Introduction
Ulcerative colitis (UC) is a non-specific, chronic,
relapsing inflammatory disorder of the colonic mucosa, which
affects the rectum and colon, and its incidence is rising worldwide
(1,2). The underlying causes of UC are complex
and have not been fully elucidated. The most accepted view is that
UC is a complex disease resulting from interactions between genes,
the gut flora, host immune system and environmental factors
(3,4). Currently, inappropriate activation of
T cells has been deemed as a crucial factor that contributes to the
pathogenesis of UC (5). The
imbalance of the immune axis formed by T helper 17 (Th17) cells
that contribute to the immune response and regulatory T cells
(Tregs) that mediate immune tolerance may play a main role in the
pathogenesis of UC (6).
Th17 cells exert a pro-inflammatory effect on the
inflammatory reaction by secreting pro-inflammatory cytokines, such
as IL-17; their excessive activation causes intestinal inflammation
and damages the intestinal mucosa (7). Tregs have immunosuppressive functions
in autoimmune diseases, regulate self-tolerance and limit excessive
immune reaction (8). An increasing
number of studies have indicated that the balance of Th17 cell and
Treg function is essential for host immunity and immune tolerance
(9,10). During the transformation of initial
T cells into Th17 cells and Tregs, the JAK/STAT pathway,
particularly STAT3, plays an important role in promoting this
transformation (11). Moreover,
some cytokines and transcription factors are essential; IL-6
signaling and TGF-β1 act synergistically to program Th17-related
genes through STAT3, thereby inducing Th17 cell development
(12). The vital transcription
factor mediating Th17 cell differentiation is retinoic acid-related
orphan receptor (RORγt). The biological function of Tregs is
controlled by the expression of the transcription factor forkhead
box protein P3 (FOXP3) (5,13). An increase in the number of Th17
cells in UC has been reported to lead to an increase in serum IL-17
levels, and the reduction of Tregs leads to weakness of
anti-inflammatory function (14).
Therefore, promoting Tregs and suppressing Th17 cells to regulate
Th17/Treg cell balance may be an efficient strategy for the
treatment of UC.
PolyADP-ribose polymerase-1 (PARP)-1 is a ribozyme
with significant biological activity in eukaryotic cells (15). It can catalyze the polyADP
ribosylation of DNA-binding proteins involved in surveillance and
genomic integrity maintenance (16). A number of studies have demonstrated
that PARP-1 can regulate the inflammatory response (17,18).
In particular, PARP-1 can regulate and enhance NF-κB
transcriptional activity (18).
Therefore, the inhibition of PARPs has been extensively studied; in
several acute models of kidney injury and organ transplantation,
PARP-1 knockout animals or pharmacological inhibitors of PARP-1
have been shown to lead to reduced inflammatory response (19,20).
Although PARP-1 does not participate in the differentiation of
natural T cells into Th17 cells, it does affect the development of
Tregs (21). It has been
demonstrated that PARP-1 negatively regulates Treg function through
FOXP3 poly(ADP-ribosyl) (22).
5-aminoisoquinolinone (5-AIQ), a water-soluble PARP-1 inhibitor,
has been demonstrated to provide important protection against
multiple forms of tissue injury induced by reperfusion injury, the
inflammatory response and neurotoxicity (23). In addition, the pharmacological
inhibition of PARP-1 by 5-AIQ has been shown to inhibit NF-κB
activity with the subsequent downregulation of the expression of
several gene products (24).
Therefore, the present study aimed to examine the regulatory
effects of 5-AIQ on Th17/Tregs in experimental colitis and to
elucidate the potential mechanisms involved.
Materials and methods
Pharmacological compounds and
reagents
The water-soluble compound 5-AIQ, was obtained from
Matrix Science, Inc. Dextran sulfate sodium (DSS) was purchased
from MP Biomedicals, LLC (molecular weight, 36-50 kDa). The
following antibodies were purchased from BD Biosciences: Anti-CD3
FITC (1:50; cat. no. 561827), anti-CD4 (1:50; cat. no. 566408) and
anti-CD25 (1:50; cat. no. 561065) phycoerythrin, anti-Foxp3 (1:50;
cat. no. 560402) and anti-IL-17A (1:50; cat. no. 560224)
allophycocyanin.
Animals
A total of 30 C57BL/6 mice (males; 6-8 weeks old;
weighing 20-25 g) were obtained from Beijing Vital River Laboratory
Animal Technology Co., Ltd.. The mice were housed under constant
environmental conditions (12-h light/dark cycle; 21±2˚C) and were
provided with standard laboratory food and water ad libitum.
All experimental procedures were approved by the Ethics Committee
at the Renmin Hospital of Wuhan University.
Experimental design
The mice were randomly divided into three groups
(n=10) following adaptive feeding for 1 week as follows: i) The
control group (control); ii) 3% DSS-induced group (DSS) and iii) 3%
DSS-induced + 5-AIQ group (5-AIQ). Apart from those in the control
group, mice were exposed to 3% DSS for 7 days to develop symptoms
of acute experimental colitis (25). Aside from the mice in the 5-AIQ
group, an intraperitoneal injection of physiological saline was
administered to the remaining mice, and 5-AIQ (1.5 mg/kg) dissolved
in water was injected intraperitoneally into mice in the 5-AIQ
group for 7 days (26). During the
experimental period, the food and water intake and the disease
activity index (DAI), including body weight, stool consistency and
stool occult blood, were evaluated each day for each animal
(Table I).
 | Table IDisease activity index score. |
Table I
Disease activity index score.
| Score | Weight loss | Stool
consistency | Bloody stool |
|---|
| 0 | None | Normal | None |
| 1 | 1-5% | Paste stools | Occult blood |
| 2 | 6-10% | Loose stools | Bleeding |
| 3 | >10% | Diarrhea | Gross bleeding |
Histopathological assessment
Mice were sacrificed by cervical dislocation on the
8th day of colitis induction. Colorectal and ileocecal tissue
sections (thickness, 4 µm) were obtained from the mice and the
length of the colon was measured. Part of the colon was fixed in 4%
paraformaldehyde at 4˚C for 24 h and embedded in paraffin, followed
by hematoxylin and eosin staining for 97 min at room temperature
and observed under a light microscope, (magnification, x100 and
x200). Intestinal inflammation was assessed in a blinded manner and
the histological score was evaluated as described in Table II.
| Table IIHistological score. |
Table II
Histological score.
| Score | Percent of tissue
damage | Extent of tissue
damage | Degree of
inflammation | Extent of crypt
damage |
|---|
| 0 | None | None | None | None |
| 1 | ≤25% | Mucosa | Slight | Basal 1/3 |
| 2 | ≤50% | Mucosa and
submucosa | Moderate | Basal 2/3 |
| 3 | ≤75% | Beyond the
submucosa | Severe | Only the surface
epithelium was intact |
| 4 | 100% | - | - | The entire crypt
and epithelium were lost |
Flow cytometry
The spleen of the mice was aseptically isolated,
filtered with a nylon mesh, and centrifuged for 10 min at 4˚C at
1,500 x g to obtain a single-cell suspension. Cells from the
single-cell suspension were seeded into 96-well plates
(1-3x106 lymphocytes/well) and stimulated for 7 h using
Leukocyte Activation Cocktail (BD Biosciences) in an incubator. The
cells were then collected, stained, fixed and permeabilized
strictly according to the instructions provided with the kit. Flow
cytometry antibodies, including anti-CD3 FITC, anti-CD4 and
anti-CD25 phycoerythrin, were then added to each tube in turn,
followed by mixing and incubation for 35 min at room temperature.
The cells were centrifuged at 1,500 x g for 3 min at 4˚C and the
supernatants were discarded. Fixation/Permeabilization working
solution (1 ml; eBioscience; Thermo Fisher Scientific, Inc.) was
added to each sample before incubation for 30 min in the dark at
room temperature. Subsequently, permeabilization buffer (2 ml;
eBioscience; Thermo Fisher Scientific, Inc.) was added to each
sample before centrifuging again at 400 x g for 5 min at 4˚C.
Intracellular cytokine antibodies anti-Foxp3 and anti-IL-17A
allophycocyanin were added. The solutions were well mixed and
incubated for 30 min in the dark at room temperature. The
proportions of Treg and Th17 cells were analyzed by flow cytometer
(BD Biosciences) and FlowJo 7.0 (FlowJo LLC) software was used to
analyze data.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) for mRNA expression
analysis
First, total RNA was isolated from colon samples
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). After isolation of RNA, total RNA was reverse
transcribed into cDNA using a First Strand cDNA Synthesis kit
(Thermo Fisher Scientific, Inc.) with the following temperature
protocol: 25˚C for 5 min, 42˚C for 60 min, 70˚C for 5 min and 4˚C
for 10 min. Following RT, the target gene was amplified, and
RT-qPCR was performed on the ABI 7500 Real-time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) using SYBR-Green PCR
Master Mix (Thermo Fisher Scientific, Inc.) under the following
thermocycling conditions: Initial denaturation at 95˚C for 10 min,
followed by 40 cycles of 95˚C for 30 sec and annealing/extension at
60˚C for 30 sec. β-actin served as the endogenous control.
Expressions were analyzed using the 2-ΔΔCq method
(27). The sequences of the primers
used are listed in Table III.
| Table IIIPCR primers. |
Table III
PCR primers.
| Name | Primer
sequences |
|---|
| β-actin | F:
5'-CACGATGGAGGGGCCGGACTCATC-3' |
| | R:
5'-TAAAGACCTCTATGCCAACACAGT-3' |
| IL-1β | F:
5'-TCAGGCAGGCAGTATCACTC-3' |
| | R:
5'-AGCTCATATGGGTCCGACAG-3' |
| TNF-α | F:
5'-ACCCTCACACTCACAAACCA-3' |
| | R:
5'-GGCAGAGAGGAGGTTGACTT-3' |
| IL-17 | F:
5'-GAAGGCCCTCAGACTACCTC-3' |
| | R:
5'-CAGCATCTTCTCGACCCTGA-3' |
| IL-10 | F:
5'-GCTGGACAACATACTGCTAACCG-3' |
| | R:
5'-CACAGGGGAGAAATCGATGACAG-3' |
| RORγt | F:
5'-CCTGGGCTCCTCGCCTGACC-3' |
| | R:
5'-TCTCTCTGCCCTCAGCCTTGCC-3' |
| Foxp3 | F:
5'-GAGAAGCTGAGTGCCATGCA-3' |
| | R:
5'-GCCACAGATGAAGCCTTGGT-3' |
| IL-6 | F:
5'-GTTGCCTTCTTGGGACTGAT-3' |
| | R:
5'-ATTAAGCCTCCGACTTGTGA-3' |
| TGF-β1 | F:
5'-GCTGAGCGCTTTTCTGATCCT-3' |
| | R:
5'-GAGTGTGCTGCAGGTAGACA-3' |
Western blot analysis
Protein for western blot analysis was extracted from
colonic tissue using RIPA Pierce™ buffer (Thermo Fisher Scientific,
Inc.) supplemented with protease inhibitor at a final 1X
concentration (Halt™ Phosphatase Inhibitor Cocktail; Thermo Fisher
Scientific, Inc.). Protein concentrations were measured using a BCA
protein assay kit (Thermo Fisher Scientific, Inc.). A total of 20
µg protein/lane were separated by 10% SDS-PAGE and electrophoresis
was performed for 1.5 h prior to protein transfer onto PVDF
membranes (EMD Millipore). Subsequently, membranes were blocked
with TBS-0.1%-Tween-20 (TBS-T) containing 5% skim milk for 2 h at
room temperature. The membranes were then incubated with the
following primary antibodies at 4˚C overnight: Anti-NF-κB p65 (cat.
no. 10745-1-AP; 1:2,000), anti-STAT3 (cat. no. 10253-2-AP;
1:1,000), anti-PARP (cat. no. 66520-1-IG; 1:1,000), anti-Foxp3
(cat. no. 22173-1-AP; 1:1,000; all, ProteinTech Group, Inc.),
anti-phosphorylated (p)-STAT3 (cat. no. AF3293; 1:500; Affinity
Biosciences, Inc.), anti-phosphorylated (p)-NF-κB p65 (cat. no.
3033; 1:1,000; Cell Signaling Technology, Inc.), anti-RORγt (cat.
no. bs-23110; 1:1,000), anti-IκB-α (cat. no. bs-1287; 1:1,000; all
from BIOSS), anti-GAPDH (cat. no. AB-P-R001; 1:1,000; Hangzhou
Goodhere Biotech Co., Ltd.). After washing five times with TBS-T
for 5 min each, membranes were further immunoblotted with an
horseradish peroxidase-conjugated AffiniPure goat anti-rabbit IgG
(cat. no. BA1054; 1:50,000; Boster Biological Technology) secondary
antibodies for 2 h at 37˚C. Membranes were then washed in TBS-T and
signals were detected using BandScan v5.0 software (Glyko
Biomedical Ltd.).
Statistical analysis
Data are presented as the mean ± SD and analyzed
using SPSS 20.0 software (IBM Corp.). One-way ANOVA followed by the
Tukey-Kramer test was used for comparisons between groups.
P<0.05 were considered to indicate a statistically significant
difference.
Results
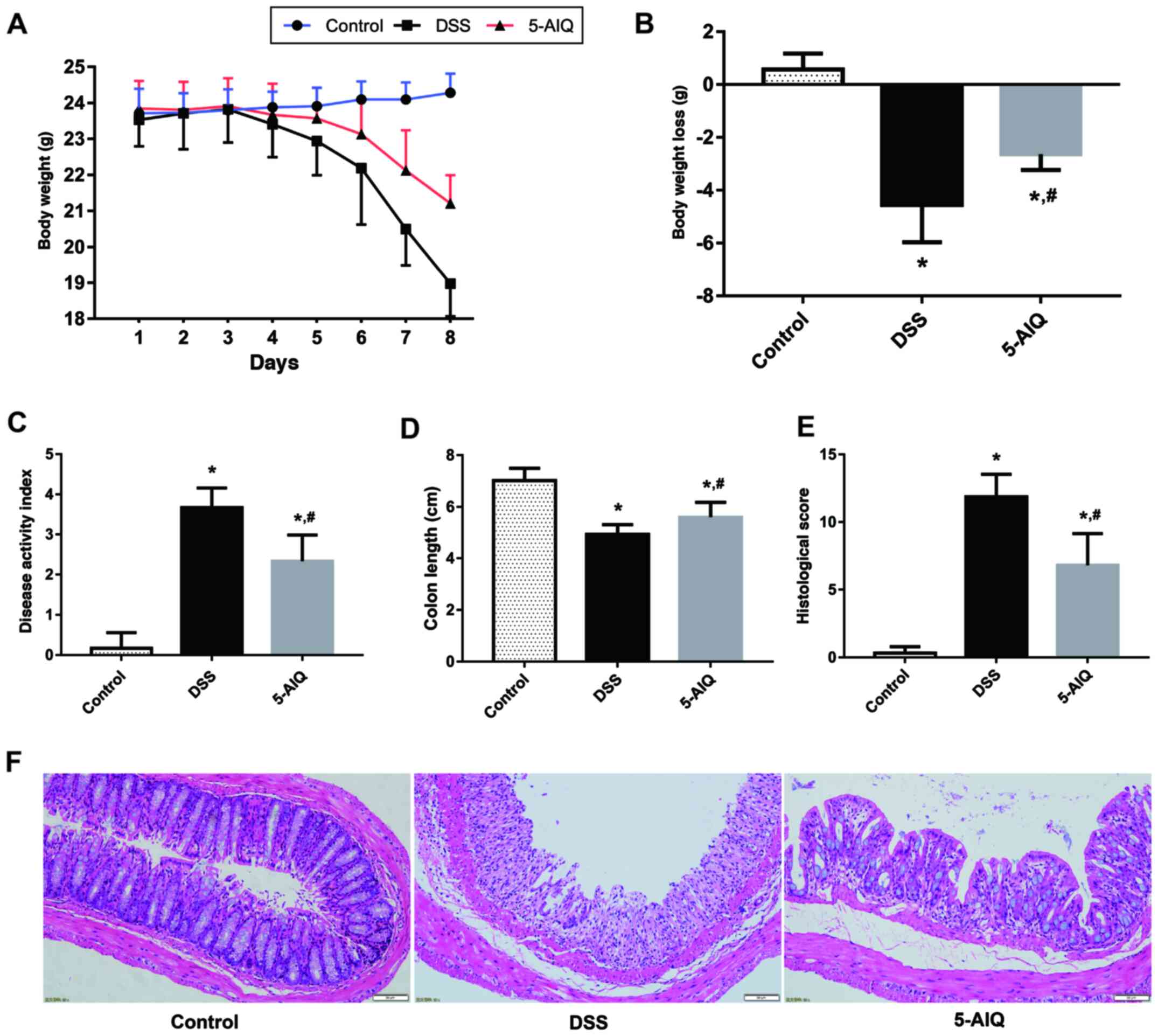
Protective effects of 5-AIQ against
DSS-induced colitis
The DAI score of mice with DSS-induced UC was
significantly higher compared with the control group. Mice with
DSS-induced UC that receiving 5-AIQ treatment exhibited
significantly lower body weight loss and DAI scores compared with
untreated mice with DSS-induced UC (Fig. 1A-C). Moreover, the colon length is a
useful index to reflect the severity of inflammation. The colon
length of the mice in the DSS group was significantly lower
compared with controls and this reduction was alleviated by the
administration of 5-AIQ (Fig.
1D).
5-AIQ attenuates histological damage
in mice with DSS-induced colitis
Colonic mucosal epithelial cells in the control
group exhibited an intact structure, normal and neatly arranged
lamina propria glands and normal crypts. By contrast, the mucosa of
the mice in the DSS group exhibited evident acute inflammatory
reaction, which was characterized by the infiltration of
neutrophils and lymphocytes edema, erosion and ulcers. However, the
damage to the colonic mucosa was alleviated following 5-AIQ
treatment (Fig. 1F). Treatment with
5-AIQ significantly lowered the histological score compared with
the DSS group (Fig. 1E).
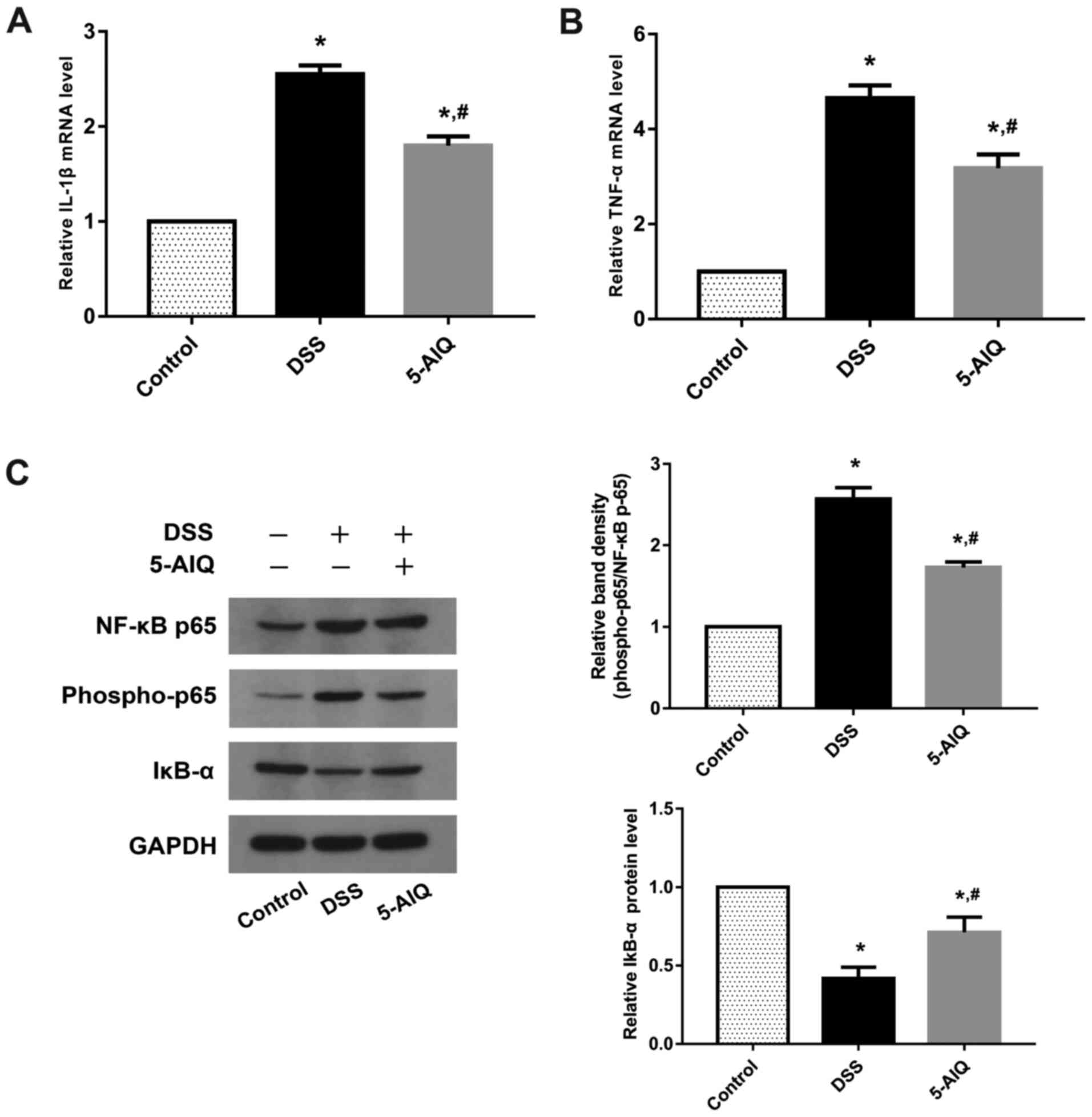
5-AIQ ameliorates the inflammatory
response in mice DSS-induced colitis
To evaluate the protective effects of 5-AIQ, RT-qPCR
analysis of pro-inflammatory cytokines, such as IL-1 and TNF-α, was
performed. Mice exposed to DSS exhibited significantly higher
levels of TNF-α and IL-1β compared with controls. By contrast,
5-AIQ treatment significantly attenuated the expression of these
cytokines (Fig. 2A and B). The levels of NF-κB p65, phosphorylated
(p)-NF-κB p65 and IκB-α was further investigated, as NF-κB p65
regulates the production of pro-inflammatory cytokines. Western
blot analysis demonstrated that 5-AIQ inhibited p-NF-κB p65
expression and suppressed the degradation of IκB-α. Treatment with
5-AIQ inhibited p-NF-κB p65/NF-κB p65 ratios compared with the DSS
group (Fig. 2C).
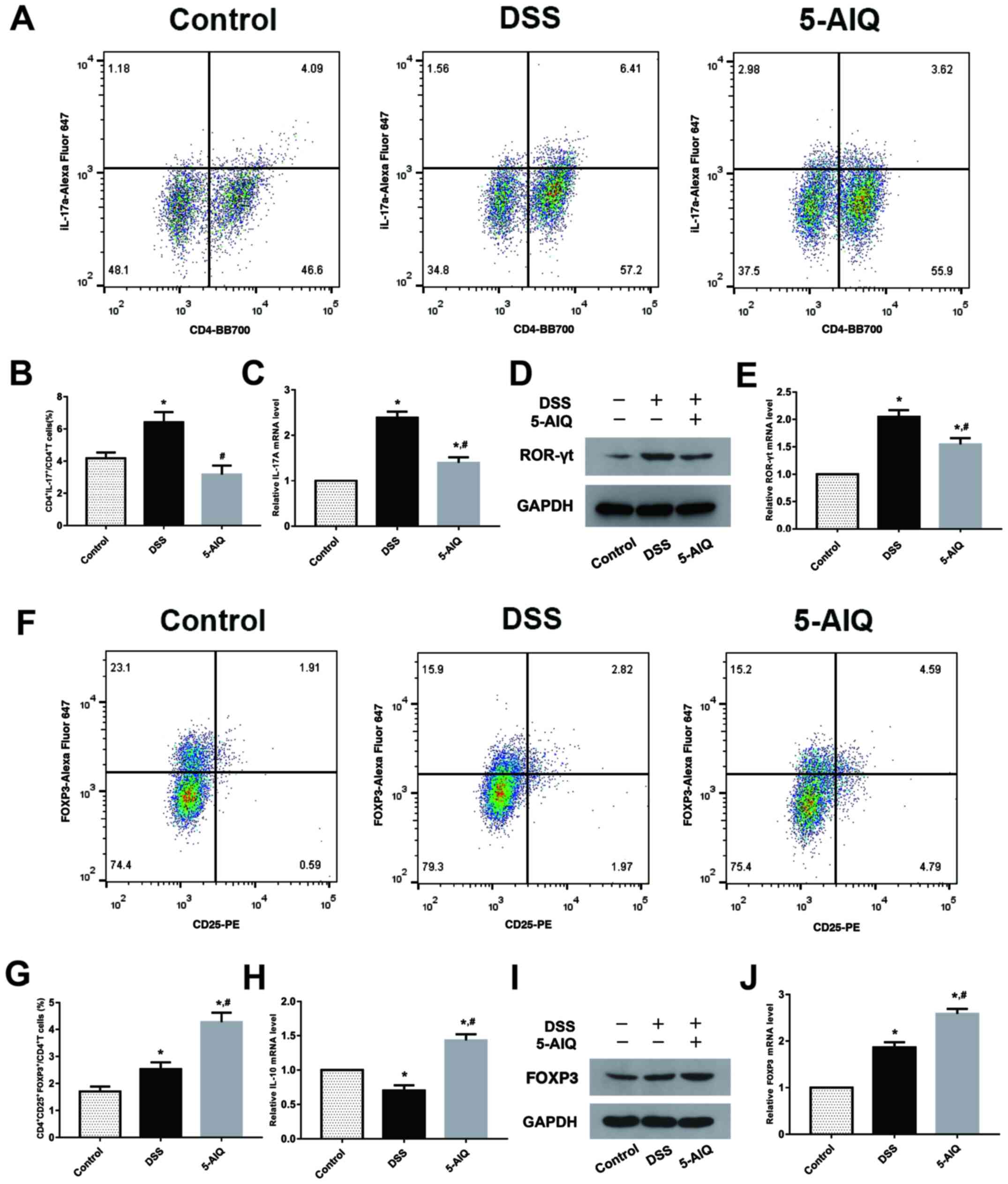
5-AIQ inhibits Th17 cell production in
mice with DSS-induced colitis
The percentage of Th17 cells in the spleen was
significantly elevated in mice exposed to DSS compared with normal
control mice. Notably, 5-AIQ significantly decreased the proportion
of Th17 cells (Fig. 3A and B). Moreover, IL-17A expression was
decreased in mice with DSS-induced UC treated with 5-AIQ (Fig. 3C). Subsequently, RORγt expression
was examined at both the mRNA and protein levels. It was found that
5-AIQ significantly reduced the expression of RORγt compared with
the DSS group (Fig. 3D and E).
5-AIQ promotes Treg development in
mice with DSS-induced colitis
The percentage of activated Tregs in the spleen was
significantly increased following 5-AIQ treatment compared with
mice with DSS-induced colitis (Fig.
3F and G). Additionally, IL-10
levels were increased in mice with DSS-induced colitis treated with
5-AIQ (Fig. 3H). Furthermore,
compared with the normal mice, Foxp3 expression was also elevated
in mice with DSS-induced colitis. As shown by the results of
RT-qPCR and western blot analysis, 5-AIQ significantly upregulated
the levels of Foxp3 (Fig. 3I and
J).
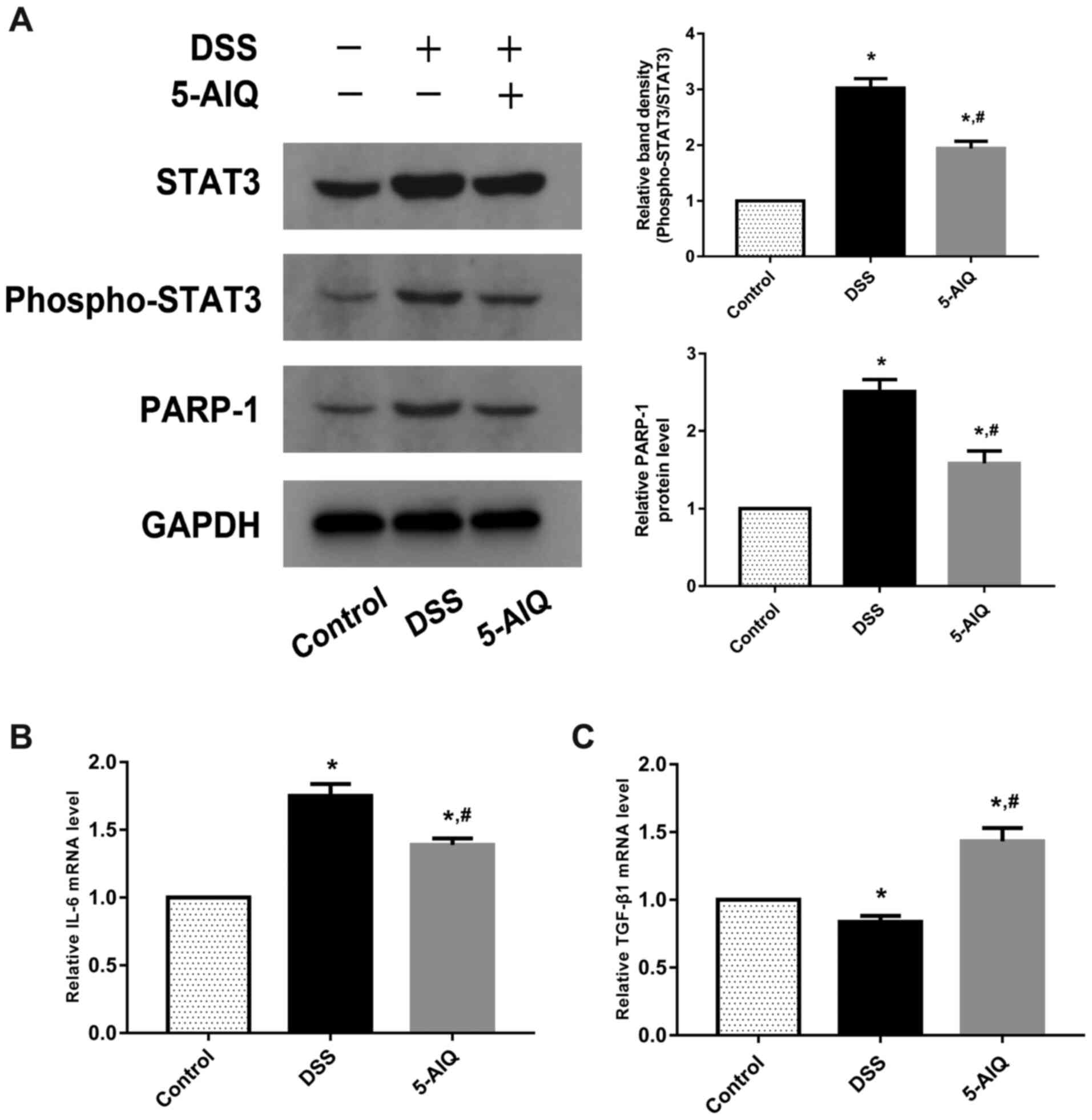
Effects of 5-AIQ on STAT3 and
PARP/NF-κB pathway activation in the colon
The ratios of p-STAT3/STAT3 were significantly
upregulated in mice with DSS-induced colitis. Following 5-AIQ
treatment, these ratios were significantly reduced compared with
the DSS group (Fig. 4A).
Furthermore, 5-AIQ significantly prevented the activation of PARP-1
(Fig. 4A). Thus, these results
indicated that 5-AIQ can downregulate the STAT3 and PARP/NF-κB
pathway in mice with colitis. Moreover, following 5-AIQ treatment,
the expression of IL-6 was significantly reduced compared with mice
with DSS-induced colitis (Fig. 4B).
Additionally, the expression of TGF-β1 was significantly
upregulated in the mice with DSS-induced colitis treated with 5-AIQ
(Fig. 4C).
Discussion
PARP is a type of ribozyme closely associated with
DNA damage repair and gene transcription (28). PARP-1, the most abundant isoform,
plays a key role in inflammatory pathways, promoting inflammatory
responses through the stimulation of pro-inflammatory signal
transduction pathways (15). Thus,
the association between PARP-1 and inflammatory responses has been
extensively investigated. Several studies have demonstrated that
PARP-1 physically interacts with NF-κB, one of the main
pro-inflammatory transcription factors, leading to the activation
of inflammatory signaling (29).
Recently, research conducted on mice revealed PARP-1 inhibitors
exerted protective effects against several inflammatory disorders
(26,30,31).
In addition, the number of Tregs increased in multiple organs of
PARP-1-deficient mice (21).
Larmonier et al (32)
demonstrated that the transcriptional reprogramming of the
intestines of PARP-1 knockout mice exerted protective effects
against experimental colitis. Moreover, the protective effects of
5-AIQ on various types of inflammation (20,33-35)
have attracted wide attention (23).
5-AIQ has been reported to exert a protective effect
against carrageenan-induced lung inflammation and rheumatoid
arthritis (26,35). In a previous study, during severe
acute pancreatitis-associated lung injury, 5-AIQ was shown to
inhibit the activity of PARP-1, reduce NF-κB signaling levels and
decrease the levels of downstream inflammatory factors, such as
IL-1β and IL-6 to attenuate injury (36). Since 5-AIQ exerts a number of
important pharmacological effects and has therapeutic benefits, its
effects in experimental colitis in mice warrant further
investigation. In the present study, the occult blood test in mice
with colitis began to yield positive results at 2 to 3 days, and
blood in the stool began to appear on the 3rd to 4th day, which
gradually became more severe. In addition, evident anal ulceration
in mice with DSS-induced colitis was observed. However, 5-AIQ
reversed these effects, and the DAI score was significantly lower
following treatment with 5-AIQ. Simultaneously, 5-AIQ intervention
reduced weight loss, maintained the colon length and attenuated
histological damage to the colon tissue. Thus, several lines of
observations support the pharmacological action of 5-AIQ in
experimental colitis.
NF-κB is a key regulatory point in downstream
inflammatory cytokines activated by PARP-1(29). It is generally known that the
abnormal activation of intestinal inflammatory cytokines is an
important mechanism of the pathogenesis of UC, and the imbalance in
the secretion of these cytokines lies in the abnormal activation of
NF-κB, which regulates their gene transcription (37). In addition, NF-κB is highly
expressed in the intestinal mucosa in UC and is significant for
disease evaluation and judgement of treatment effects (38). Therefore, NF-κB activation is one of
the key factors involved in the development of UC. To elucidate the
molecular mechanisms of 5-AIQ in improving the inflammatory
pathology of UC, the present study evaluated the effects of 5-AIQ
on the expression of key molecules (IκB-α and NF-κB p65) in the
NF-κB signaling pathway. IκB/NF-κB is one of the most classic
signaling pathways, with IκB being an inhibitory protein. Activated
NF-κB can translocate to the nucleus, where it can activate or
inhibit the transcription of various target genes, such as IL-1β,
IL-6 and TNF-α (39). Subsequently,
the inflammatory process can be amplified and sustained, thereby
damaging the intestinal mucosa, ultimately leading to the
occurrence of UC (39,40). Therefore, the levels of IL-1β,
TNF-α, IκB-α, NF-κB p65, and p-NF-κB p65 are worthy of observation.
Following intervention with 5-AIQ, the present study observed an
increase in the expression of IκB-α in the colon, while the levels
of the other aforementioned parameters were all decreased. Based on
these findings, not only does 5-AIQ treatment reduce the
recruitment of pro-inflammatory factors to the colon and the
production of pro-inflammatory mediators, but it also results in a
reduction in overall inflammation and colonic injury.
An abnormal intestinal mucosal immune system is a
key factor in the pathogenesis of UC (41). Moreover, the activation of effector
T cells is the starting point for intestinal mucosal immunity and
subsequent inflammation (42,43).
Following inflammation, naïve T cells differentiate into various
subsets, such as Th17 and Treg cells (44). A study suggested that the imbalance
of these cells is essential for the pathogenesis of UC (45). The transcription factor Foxp3
expressed by Tregs acts decisively in maintaining Treg cell
maturation and controlling inflammatory processes (46). Furthermore, the transcription factor
RORγt controls the development and function of Th17 cells (46,47).
Th17 cells in patients with UC are mainly concentrated in the
lamina propria of the colon and secrete IL-17, which mediates the
local infiltration of inflammatory cells, resulting in intestinal
mucosal tissue damage (7). A study
indicated that zinc deficiency activates the IL-23/Th17 axis,
aggravating experimental colitis in mice (48). In addition, in a model of colitis,
the transplantation of defined microbial flora has been shown to
restore the balance of Th17/Tregs (49). Moreover, studies have confirmed that
both Compound Sophorae Decoction and Rhubarb Peony Decoction exert
protective effects against DSS-induced colitis in mice, and the
mechanisms are related to the regulation of the Th17/Treg balance
(50,51). Therefore, maintaining the Treg/Th17
balance may provide a treatment strategy for DSS-induced UC. In the
present study, the results revealed that intervention with 5-AIQ
reduced the production of Th17 cells and upregulated the proportion
of Tregs. In addition, the imbalance between the two was restored.
Furthermore, the results revealed that 5-AIQ inhibited the
expression of RORγt, which led to a significant reduction in IL-17
secretion. Compared with mice with DSS-induced colitis, 5-AIQ
upregulated the expression of IL-10 by increasing Foxp3
production.
The critical role of IL-6/STAT3 and NF-κB signaling
has been well-established in recent years and is considered as a
primary target in the treatment of colonic inflammation (52). The IL-6/STAT3 pathway exerts potent
anti-apoptotic effects on T cells in colonic inflammation (53). It has been demonstrated that the
cytokine TGF-β1 exerts anti-inflammatory effects, and a high
concentration of this cytokine can result in Treg cell
differentiation (54). However,
when naïve T cells are exposed to a high concentration of IL-6 and
a low concentration of TGF-β1, the specific transcription factor
RORγt of Th17 cells is activated via the STAT3 pathway, and naïve T
cells then differentiate into Th17 cells (55). A recent study indicated that the
maintenance of Th17 cells requires a continuous IL-6 signal, which
is significant for the treatment of Th17-mediated diseases
(12). It was also demonstrated
that STAT3 and NF-κB cooperatively regulate the expression of
several gene products (56). In
addition, the PARP-1/NF-κB interaction contributes to the
development of inflammation (29).
It is worth noting that the lack of PARP-1 inhibits the activation
of NF-κB and leads to the suppression of innate immunity (57). In the T cell immune response, PARP-1
controls the immunosuppressive function of Tregs by destabilizing
Foxp3(58). In the present study,
it was found that the levels of PARP-1 and phosphorylated STAT3
decreased following intervention with 5-AIQ. Therefore, the present
study further quantified indicators, such as IL-6 and TGF-β1. Of
particular interest is that the concentration of IL-6 decreased in
mice with DSS-induced colitis treated with 5-AIQ. 5-AIQ upregulated
the expression of TGF-β1 by inhibiting PARP-1 and NF-κB production,
compared with the DSS group.
In conclusion, the present study indicated that
5-AIQ exerts a pharmacologically protective effect against acute
experimental colitis in mice, and the mechanisms are related to
regulating the balance between Th17 and Tregs, as well as
inhibition of PARP-1/NF-κB and STAT3 signaling. Therefore, 5-AIQ,
an inhibitor of PARP-1, may prove to be a novel therapeutic agent
for UC.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed in the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LS and SSW designed the experiments, and interpreted
and analyzed the data. SP, MXT and HML performed the experiments
and statistical analysis. SP drafted and revised the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All experimental procedures were approved by the
Ethics Committee at the Renmin Hospital of Wuhan University
(approval no. 20190330).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ungaro R, Mehandru S, Allen PB,
Peyrin-Biroulet L and Colombel JF: Ulcerative colitis. Lancet.
389:1756–1770. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kaplan GG and Ng SC: Understanding and
preventing the global increase of inflammatory bowel disease.
Gastroenterology. 152:313–321.e2. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
de Souza HSP, Fiocchi C and Iliopoulos D:
The IBD interactome: An integrated view of aetiology, pathogenesis
and therapy. Nat Rev Gastroenterol Hepatol. 14:739–749.
2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Torres J and Colombel JF: Genetics and
phenotypes in inflammatory bowel disease. Lancet. 387:98–100.
2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Silva FA, Rodrigues BL, Ayrizono ML and
Leal RF: The immunological basis of inflammatory bowel disease.
Gastroenterol Res Pract. 2016(2097274)2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Fasching P, Stradner M, Graninger W,
Dejaco C and Fessler J: Therapeutic potential of targeting the
Th17/Treg axis in autoimmune disorders. Molecules.
22(134)2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Ueno A, Jeffery L, Kobayashi T, Hibi T,
Ghosh S and Jijon H: Th17 plasticity and its relevance to
inflammatory bowel disease. J Autoimmun. 87:38–49. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Yamada A, Arakaki R, Saito M, Tsunematsu
T, Kudo Y and Ishimaru N: Role of regulatory T cell in the
pathogenesis of inflammatory bowel disease. World J Gastroenterol.
22:2195–2205. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Zhang L, Zhang Y, Zhong W, Di C, Lin X and
Xia Z: Heme oxygenase-1 ameliorates dextran sulfate sodium-induced
acute murine colitis by regulating Th17/Treg cell balance. J Biol
Chem. 289:26847–26858. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Yao J, Wei C, Wang JY, Zhang R, Li YX and
Wang LS: Effect of resveratrol on Treg/Th17 signaling and
ulcerative colitis treatment in mice. World J Gastroenterol.
21:6572–6581. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Salas A, Hernandez-Rocha C, Duijvestein M,
Faubion W, McGovern D, Vermeire S, Vetrano S and Vande Casteele N:
JAK-STAT pathway targeting for the treatment of inflammatory bowel
disease. Nat Rev Gastroenterol Hepatol. 17:323–337. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Harbour SN, DiToro DF, Witte SJ, Zindl CL,
Gao M, Schoeb TR, Jones GW, Jones SA, Hatton RD and Weaver CT: TH17
cells require ongoing classic IL-6 receptor signaling to retain
transcriptional and functional identity. Sci Immunol.
5(eaaw2262)2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Britton GJ, Contijoch EJ, Mogno I, Vennaro
OH, Llewellyn SR, Ng R, Li Z, Mortha A, Merad M, Das A, et al:
Microbiotas from humans with inflammatory bowel disease alter the
balance of gut Th17 and RORγt+ regulatory T cells and exacerbate
colitis in mice. Immunity. 50:212–224.e4. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Gong Y, Lin Y, Zhao N, He X, Lu A, Wei W
and Jiang M: The Th17/Treg immune imbalance in ulcerative colitis
disease in a Chinese han population. Mediators Inflamm.
2016(7089137)2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Gupte R, Liu Z and Kraus WL: PARPs and
ADP-ribosylation: Recent advances linking molecular functions to
biological outcomes. Genes Dev. 31:101–126. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Rosado MM, Bennici E, Novelli F and Pioli
C: Beyond DNA repair, the immunological role of PARP-1 and its
siblings. Immunology. 139:428–437. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Abd Elmageed ZY, Naura AS, Errami Y and
Zerfaoui M: The poly(ADP-ribose) polymerases (PARPs): New roles in
intracellular transport. Cell Signal. 24:1–8. 2012.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ba X and Garg NJ: Signaling mechanism of
poly(ADP-ribose) polymerase-1 (PARP-1) in inflammatory diseases. Am
J Pathol. 178:946–955. 2011.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Sodhi RK, Singh N and Jaggi AS:
Poly(ADP-ribose) polymerase-1 (PARP-1) and its therapeutic
implications. Vascul Pharmacol. 53:77–87. 2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Quesada A, O'Valle F, Montoro-Molina S,
Gómez-Morales M, Caba-Molina M, González JF, de Gracia MC, Osuna A,
Vargas F and Wangensteen R: 5-aminoisoquinoline improves renal
function and fibrosis during recovery phase of cisplatin-induced
acute kidney injury in rats. Biosci Rep.
38(BSR20171313)2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Fehr AR, Singh SA, Kerr CM, Mukai S,
Higashi H and Aikawa M: The impact of PARPs and ADP-ribosylation on
inflammation and host-pathogen interactions. Genes Dev. 34:341–359.
2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Luo X, Nie J, Wang S, Chen Z, Chen W, Li
D, Hu H and Li B: Poly(ADP-ribosyl)ation of FOXP3 protein mediated
by PARP-1 protein regulates the function of regulatory T cells. J
Biol Chem. 290:28675–28682. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Threadgill MD: 5-Aminoisoquinolin-1-one
(5-AIQ), a water-soluble inhibitor of the
poly(ADP-Ribose)polymerases (PARPs). Curr Med Chem. 22:3807–3829.
2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Brady PN, Goel A and Johnson MA:
Poly(ADP-Ribose) polymerases in host-pathogen interactions,
inflammation, and immunity. Microbiol Mol Biol Rev. 83:e00038–18.
2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wirtz S, Popp V, Kindermann M, Gerlach K,
Weigmann B, Fichtner-Feigl S and Neurath MF: Chemically induced
mouse models of acute and chronic intestinal inflammation. Nat
Protoc. 12:1295–1309. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ahmad SF, Zoheir KM, Bakheet SA, Ashour AE
and Attia SM: Poly(ADP-ribose) polymerase-1 inhibitor modulates T
regulatory and IL-17 cells in the prevention of adjuvant induced
arthritis in mice model. Cytokine. 68:76–85. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Gibson BA and Kraus WL: New insights into
the molecular and cellular functions of poly(ADP-ribose) and PARPs.
Nat Rev Mol Cell Biol. 13:411–424. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
29
|
Hassa PO and Hottiger MO: The functional
role of poly(ADP-ribose)polymerase 1 as novel coactivator of
NF-kappaB in inflammatory disorders. Cell Mol Life Sci.
59:1534–1553. 2002.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Niyazoglu M, Baykara O, Koc A, Aydoğdu P,
Onaran I, Dellal FD, Tasan E and Sultuybek GK: Association of
PARP-1, NF-κB, NF-κBIA and IL-6, IL-1β and TNF-α with graves
disease and graves ophthalmopathy. Gene. 547:226–232.
2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Dharwal V and Naura AS: PARP-1 inhibition
ameliorates elastase induced lung inflammation and emphysema in
mice. Biochem Pharmacol. 150:24–34. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Larmonier CB, Shehab KW, Laubitz D, Jamwal
DR, Ghishan FK and Kiela PR: Transcriptional reprogramming and
resistance to colonic mucosal injury in poly(ADP-ribose) polymerase
1 (PARP1)-deficient mice. J Biol Chem. 291:8918–8930.
2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Cuzzocrea S, McDonald MC, Mazzon E, Dugo
L, Serraino I, Threadgill M, Caputi AP and Thiemermann C: Effects
of 5-aminoisoquinolinone, a water-soluble, potent inhibitor of the
activity of poly(ADP-ribose) polymerase, in a rodent model of lung
injury. Biochem Pharmacol. 63:293–304. 2002.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Di Paola R, Genovese T, Caputi AP,
Threadgill M, Thiemermann C and Cuzzocrea S: Beneficial effects of
5-aminoisoquinolinone, a novel, potent, water-soluble, inhibitor of
poly(ADP-ribose) polymerase, in a rat model of splanchnic artery
occlusion and reperfusion. Eur J Pharmacol. 492:203–210.
2004.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Ahmad SF, Zoheir KM, Ansari MA, Korashy
HM, Bakheet SA, Ashour AE, Al-Shabanah OA, Al-harbi MM and Attia
SM: The role of poly(ADP-ribose) polymerase-1 inhibitor in
carrageenan-induced lung inflammation in mice. Mol Immunol.
63:394–405. 2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Yang B, Guo WY, Yu J, Zhao KL, Shi Q, Zuo
T and Wang WX: Expression of PARP/NF-κB and intervention effect of
5-AIQ/PDTC in SAP rats with adrenal damage. Zhonghua Yi Xue Za Zhi.
93:3063–3067. 2013.PubMed/NCBI(In Chinese).
|
|
37
|
Neurath MF: Cytokines in inflammatory
bowel disease. Nat Rev Immunol. 14:329–342. 2014.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Fonseca-Camarillo G and Yamamoto-Furusho
JK: Immunoregulatory pathways involved in inflammatory bowel
disease. Inflamm Bowel Dis. 21:2188–2193. 2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Hayden MS and Ghosh S: NF-kappaB in
immunobiology. Cell Res. 21:223–244. 2011.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Atreya I, Atreya R and Neurath MF:
NF-kappaB in inflammatory bowel disease. J Intern Med. 263:591–596.
2008.PubMed/NCBI View Article : Google Scholar
|
|
41
|
de Souza HS and Fiocchi C:
Immunopathogenesis of IBD: Current state of the art. Nat Rev
Gastroenterol Hepatol. 13:13–27. 2016.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Wallace KL, Zheng LB, Kanazawa Y and Shih
DQ: Immunopathology of inflammatory bowel disease. World J
Gastroenterol. 20:6–21. 2014.PubMed/NCBI View Article : Google Scholar
|
|
43
|
van Wijk F and Cheroutre H: Intestinal T
cells: Facing the mucosal immune dilemma with synergy and
diversity. Semin Immunol. 21:130–138. 2009.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Zenewicz LA, Antov A and Flavell RA: CD4
T-cell differentiation and inflammatory bowel disease. Trends Mol
Med. 15:199–207. 2009.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Liu TC and Stappenbeck TS: Genetics and
pathogenesis of inflammatory bowel disease. Annu Rev Pathol.
11:127–148. 2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Yang BH, Hagemann S, Mamareli P, Lauer U,
Hoffmann U, Beckstette M, Föhse L, Prinz I, Pezoldt J, Suerbaum S,
et al: Foxp3(+) T cells expressing RORγt represent a stable
regulatory T-cell effector lineage with enhanced suppressive
capacity during intestinal inflammation. Mucosal Immunol.
9:444–457. 2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Ohnmacht C, Park JH, Cording S, Wing JB,
Atarashi K, Obata Y, Gaboriau-Routhiau V, Marques R, Dulauroy S,
Fedoseeva M, et al: Mucosal immunology. The microbiota regulates
type 2 immunity through RORγt+ T cells. Science.
349:989–993. 2015.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Higashimura Y, Takagi T, Naito Y, Uchiyama
K, Mizushima K, Tanaka M, Hamaguchi M and Itoh Y: Zinc deficiency
activates the IL-23/Th17 axis to aggravate experimental colitis in
mice. J Crohns Colitis. 14:856–866. 2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Britton GJ, Contijoch EJ, Spindler MP,
Aggarwala V, Dogan B, Bongers G, San Mateo L, Baltus A, Das A,
Gevers D, et al: Defined microbiota transplant restores
Th17/RORγt+ regulatory T cell balance in mice colonized
with inflammatory bowel disease microbiotas. Proc Natl Acad Sci
USA. 117:21536–21545. 2020.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Xu M, Duan XY, Chen QY, Fan H, Hong ZC,
Deng SJ, Nan Z, Wu H, Dong YL, Liu YJ and Zhou CZ: Effect of
compound sophorae decoction on dextran sodium sulfate (DSS)-induced
colitis in mice by regulating Th17/Treg cell balance. Biomed
Pharmacother. 109:2396–2408. 2019.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Luo S, Wen R, Wang Q, Zhao Z, Nong F, Fu
Y, Huang S, Chen J, Zhou L and Luo X: Rhubarb peony decoction
ameliorates ulcerative colitis in mice by regulating gut microbiota
to restoring Th17/Treg balance. J Ethnopharmacol. 231:39–49.
2019.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Serrano C, Galán S, Rubio JF,
Candelario-Martínez A, Montes-Gómez AE, Chánez-Paredes S,
Cedillo-Barrón L, Schnoor M, Meraz-Ríos MA, Villegas-Sepúlveda N,
et al: Compartmentalized response of IL-6/STAT3 signaling in the
colonic mucosa mediates colitis development. J Immunol.
202:1239–1249. 2019.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Coskun M, Salem M, Pedersen J and Nielsen
OH: Involvement of JAK/STAT signaling in the pathogenesis of
inflammatory bowel disease. Pharmacol Res. 76:1–8. 2013.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Hadaschik EN and Enk AH: TGF-β1-induced
regulatory T cells. Hum Immunol. 76:561–564. 2015.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Ghoreschi K, Laurence A, Yang XP, Tato CM,
McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N,
et al: Generation of pathogenic T(H)17 cells in the absence of
TGF-β signalling. Nature. 467:967–971. 2010.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Martincuks A, Andryka K, Küster A,
Schmitz-Van de Leur H, Komorowski M and Müller-Newen G: Nuclear
translocation of STAT3 and NF-κB are independent of each other but
NF-κB supports expression and activation of STAT3. Cell Signal.
32:36–47. 2017.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Pazzaglia S and Pioli C: Multifaceted Role
of PARP-1 in DNA repair and inflammation: Pathological and
therapeutic implications in cancer and non-cancer diseases. Cells.
9(41)2019.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Zhang P, Maruyama T, Konkel JE, Abbatiello
B, Zamarron B, Wang ZQ and Chen W: PARP-1 controls
immunosuppressive function of regulatory T cells by destabilizing
Foxp3. PLoS One. 8(e71590)2013.PubMed/NCBI View Article : Google Scholar
|