment. Similarly, compared with the control, pretreatment with
1 µM DEX significantly inhibited the release of LDH from H9c2 cells
by 13.7% (P<0.05).
DEX reduces the apoptosis of H9c2
cells
To further investigate the role of DEX in H9c2 cells
during H/R, several different treatments were applied (Figs. 4 and 5). Cell viability and LDH release in the
control group, the DEX pretreatment group incubated under normal
conditions and the 4-PBA pretreatment group incubated under normal
conditions exhibited similar and comparable results (P>0.05).
However, the H/R group exhibited significantly decreased cell
viability and increased LDH release compared with the control group
(P<0.05). The 1 µM DEX + H/R group exhibited an attenuation of
cellular injury, and 4-PBA pretreatment successfully reversed the
protective effect of DEX against H/R injury in terms of cell
viability and LDH release (Fig.
4).
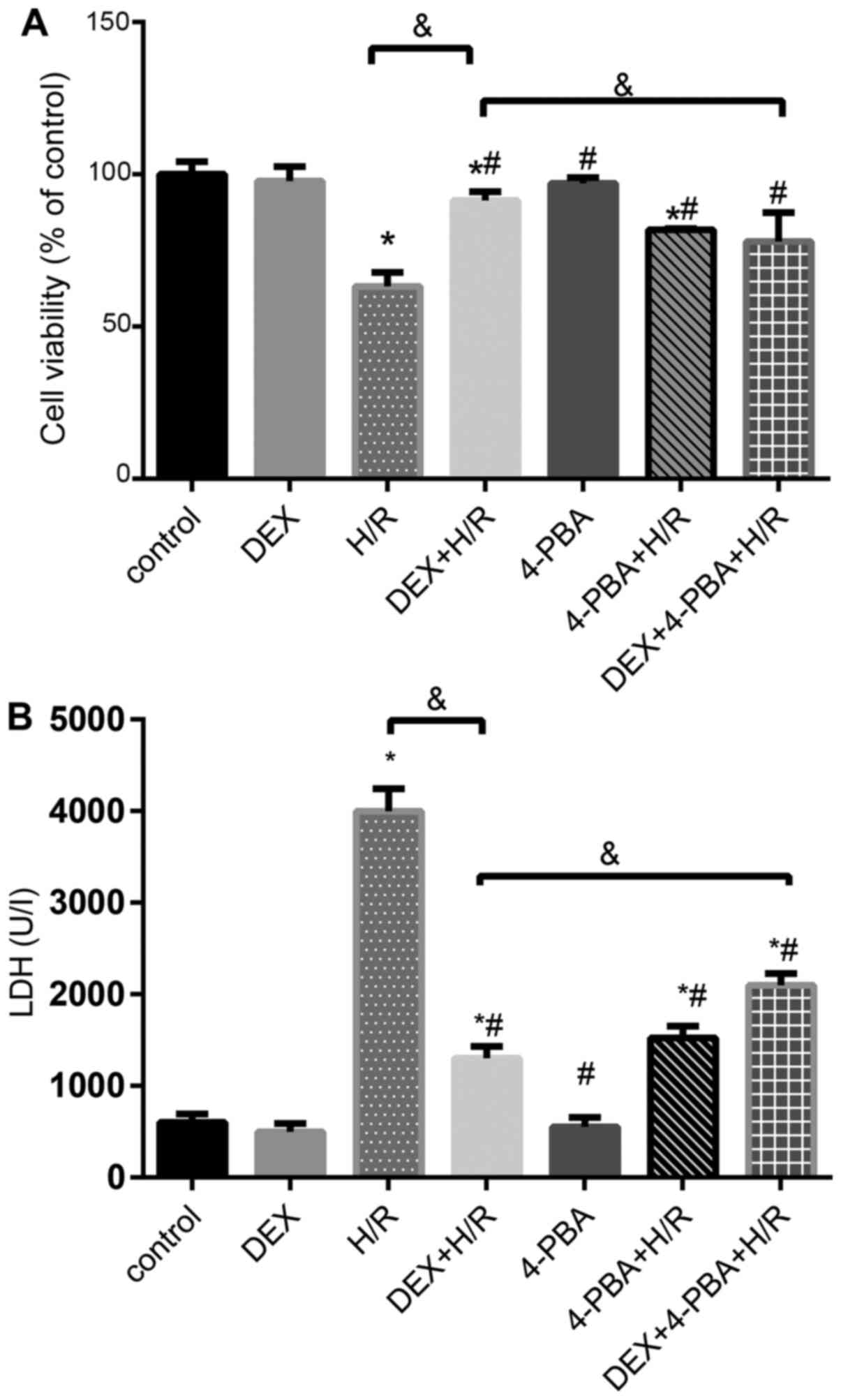 | Figure 4Effects of DEX on the levels of
myocardial cell injury induced by H/R under different conditions.
(A) Cell viability percentages and (B) LDH release. Results are
expressed as the mean ± standard deviation. *P<0.05
vs. Con; #P<0.05 vs. the H/R group;
&P<0.05 vs. the DEX + H/R group. Con, control
group; 1, normoxic incubation with DEX; 2, H/R incubation; 3, H/R
incubation with DEX; 4, normoxic incubation with 4-PBA; 5, H/R
incubation with 4-PBA; 6, H/R incubation with DEX and 4-PBA; DEX,
dexmedetomidine; H/R, hypoxia/reoxygenation; LDH, lactate
dehydrogenase. |
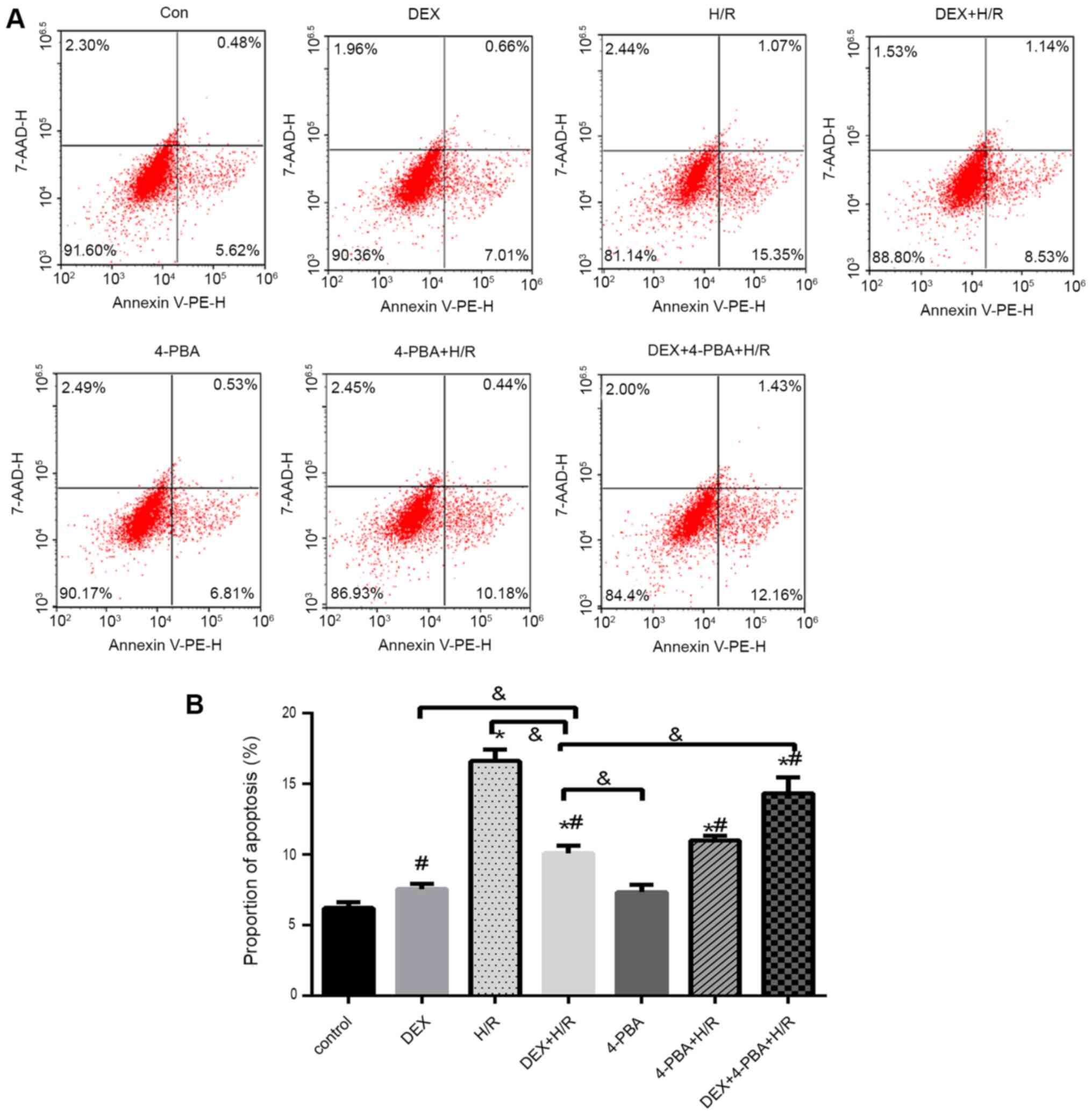 | Figure 5H9c2 cardiomyocyte apoptosis under
different conditions. (A) Representative flow cytometry plots and
(B) percentages of apoptotic H9c2 cardiomyocytes. Results are
expressed as the mean ± standard deviation. *P<0.05
vs. Con; #P<0.05 vs. the H/R group;
&P<0.05 vs. the DEX+H/R group. Con, control
group; 1, normoxic incubation with DEX; 2, H/R incubation; 3, H/R
incubation with DEX; 4, normoxic incubation with 4-PBA; 5, H/R
incubation with 4-PBA; 6, H/R incubation with DEX and 4-PBA; DEX,
dexmedetomidine; H/R, hypoxia/reoxygenation; 4-PBA, 4-phenylbutyric
acid. |
In the cell apoptosis assay, the percentage of
apoptotic cells increased by 10% in the H/R group (mean, 16%)
compared with the control group (mean, 6%) (Fig. 5). However, apoptosis was reduced in
the DEX + H/R (mean, 9%) and 4-PBA + H/R (mean, 10%) pretreatment
groups, which was significantly attenuated by 7 and 6%,
respectively, compared with that in the H/R group (P<0.05).
Additionally, 4-PBA reversed the anti-apoptotic effect of DEX and
resulted in a significant increase of ~4% in the 4-PBA + DEX + H/R
group (mean, 13%) (Fig. 5).
DEX reduces the expression levels of
GRP78, CHOP and caspase-12 in H9c2 cells after H/R
To investigate the role of DEX, three molecules
mediating ERS and the associated signaling pathways were examined
(Fig. 6). Compared with the control
group, the H/R group exhibited the marked and significant
upregulation of indicators of ERS and apoptosis. Pretreatment with
either DEX or 4-PBA significantly reduced the expression of GRP78,
CHOP and caspase-12 in response to H/R (P<0.05). Moreover, these
reductions, which are indicative of the alleviation of ERS and
apoptosis, observed in the DEX + H/R group were significantly
attenuated by 4-PBA, as seen in the DEX + H/R + 4-PBA group.
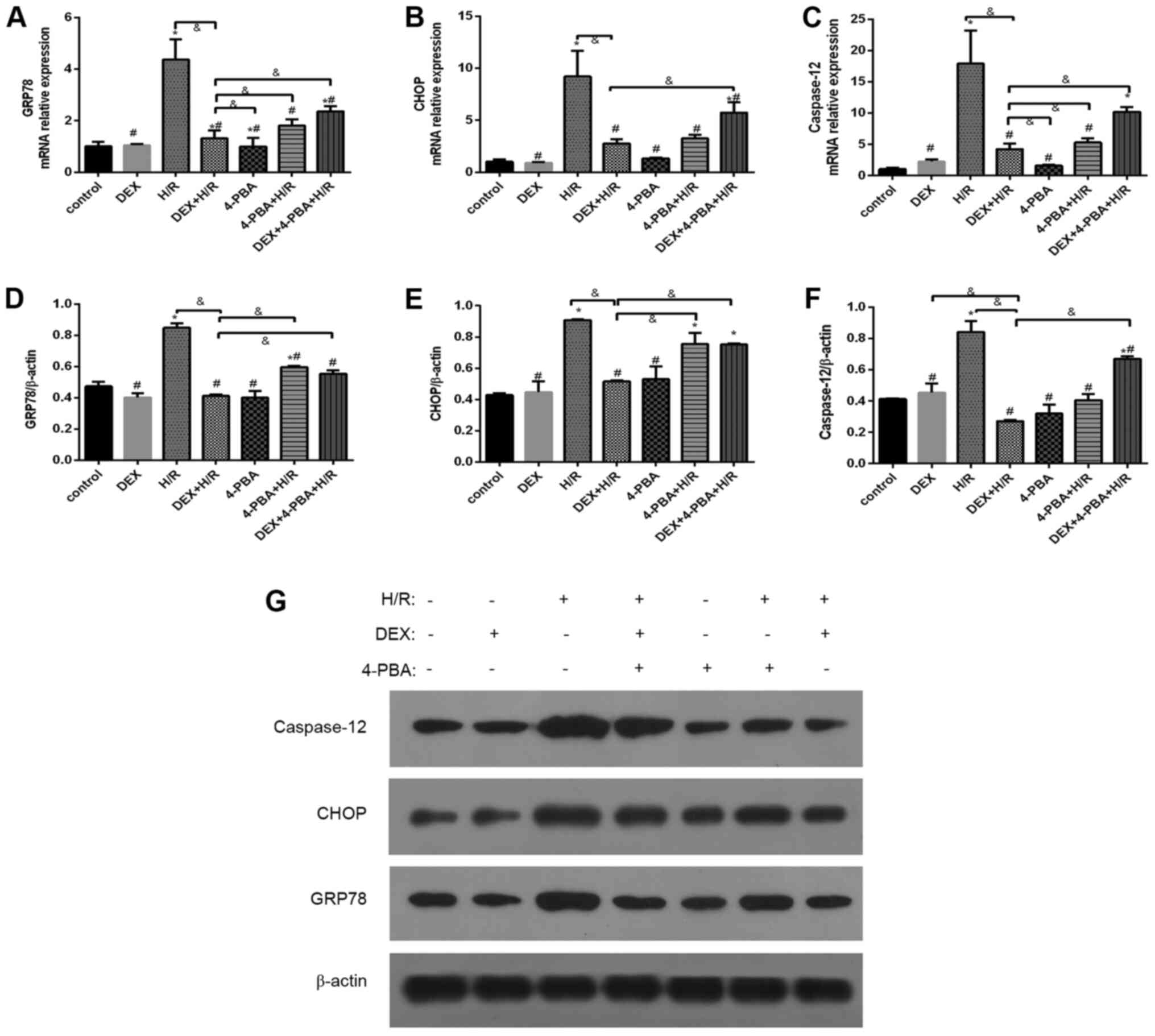 | Figure 6Effects of DEX on the expression of
GRP78, CHOP and caspase-12 in myocardial cells treated under
different conditions. mRNA expression levels of (A) GRP78, (B) CHOP
and (C) caspase-12, and protein expression levels of (D) GRP78, (E)
CHOP and (F) caspase-12. (G) Representative western blots. Results
are expressed as the mean ± standard deviation.
*P<0.05 vs. Con; #P<0.05 vs. the H/R
group; &P<0.05 vs. the DEX+H/R group. Con,
control group; 1, normoxic incubation with DEX; 2, H/R incubation;
3, H/R incubation with dexmedetomidine; 4, normoxic incubation with
4-PBA; 5, H/R incubation with 4-PBA; 6, H/R incubation with DEX and
4-PBA; DEX, dexmedetomidine; GRP78, glucose-regulated protein 78;
CHOP, C/EBP homologous protein; H/R, hypoxia/reoxygenation; 4-PBA,
4-phenylbutyric acid. |
Discussion
DEX has been demonstrated to exert protective
effects on the myocardium under IRI or H/R conditions. The present
study aimed to determine whether DEX treatment affects ERS in the
complex state of H/R and to identify the optimal experimental
conditions for analyzing this. The results confirmed that DEX
attenuated H/R-induced myocardial damage through the downregulation
of several key molecules associated with ERS and apoptosis, and
indicated that its cardioprotective effects might be connected with
the regulation of ERS.
As a common clinical phenomenon, MIRI is associated
with a variety of processes. To date, no clinical therapy has been
effective in ameliorating MIRI, including therapies that were
successful in experimental studies, such as cyclosporine A
(34). The limitations of animal
experiments, with the exception of those in large animals, underlie
this discrepancy due to numerous differences between animal and
human cardiac physiology. Further clarification of the protective
mechanisms of numerous approaches is necessary (35), and this could be accomplished by the
establishment of well-designed in vitro models with isolated
cardiomyocytes that allow the independent control of external
factors (36). Thus, it is crucial
to use validated in vitro models to draw conclusions and
clarify the important mechanisms.
Among previous studies, the durations of hypoxia and
reoxygenation used vary, which has led to disagreement regarding
the experimental strategy. In addition, the composition of the
culture medium, including the nutrients, extracellular pH and
calcium concentration, at the time of reoxygenation is a major
factor that requires consideration (36). A previous study indicated that 30
min is sufficient to establish IRI in animal experiments (37); however, no consistent duration has
been established for cardiomyocytes because of their different
levels of maturity and oxygen dependency based on the cell source
used. For example, Xie et al (38) determined that an ischemic period
ranging from 2 to 5 h was optimal in an I/R model of primary adult
rat ventricular myocytes.
The H9c2 cell line, as an immortalized cell line, is
currently considered to be the most suitable cardiomyocyte line for
IRI and toxicity experiments if no cellular contraction is
necessary (39,40). Additionally, the European Society of
Cardiology Working Group Cellular Biology of the Heart Position
Paper has clarified that the optimal duration for combined ischemic
and reperfusion is that which results in 50% cell death (41) but is not too long to affect the
possible intervention effect.
Based on all of the above considerations, H9c2 cells
were used in the present study to conduct experiments with exposure
to hypoxia for 3 h, a duration that has previously been used by
other researchers (31,42), and several reoxygenation durations
were evaluated. A cell death rate of ~50% was achieved with 3 h of
reoxygenation, although cell death peaked at 6 h, with a reduction
of 64%.
A number of studies have been conducted to test the
cardioprotective function of DEX in the context of pretreatment or
postconditioning (1,6,24,32).
Several animal studies have identified that DEX exerts a protective
effect on the myocardium by reducing ERS after myocardial IRI or by
regulating myocardial apoptosis, which proceeds via intrinsic and
extrinsic apoptotic pathways (1,6,24,43).
To further understand the underlying mechanism of IRI, some
cell-level experiments (17,31,32,44)
have been carried out with different cardiomyocytes or H/R
protocols in which DEX was infused 1-2 h prior to the H/R
procedure, as in the present study. With the exception of Wang
et al (31) and Yuan et
al (32), who reoxygenated
cells for 2-3 h, all other researchers cultured cells in
high-glucose medium for >12 h after hypoxia, which is much
longer than the duration of reoxygenation used in the present
study. From the aforementioned studies of IRI mechanism, it was
concluded that DEX affects, for example, calcium overload, small
non-coding RNAs and inflammation.
In the present research, a wide range of
concentrations of DEX (1 nM to 10 µM) was adopted for pretreatment
to determine the optimal concentration, and the effect of DEX on
certain indicators of ERS, namely GRP78, CHOP and caspase-12, was
tested. To the best of our knowledge, this study is the first to
examine the protective function of DEX over such a broad range of
concentrations. According to a study by Peng et al (44), concentrations of DEX >30 µM can
reduce the viability of cardiomyocytes and induce cytotoxicity.
Thus, 10 µM was selected as the highest concentration of DEX to
study. The results indicated that DEX was not cytotoxic to H9c2
cells at any concentration ≤10 µM; indeed, a significant increase
in cell proliferation was observed at the higher concentrations of
1, 5 and 10 µM. Furthermore, 1 µM DEX attenuated the H/R-induced
injury of H9c2 cells. This result is similar to that of other
studies conducted by Yuan et al (32) and Gao et al (17), who used the same dose of DEX but
examined the involvement of non-ERS pathways.
MIRI can lead to severe ERS, which is associated
with GRP78 upregulation (45,46).
If ERS increases, apoptotic cascades are considered an underlying
mechanism of MIRI, and the transcription of specific molecules,
such as CHOP and caspase-12, is upregulated in an ERS-dependent
manner. Most researchers concur that these molecules, as downstream
markers of the ERS signaling pathway, are able to represent the ERS
status and even the developmental direction of cell survival
(47-49).
In the present in vitro study, these three
molecules were highly expressed under H/R conditions. However, the
increases in their expression levels were strongly reduced by DEX,
and 4-PBA attenuated this effect of DEX. These findings indicate
that DEX attenuates ERS-associated apoptosis and regulates ERS via
an unknown mechanism that is inhibited by 4-PBA, as reflected by the
decreased expression of CHOP, GRP78 and caspase-12 at the mRNA and
protein levels. Consistent results were observed in a study by Liu
et al (26), in which DEX
intervention led to a significant reduction in the expression level
of GRP78, a marker of ERS, and to ER-phagy, while treatment with
4-PBA successfully elevated the expression of GRP78. In a study by
Liu et al (30), DEX exerted
a similar effect in an animal model of cerebral
ischemia-reperfusion injury, decreasing the levels of CHOP and
GRP78. Additionally, in the present study, DEX exerted stronger
protective effects against ERS-related apoptosis than can be
accounted for by the inhibition of ERS in the DEX + H/R + 4-PBA
group compared with the H/R+4-PBA group, which exhibited lower
expression levels of CHOP, GRP78 and caspase-12. It is possible
that DEX intervenes in mitochondria-dependent or death
receptor-dependent apoptosis in addition to the ERS-associated
apoptotic signaling pathway. Considering this possibility, the
findings of the present study are compatible with those of Davidson
et al (18), who found that
both higher and lower concentrations of DEX might perform multiple
functions to alleviate IRI. Future studies should be conducted to
focus on other functions of DEX potentially involved in this
process.
Undoubtedly, several limitations of the present
study require consideration. First, this study was a pretreatment
experiment that did not establish a connection with any signaling
pathway, which is a clear direction for future fundamental
research. Second, the purpose of any in vitro H/R model is
to mimic clinical IRI, and condition-dependent models may vary.
Furthermore, human cells or stem cells that can more accurately
reflect the clinical scenario were not investigated in the present
study. Therefore, it is necessary to test the hypothesis of the
present study using different types of cardiomyocytes, such as
primary cells. Finally, the study investigated only the effects of
DEX pretreatment, and the effects of post-event treatments as
applied in clinical trials were not investigated. In addition, the
precise mechanisms identified in this study merit further
investigation.
In conclusion, 1 µM DEX was confirmed at the
cellular level to protect H9c2 cells against injury induced by 3 h
of hypoxia and 3 h of reoxygenation. Furthermore, these results
indicate that the effects of DEX were mediated via intervention
with ERS and subsequent apoptosis.
Acknowledgements
Not applicable.
Funding
This research was supported by the Zhejiang
Provincial Natural Science Foundation of China (grant no.
2017C33185) and the Project of the Health Commission of Zhejiang
Province (grant no. 2019ZD053).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZZ designed the protocol and prepared and revised
the manuscript. XL acquired the data and revised the manuscript. HZ
performed experiments and analyzed the data. CZ interpreted the
data and plotted the graphs. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bunte S, Behmenburg F, Majewski N,
Stroethoff M, Raupach A, Mathes A, Heinen A, Hollmann MW and Huhn
R: Characteristics of dexmedetomidine postconditioning in the field
of myocardial ischemia-reperfusion injury. Anesth Analg. 130:90–98.
2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Li L, Li X, Zhang Z, Liu L, Zhou Y and Liu
F: Protective mechanism and clinical application of hydrogen in
myocardial ischemia-reperfusion injury. Pak J Biol Sci. 23:103–112.
2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Xi J, Li QQ, Li BQ and Li N: miR-155
inhibition represents a potential valuable regulator in mitigating
myocardial hypoxia/reoxygenation injury through targeting BAG5 and
MAPK/JNK signaling. Mol Med Rep. 21:1011–1020. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Li J, Zhou W, Chen W, Wang H, Zhang Y and
Yu T: Mechanism of the hypoxia inducible factor 1/hypoxic response
element pathway in rat myocardial ischemia/diazoxide
post-conditioning. Mol Med Rep. 21:1527–1536. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kitazume-Taneike R, Taneike M, Omiya S,
Misaka T, Nishida K, Yamaguchi O, Akira S, Shattock MJ, Sakata Y
and Otsu K: Ablation of Toll-like receptor 9 attenuates myocardial
ischemia/reperfusion injury in mice. Biochem Biophys Res Commun.
515:442–447. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Li J, Zhao Y, Zhou N, Li L and Li K:
Dexmedetomidine attenuates myocardial ischemia-reperfusion injury
in diabetes mellitus by inhibiting endoplasmic reticulum stress. J
Diabetes Res. 2019(7869318)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Heusch G and Gersh BJ: The pathophysiology
of acute myocardial infarction and strategies of protection beyond
reperfusion: A continual challenge. Eur Heart J. 38:774–784.
2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Heusch G: Cardioprotection research must
leave its comfort zone. Eur Heart J. 39:3393–3395. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Heusch G: Critical issues for the
translation of cardioprotection. Circ Res. 120:1477–1486.
2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wu H, Ye M, Yang J and Ding J: Endoplasmic
reticulum stress-induced apoptosis: A possible role in myocardial
ischemia-reperfusion injury. Int J Cardiol. 208:65–66.
2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Li W, Li W, Leng Y, Xiong Y and Xia Z:
Ferroptosis is involved in diabetes myocardial ischemia/reperfusion
injury through endoplasmic reticulum stress. DNA Cell Biol.
39:210–225. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Gao J, Guo Y, Liu Y, Yan J, Zhou J, An X
and Su P: Protective effect of FBXL10 in myocardial ischemia
reperfusion injury via inhibiting endoplasmic reticulum stress.
Respir Med. 161(105852)2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Guo C, Zhang J, Zhang P, Si A, Zhang Z,
Zhao L, Lv F and Zhao G: Ginkgolide B ameliorates myocardial
ischemia reperfusion injury in rats via inhibiting endoplasmic
reticulum stress. Drug Des Devel Ther. 13:767–774. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wang X, Yuan B, Cheng B, Liu Y, Zhang B,
Wang X, Lin X, Yang B and Gong G: Crocin alleviates myocardial
ischemia/reperfusion-induced endoplasmic reticulum stress via
regulation of miR-34a/Sirt1/Nrf2 pathway. Shock. 51:123–130.
2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hou X, Fu M, Cheng B, Kang Y and Xie D:
Galanthamine improves myocardial ischemia-reperfusion-induced
cardiac dysfunction, endoplasmic reticulum stress-related
apoptosis, and myocardial fibrosis by suppressing AMPK/Nrf2 pathway
in rats. Ann Transl Med. 7(634)2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhang BF, Jiang H, Chen J, Guo X, Li Y, Hu
Q and Yang S: Nobiletin ameliorates myocardial ischemia and
reperfusion injury by attenuating endoplasmic reticulum
stress-associated apoptosis through regulation of the PI3K/AKT
signal pathway. Int Immunopharmacol. 73:98–107. 2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Gao JM, Meng XW, Zhang J, Chen WR, Xia F,
Peng K and Ji FH: Dexmedetomidine protects cardiomyocytes against
hypoxia/reoxygenation injury by suppressing TLR4-MyD88-NF-κB
signaling. Biomed Res Int. 2017(1674613)2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Davidson SM, Ferdinandy P, Andreadou I,
Bøtker HE, Heusch G, Ibáñez B, Ovize M, Schulz R, Yellon DM,
Hausenloy DJ, et al: Multitarget strategies to reduce myocardial
ischemia/reperfusion injury: JACC review topic of the week. J Am
Coll Cardiol. 73:89–99. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kong Q, Wu X, Qiu Z, Huang Q, Xia Z and
Song X: Protective effect of dexmedetomidine on acute lung injury
via the upregulation of tumour necrosis factor-α-induced
protein-8-like 2 in septic mice. Inflammation. 43:833–846.
2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhang Y, Liu M, Yang Y, Cao J and Mi W:
Dexmedetomidine exerts a protective effect on ischemia-reperfusion
injury after hepatectomy: A prospective, randomized, controlled
study. J Clin Anesth. 61(109631)2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Xiong J, Quan J, Qin C, Wang X, Dong Q and
Zhang B: Dexmedetomidine exerts brain-protective effects under
cardiopulmonary bypass through inhibiting the janus kinase 2/signal
transducers and activators of transcription 3 pathway. J Interferon
Cytokine Res. 40:116–124. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Gong J, Zhang R, Shen L, Xie Y and Li X:
The brain protective effect of dexmedetomidine during surgery for
paediatric patients with congenital heart disease. J Int Med Res.
47:1677–1684. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Oh JE, Jun JH, Hwang HJ, Shin EJ, Oh YJ
and Choi YS: Dexmedetomidine restores autophagy and cardiac
dysfunction in rats with streptozotocin-induced diabetes mellitus.
Acta Diabetol. 56:105–114. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
He L, Hao S, Wang Y, Yang W, Liu L, Chen H
and Qian J: Dexmedetomidine preconditioning attenuates
ischemia/reperfusion injury in isolated rat hearts with endothelial
dysfunction. Biomed Pharmacother. 114(108837)2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Riquelme JA, Westermeier F, Hall AR,
Vicencio JM, Pedrozo Z, Ibacache M, Fuenzalida B, Sobrevia L,
Davidson SM, Yellon DM, et al: Dexmedetomidine protects the heart
against ischemia-reperfusion injury by an endothelial eNOS/NO
dependent mechanism. Pharmacol Res. 103:318–327. 2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Liu Y, Wang S, Wang Z, Ding M, Li X, Guo
J, Han G and Zhao P: Dexmedetomidine alleviated endoplasmic
reticulum stress via inducing ER-phagy in the spinal cord of
neuropathic pain model. Front Neurosci. 14(90)2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Chai Y, Zhu K, Li C, Wang X, Shen J, Yong
F and Jia H: Dexmedetomidine alleviates cisplatin-induced acute
kidney injury by attenuating endoplasmic reticulum stress-induced
apoptosis via the α2AR/PI3K/AKT pathway. Mol Med Rep. 21:1597–1605.
2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Sun D, Wang J, Liu X, Fan Y, Yang M and
Zhang J: Dexmedetomidine attenuates endoplasmic reticulum
stress-induced apoptosis and improves neuronal function after
traumatic brain injury in mice. Brain Res.
1732(146682)2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhao L, Zhai M, Yang X, Guo H, Cao Y, Wang
D, Li P and Liu C: Dexmedetomidine attenuates neuronal injury after
spinal cord ischaemia-reperfusion injury by targeting the
CNPY2-endoplasmic reticulum stress signalling. J Cell Mol Med.
23:8173–8183. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Liu C, Fu Q, Mu R, Wang F, Zhou C, Zhang
L, Yu B, Zhang Y, Fang T and Tian F: Dexmedetomidine alleviates
cerebral ischemia-reperfusion injury by inhibiting endoplasmic
reticulum stress dependent apoptosis through the
PERK-CHOP-Caspase-11 pathway. Brain Res. 1701:246–254.
2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Wang Z, Yang Y, Xiong W, Zhou R, Song N,
Liu L and Qian J: Dexmedetomidine protects H9C2 against
hypoxia/reoxygenation injury through miR-208b-3p/Med13/Wnt
signaling pathway axis. Biomed Pharmacother.
125(110001)2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yuan M, Meng XW, Ma J, Liu H, Song SY,
Chen QC, Liu HY, Zhang J, Song N, Ji FH and Peng K: Dexmedetomidine
protects H9c2 cardiomyocytes against oxygen-glucose
deprivation/reoxygenation-induced intracellular calcium overload
and apoptosis through regulating FKBP12.6/RyR2 signaling. Drug Des
Devel Ther. 13:3137–3149. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Cung TT, Morel O, Cayla G, Rioufol G,
Garcia-Dorado D, Angoulvant D, Bonnefoy-Cudraz E, Guérin P, Elbaz
M, Delarche N, et al: Cyclosporine before PCI in patients with
acute myocardial infarction. N Engl J Med. 373:1021–1031.
2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Rossello X and Yellon DM:
Cardioprotection: The disconnect between bench and bedside.
Circulation. 134:574–575. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Chen T and Vunjak-Novakovic G: In vitro
models of ischemia-reperfusion injury. Regen Eng Transl Med.
4:142–153. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
He X, Li S, Liu B, Susperreguy S, Formoso
K, Yao J, Kang J, Shi A, Birnbaumer L and Liao Y: Major
contribution of the 3/6/7 class of TRPC channels to myocardial
ischemia/reperfusion and cellular hypoxia/reoxygenation injuries.
Proc Natl Acad Sci USA. 114:E4582–E4591. 2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Xie M, Kong Y, Tan W, May H, Battiprolu
PK, Pedrozo Z, Wang ZV, Morales C, Luo X, Cho G, et al: Histone
deacetylase inhibition blunts ischemia/reperfusion injury by
inducing cardiomyocyte autophagy. Circulation. 129:1139–1151.
2014.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Oh JG, Kho C, Hajjar RJ and Ishikawa K:
Experimental models of cardiac physiology and pathology. Heart Fail
Rev. 24:601–615. 2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Kuznetsov AV, Javadov S, Sickinger S,
Frotschnig S and Grimm M: H9c2 and HL-1 cells demonstrate distinct
features of energy metabolism, mitochondrial function and
sensitivity to hypoxia-reoxygenation. Biochim Biophys Acta.
1853:276–284. 2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Lecour S, Bøtker HE, Condorelli G,
Davidson SM, Garcia-Dorado D, Engel FB, Ferdinandy P, Heusch G,
Madonna R, Ovize M, et al: ESC working group cellular biology of
the heart: Position paper: Improving the preclinical assessment of
novel cardioprotective therapies. Cardiovasc Res. 104:399–411.
2014.PubMed/NCBI View Article : Google Scholar
|
|
42
|
He S, Wang X, Zhong Y, Tang L, Zhang Y,
Ling Y, Tan Z, Yang P and Chen A: Hesperetin post-treatment
prevents rat cardiomyocytes from hypoxia/reoxygenation injury in
vitro via activating PI3K/Akt signaling pathway. Biomed
Pharmacother. 91:1106–1112. 2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Yang YF, Peng K, Liu H, Meng XW, Zhang JJ
and Ji FH: Dexmedetomidine preconditioning for myocardial
protection in ischaemia-reperfusion injury in rats by
downregulation of the high mobility group box 1-toll-like receptor
4-nuclear factor κB signalling pathway. Clin Exp Pharmacol Physiol.
44:353–361. 2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Peng K, Qiu Y, Li J, Zhang ZC and Ji FH:
Dexmedetomidine attenuates hypoxia/reoxygenation injury in primary
neonatal rat cardiomyocytes. Exp Ther Med. 14:689–695.
2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Wang R, Yang M, Wang M, Liu X, Xu H, Xu X,
Sun G and Sun X: Total saponins of aralia elata (Miq) seem
alleviate calcium homeostasis imbalance and endoplasmic reticulum
stress-related apoptosis induced by myocardial ischemia/reperfusion
injury. Cell Physiol Biochem. 50:28–40. 2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Bi X, Zhang G, Wang X, Nguyen C, May HI,
Li X, Al-Hashimi AA, Austin RC, Gillette TG, Fu G, et al:
Endoplasmic reticulum chaperone GRP78 protects heart from
ischemia/reperfusion injury through Akt activation. Circ Res.
122:1545–1554. 2018.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Li H, Chen H, Li R, Xin J, Wu S, Lan J,
Xue K, Li X, Zuo C, Jiang W and Zhu L: Cucurbitacin I induces
cancer cell death through the endoplasmic reticulum stress pathway.
J Cell Biochem, Sep 11, 2018 (Online ahead of print).
|
|
48
|
Huang ZH, Zhang SX, Wang C, Zhao R, Qiao
J, Bai WQ, Lu JF, Lu XQ and Zhang HH: Downregulated long non-coding
RNA FOXD3-AS1 promotes endoplasmic reticulum stress-induced
apoptosis by inhibiting RCN1 via let-7e-5p in nasopharyngeal
carcinoma. Am J Physiol Cell Physiol. 319(C455)2020.
|
|
49
|
Chen J, Chen J, Cheng Y, Fu Y, Zhao H,
Tang M, Zhao H, Lin N, Shi X, Lei Y, et al: Mesenchymal stem
cell-derived exosomes protect beta cells against hypoxia-induced
apoptosis via miR-21 by alleviating ER stress and inhibiting p38
MAPK phosphorylation. Stem Cell Res Ther. 11(97)2020.PubMed/NCBI View Article : Google Scholar
|