Introduction
Hypercoagulability and fibrinolytic inhibition,
either systemic or in lung tissue, are important characteristics of
acute respiratory distress syndrome (ARDS) (1-4).
Both are associated with decreased lung compliance, diffusion
dysfunction and imbalance of the ventilation/perfusion ratio,
resulting in refractory hypoxemia, very small lung volume or even
pulmonary fibrosis (3). It has been
proposed that correcting coagulation defects would benefit
critically-ill patients who develop lung injury (5). A previous study demonstrated that
alveolar hypercoagulation and fibrinolytic inhibition are
associated with pulmonary inflammation in ARDS (6). Pulmonary inflammation can damage
pulmonary vascular endothelial cells and type-II alveolar
epithelial cells (AECII), which further increases the expression of
tissue factor (TF) and plasminogen activator inhibitor-1 (PAI-1) in
these cells, thus activating the exogenous coagulation system.
Moreover, activation of the coagulation system can in turn result
in or aggravate pulmonary inflammatory responses (3,7).
NF-κB is a nuclear transcription factor that
participates in several physiopathological processes, such as
apoptosis, inflammation and various autoimmune diseases (8,9).
Previous studies suggested that the NF-κB pathway could mediate
hypercoagulation, fibrinolysis inhibition and pulmonary
inflammation (10-13).
It was suggested that AECII serves a pivotal role in alveolar
hypercoagulation and fibrinolysis inhibition in ARDS (14). Our previous studies indicated that
knockdown of the p65 gene or IKKβ inhibited lipopolysaccharide
(LPS)-induced upregulation of TF and PAI-1 in rat AECII (15,16),
indicating that the NF-κB signaling pathway participates in
coagulation and fibrinolysis regulation in LPS-treated AECⅡ cells.
Given that pulmonary inflammation is a strong inducer for
hypercoagulation and fibrinolytic inhibition and that NF-κB is also
involved in the onset of inflammation in ARDS, it was hypothesized
that the NF-κB signaling pathway is the mechanism underlying the
crosstalk between hypercoagulation, fibrinolysis inhibition and
pulmonary inflammation in ARDS. To confirm the present hypothesis,
AECII were pre-treated with BAY11-7082, an inhibitor of IκBα
(17), then stimulated with the
pro-inflammatory factor TNF-α. The expressions of coagulants, such
as TF, PAI-I and fibrinolysis inhibition factor, and activation of
the NF-κB pathway were also determined in AECII.
Materials and methods
Cell culture
The RLE-6TN rat ACEII cell line was cultured in M199
medium (Gibco; Thermo Fisher Scientific, Inc.), supplemented with
10% FBS (Hyclone; Cytiva), penicillin (100 units/ml) and
streptomycin (100 µg/ml) (Hyclone; Cytiva) at 37˚C with 5%
CO2. For experiments, cells in the control group
received culture medium, whereas cells in the BAY group were
treated BAY11-7082 (5 µM; purity: 99.73%; cat. no. 19542-67-7;
MedChem Express) for 24 h. In the model group, cells were
stimulated with TNF-α (10 ng/ml; Peprotech, Inc.) for 1 h. In the
BAY + TNF group, cells were treated with BAY11-7082 for 24 h, then
with TNF-α for 1 h.
Detection of BAY11-7082 cytotoxicity
using cell counting Kit-8 (CCK-8)
RLE-6TN cells were seeded into a 96-well plate
(5x103 cells/well in 100 µl volume) and pre-incubated
for 24 h at 37˚C with 5% CO2. Different concentrations
of BAY11-7082 were added into the wells (0, 1, 2, 3, 4 and 5 µM).
PBS was used as a blank control. The cells were cultured for 24 h
in the incubator, and CCK-8 reagent (Dojindo Molecular
Technologies, Inc.) was added at 10 µl/well for 1 h. The absorbance
was measured at a wavelength of 450 nm using a microplate
reader.
Reverse transcription-quantitative
(RT-q) PCR
The mRNA expression of p65, TF and PAI-1 were
measured using RT-qPCR. GAPDH was used as an internal reference.
Briefly, cells were collected, and total RNA was extracted using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). RNA concentration was assessed using a NanoDrop™
2000 spectrophotometer (Thermo Fisher Scientific, Inc.). The
A260/A280 ratio of the extracted RNA was adjusted to 1.8-2.0, then
RT was performed on 2 µg RNA with oligo (dT) primers in 20-µl
reactions using the RevertAid First Strand cDNA Synthesis kit
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. Primers were designed according to the sequence of
the rat NF-κB p65 (Rela) gene in the National Center for
Biotechnology Information database. The primers were synthesized by
Shanghai Bioengineering Co., Ltd. (Table I). The reactions were set up as
follows: 10 µl SYBR Green mix (Thermo Fisher Scientific, Inc.), 0.8
µl forward primer, 0.8 µl reverse primer, 0.8 µl cDNA template and
7.6 µl ddH2O, for a total reaction volume of 20 µl. The
entire reaction system was preheated at 95˚C for 10 min. qPCR was
then performed using the following thermo-cycling procedure: 95˚C
for 15 sec, 60˚C for 30 sec and 72˚C for 30 sec, for 40 cycles.
After that, dissolution and amplification curves of the target gene
were recorded following gene amplification. Specificity of the
reaction was evaluated, and the Ct value was calculated according
to the dissolution and amplification curve. Expressions of target
genes were calculated using the 2-ΔΔCt method, where
ΔΔCt = (Ct, target - Ct, GAPDH)sample - (Ct, target -
Ct, GAPDH)control (18).
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Gene | Forward (5'-3') | Reverse (3'-5') | Size (bp) |
|---|
| p65 | AGCAAGCCATTAGCC | ACCGCATTCAAGTCAT | 91 |
| TF |
AATGGGCAGATAGAGTGT |
TCTGATTGTGGGTTTGTA | 182 |
| PAI-1 |
ACCAACTTCGGAGTAAAA |
TTGAATCCCATAGCATCT | 158 |
| GAPDH |
CAAGTTCAACGGCACAG |
CCAGTAGACTCCACGACAT | 138 |
Western blotting
The levels of p65, phosphorylated (p)-p65, IκBα,
p-IκBα, TF and PAI-1 were determined by western blot analysis.
Following treatment with TNF-α for 1 h, the cells were washed with
cold PBS. Total protein from the cells was extracted using RIPA
buffer (Beijing Solarbio Science & Technology Co., Ltd.).
Protein concentration measured with a BCA assay kit according to
the manufacturer's instructions. 10 µg of protein from each sample
was resolved on 12% Tris-glycine gel using SDS-PAGE. Subsequently,
protein bands were blotted into nitrocellulose membranes. After
incubation for 3 h in blocking solution (5% skim milk powder
diluted with TBST solution, containing 0.05% Tween-20) at room
temperature, the membrane was incubated for 24 h with antibodies
targeting p65 (1:1,000; cat. no. ab16502; Abcam), p-p65 (1:1,000;
cat. no. ab86299; Abcam), IκBα (1:1,000; cat. no. ab32518; Abcam),
TF (1:1,000; cat. no. ab151748; Abcam) and PAI-1 (1:1,000; cat. no.
ab66705; Abcam) at 4˚C. The secondary antibody (horseradish
peroxidase-conjugated goat ant-rabbit immuno-globulin; 1:5,000;
cat. no. ZB-2301; OriGene Technologies, Inc.) was added and
incubated with horseradish blocking solution for 1 h at room
temperature using the membrane chemiluminescence detection system
(EMD Millipore). Relative band densities were quantified using
ImageJ software 1.4.3 (National Institutes of Health).
ELISA
Cell supernatants were harvested and stored at
-80˚C. TF (Rat; cat. no. CSB-E07914r; Cusabio Technology LLC) and
PAI-1 (Rat; cat. no. CSB-E07948r; Cusabio Technology LLC) levels in
cell supernatants were determined using ELISA kits according to the
manufacturer's instructions.
Immunofluorescence staining
Briefly, cells of each groups were fixed at room
temperature with 4% formaldehyde in PBS for 30 min, permeabilised
with 0.5% Triton X-100 for 30 min and blocked with 1% BSA (Beijing
Solarbio Science & Technology Co., Ltd.) at room temperature
for 30 min. After that, these cells were incubated with primary
antibodies targeting rabbit anti-rat p65 (1:100; cat. no. ab16502;
Abcam) overnight at 4˚C. Subsequently, they were incubated with
FITC-labeled secondary antibodies (1:200; cat. no. ZF-0311; OriGene
Technologies, Inc.) for 1 h at room temperature. Each step was
followed with 5 min of washes in PBS, three times. The prepared
specimens were counterstained with DAPI for 10 min at room
temperature and observed with a fluorescence microscope (Carl Zeiss
AG) and were captured under an original magnification of x20.
Statistical analysis
Data are presented as the mean ± SEM. Statistical
significance was determined using one-way ANOVA followed by Tukey's
post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
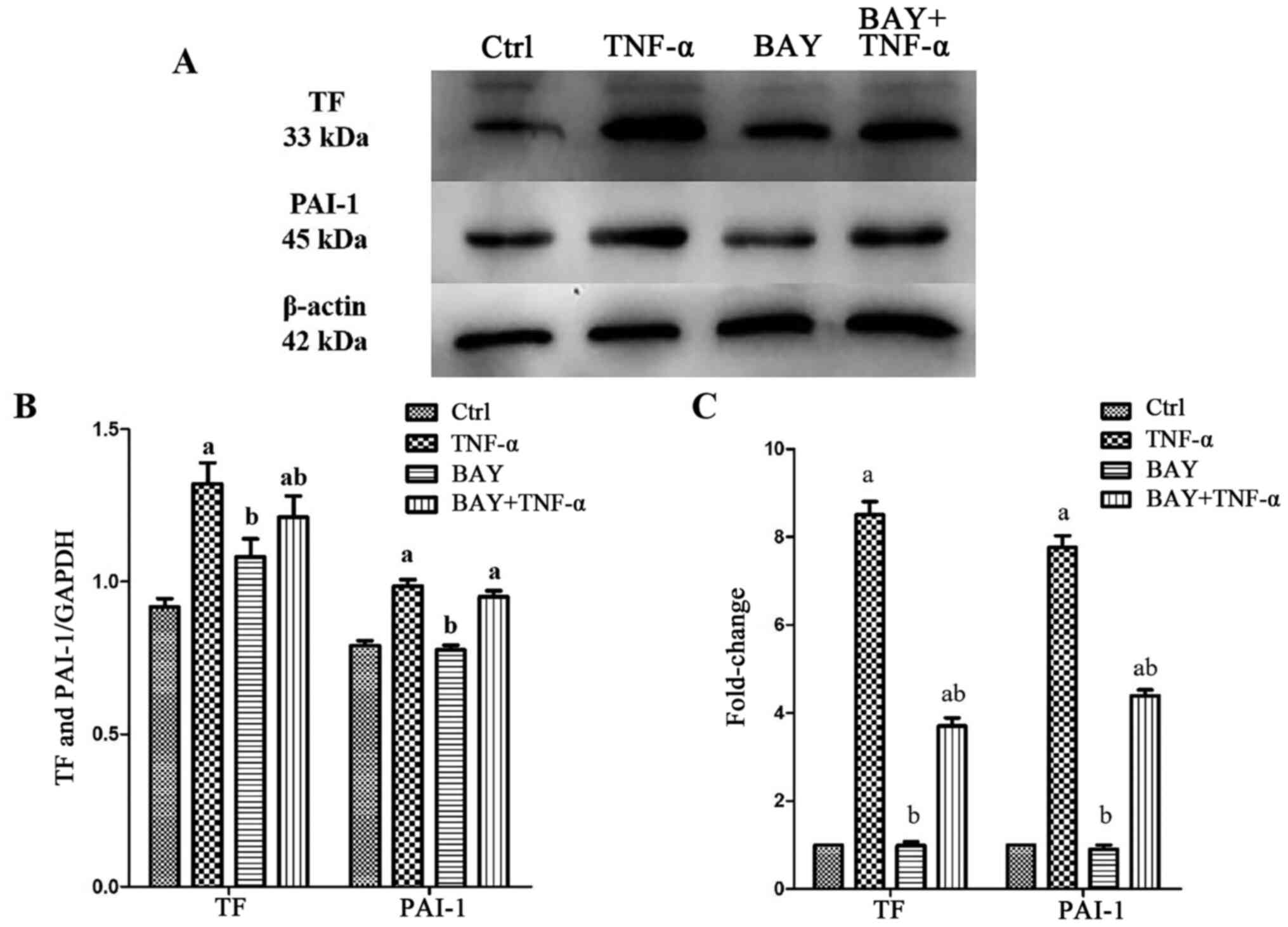
BAY11-7082 inhibits the expressions of
TF and PAI-1 in TNF-α-stimulated AECII cells
To determine the effects of BAY11-7082 on TF and
PAI-1 expression in AECII following TNF-α stimulation, TF and PAI-1
mRNA and protein levels were determined using RT-qPCR and western
blotting, respectively. The expressions of TF and PAI-1, both at
the mRNA and the protein levels, was significantly upregulated in
AECII after TNF-α stimulation compared with controls. However, the
expression levels of TF and PAI-1 significantly decreased in the
BAY and BAY + TNF-α groups (Fig.
1).
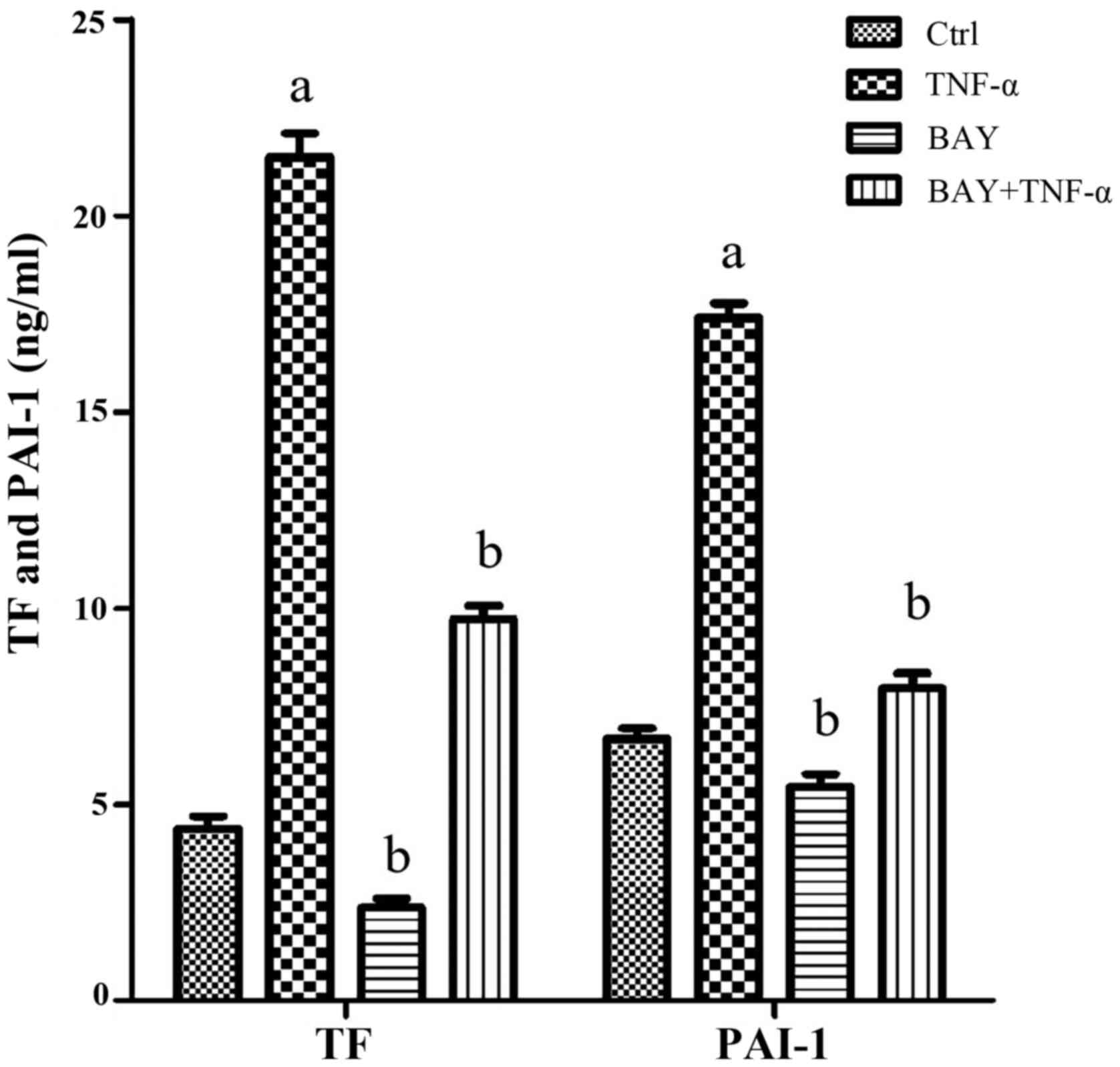
BAY11-7082 inhibits TF and PAI-1
secretion from TNF-α stimulated AECII
The levels of TF and PAI-1 were measured in cell
supernatants using ELISA. The secretion of TF and PAI-1
significantly increased following TNF-α stimulation compared with
controls. In the cells in the BAY group, this difference was not
obvious. However, pretreatment with BAY11-7082 (i.e, BAY + TNF-α
group), partially reduced the increase of TF and PAI-1 secretions
induced by TNF-α stimulation, although they are significantly
higher than the control group (Fig.
2).
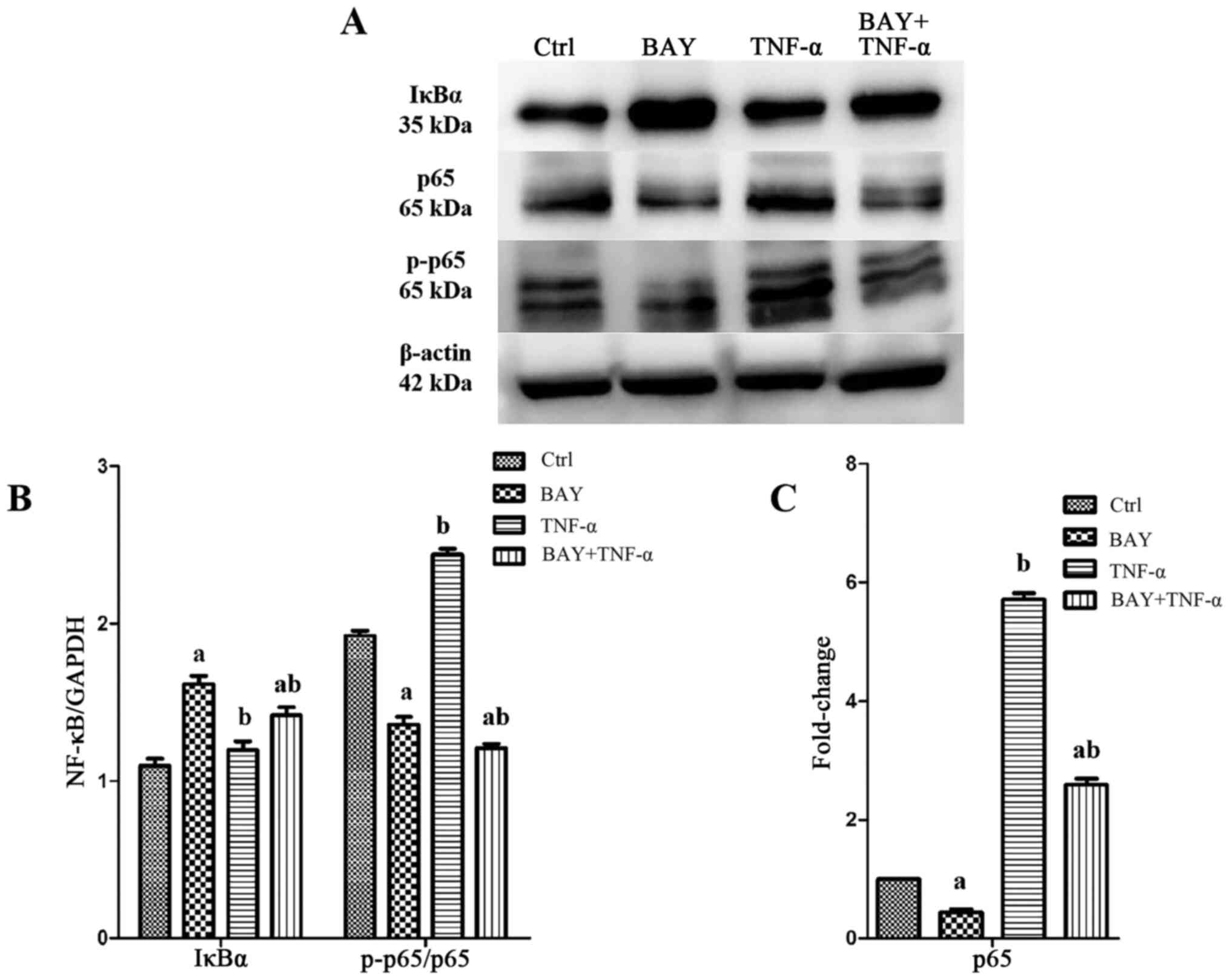
BAY11-7082 suppresses NF-κB pathway
activation following TNF-α stimulation in AECII
TNF-α stimulation caused significant activation of
the NF-κB pathway in AECII, as evidenced by the significantly
increased ratio of p-p65/p65. However, BAY11-7082 pretreatment
significantly inhibited p65 mRNA, decreased the p-p65/p65 ratio and
increased the IκBα levels, indicating that the activation of NF-κB
signaling pathway was inhibited by BAY11-7082 (Fig. 3).
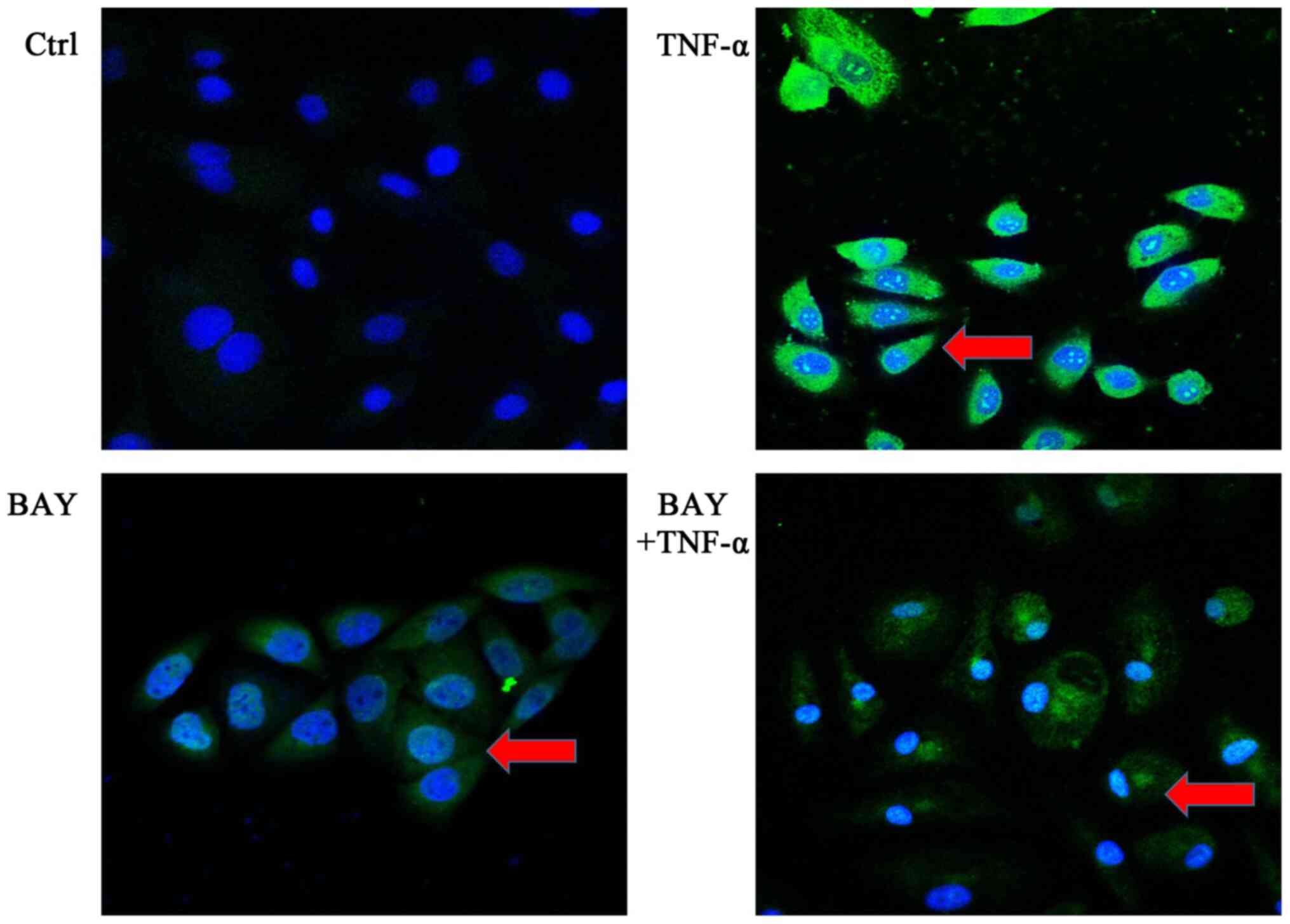
BAY11-7082 blocks the translocation of
p65 from the cytoplasm to the nucleus in TNF-α-stimulated
AECII
Immunofluorescence staining was used to determine
the cellular localization of p65. The results indicated that TNF-α
stimulation resulted in increased green fluorescence staining in
the nucleus, indicating an increase in translocation of p65 from
the cytoplasm to the nucleus. In the cells in the BAY group, this
difference was not obvious compared with cells in the ctrl group.
However, in the cells that were pretreated with BAY11-7082 (the BAY
+ TNF-α group), the degree of green fluorescence staining of p65 in
the nucleus of AECII was weakened, demonstrating that nuclear
translocation of p65 was inhibited by BAY11-7082 pre-treatment
(Fig. 4).
Discussion
AECII plays a pivotal role in the regulation of
alveolar hypercoagulation and fibrinolytic inhibition through
several important mediators, such as TF and PAI-1 (14,19-22).
In the present study, following TNF-α stimulation, AECⅡ either
expressed or secreted large concentrations of TF and PAI-1, which
was consistent with our previous studies on LPS stimulation
(15,16).
TF is a key coagulation factor that initiates the
exogenous coagulation pathway and plays an important role in
regulating coagulation during ARDS (23). PAI-1 is a key factor regulating
fibrinolysis inhibition (24).
These two molecules were used as indicators of coagulation and
fibrinolytic inhibition respectively in the present study. TF and
PAI-1 were not only highly expressed in AECII cells, but also
excessively secreted from AECII following TNF-α stimulation,
indicating that TNF-α induced a state of hypercoagulation and
fibrinolytic inhibition in AECⅡ.
TNF-α is a potent mediator of the inflammatory
response (25). Given the
functional characteristics of TF and PAI-1, the present findings
suggested that TNF-α induced a hypercoagulation and fibrinolysis
inhibition mediated by AECⅡ, which confirmed that inflammatory
stimuli can provoke hypercoagulation and fibrinolysis inhibition in
ARDS.
Preclinical and clinical studies have demonstrated
abnormalities in coagulation and fibrinolytic activity in pulmonary
tissue during ARDS (3,21). However, no satisfactory therapeutic
drugs for alveolar hypercoagulation and fibrinolysis inhibition are
available. This may be related to the crosstalk between alveolar
hypercoagulation, fibrinolysis inhibition and pulmonary
inflammation, three pivotal characteristics in ARDS (26). As a result, treatments targeting
coagulation or inflammation alone often do not achieve satisfactory
therapeutic results. Thus, it is crucial to explore the mechanism
simultaneously regulating coagulation/fibrinolysis inhibition and
inflammation, which is the basis of effective treatment for ARDS.
Given that the NF-κB pathway participates in alveolar
hypercoagulation, fibrinolytic inhibition and in pulmonary
inflammation (15,27), disrupting the NF-κB pathway may
prove beneficial. The present study showed that BAY11-7082
significantly decreased TF and PAI-1 expression, as well as their
production in cell culture supernatant following TNF-α stimulation
in AECⅡ. In addition, BAY11-7082 reduced the levels of p65 and
p-p65 in the cytoplasm and prevented p65 nuclear translocation.
Therefore, BAY11-7082 might attenuate TNFα-induced expression and
secretion of TF and PAI-1 in AECⅡ through NF-κB pathway
inactivation. These research findings demonstrated that BAY11-7082
may prove potentially effective in ARDS treatment.
BAY11-7082 is a commonly used inhibitor of the NF-κB
pathway, which irreversibly and specifically inhibits TNF-α-induced
phosphorylation and degradation of IκBα (28), preventing NF-κB from entering the
nucleus, thus inhibiting the transcription of target genes
(29,30). IκBs are essential upstream
regulatory molecules of the NF-κB pathway, bind to NF-κB p65 and
maintain the pathway in an inactivated state. IκBs are activated by
IκB kinase complex (IKKs) inducing degradation and dissociation of
IκBs from p65(31). IκBs consist of
IκBα, IκBβ and IκBγ, among which IκBα is the key regulating
molecule (32). In the present
study, BAY11-7082 promoted the upregulation of IκBα in the
cytoplasm, which was consistent with the hypothesis that BAY11-7082
inhibited IκBα phosphorylation and degradation, whereby this is the
mechanism in which BAY11-7082 affects the NF-κB pathway. However,
whether BAY11-7082 also inhibits TF and PAI-1 expression and
secretion through other mechanisms remains to be determined.
The present study has its limitations. First, only
one cell line and one NF-κB pathway inhibitor were used. Second,
the inflammatory response caused by excessive coagulation and
fibrinolysis inhibition was not evaluated in the present study.
Third, only a single observation timepoint was used in the present
study, resulting in a lack of dynamic results. Finally, the
experiments were only performed at the cellular level, and the
results of the present study still needs to be further verified
using ARDS animal models in the future.
In summary, BAY11-7082 ameliorated the expression
and secretion of TF and PAI-1 in AECⅡ induced by TNF-α via NF-κB
signaling pathway inactivation. BAY11-7082 is expected to be an
effective therapeutic target in ARDS.
Acknowledgements
Thanks is given to Mrs. Yan Zhao and Professor Yujie
Tan, who provided significant help to the experiments, including
good suggestions and guidance in the experimental operation.
Funding
This study was supported by grants from the Major
Research Project of Innovation Group in Education Department of
Guizhou Province [grant no. (2016) 034], the Science and Technology
supportive plan project of Guizhou Province [grant no. (2017) 2876]
and the Natural Science Foundation of Guizhou Province [grant no.
(2017) 7217].
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YC and BL performed the experiments, analyzed the
data and wrote the manuscript. HQ, HY, YWa and YWu analyzed the
data. FS designed the study, analyzed the data and organized the
final manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All experiments in the present study conformed to
the Guide for the Care and Use of Laboratory Regulations and were
approved by the Institutional Experiment Committee of Guizhou
Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Frantzeskaki F, Armaganidis A and Orfanos
SE: Immunothrombosis in acute respiratory distress syndrome: Cross
talks between inflammation and coagulation. Respiration.
93:212–225. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Tuinman PR, Dixon B, Levi M, Juffermans NP
and Schultz MJ: Nebulized anticoagulants for acute lung injury-a
systematic review of preclinical and clinical investigations. Crit
Care. 16(R70)2012.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Ozolina A, Sarkele M, Sabelnikovs O,
Skesters A, Jaunalksne I, Serova J, Ievins T, Bjertnaes LJ and
Vanags I: Activation of coagulation and fibrinolysis in acute
respiratory distress syndrome: A prospective pilot study. Front Med
(Lausanne). 3(64)2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Bastarache JA, Sebag SC, Clune JK, Grove
BS, Lawson WE, Janz DR, Roberts LJ II, Dworski R, Mackman N and
Ware LB: Low levels of tissue factor lead to alveolar haemorrhage,
potentiating murine acute lung injury and oxidative stress. Thorax.
67:1032–1039. 2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wang X, Zhang L, Duan W, Liu B, Gong P,
Ding Y and Wu X: Anti-inflammatory effects of triptolide by
inhibiting the NF-κB signalling pathway in LPS-induced acute lung
injury in a murine model. Mol Med Rep. 10:447–452. 2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Sebag SC, Bastarache JA and Ware LB:
Therapeutic modulation of coagulation and fibrinolysis in acute
lung injury and the acute respiratory distress syndrome. Curr Pharm
Biotechnol. 12:1481–1496. 2011.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Christiaans SC, Wagener BM, Esmon CT and
Pittet JF: Protein C and acute inflammation: A clinical and
biological perspective. Am J Physiol Lung Cell Mol Physiol.
305:L455–L466. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bonizzi G and Karin M: The two NF-kappaB
activation pathways and their role in innate and adaptive immunity.
Trends Immunol. 25:280–288. 2004.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hayden MS and Ghosh S: Signaling to
NF-kappaB. Genes Dev. 18:2195–2224. 2004.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Gao MY, Chen L, Yang L, Yu X, Kou JP and
Yu BY: Berberine inhibits LPS-induced TF procoagulant activity and
expression through NF-κB/p65, Akt and MAPK pathway in THP-1 cells.
Pharmacol Rep. 66:480–484. 2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Jeffers A, Owens S, Koenig K, Quaid B,
Pendurthi UR, Rao VM, Idell S and Tucker TA: Thrombin
down-regulates tissue factor pathway inhibitor expression in a
PI3K/nuclear factor-κB-dependent manner in human pleural
mesothelial cells. Am J Respir Cell Mol Biol. 52:674–682.
2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
El Kebir D, Damlaj A, Makhezer N and Filep
JG: Toll-like receptor 9 signaling regulates tissue factor and
tissue factor pathway inhibitor expression in human endothelial
cells and coagulation in mice. Crit Care Med. 43:e179–e189.
2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Deng X, Jin K, Li Y, Gu W, Liu M and Zhou
L: Platelet-derived growth factor and transforming growth factor β1
regulate ARDS-associated lung fibrosis through distinct signaling
pathways. Cell Physiol Biochem. 36:937–946. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Bastarache JA, Wang L, Geiser T, Wang Z,
Albertine KH, Matthay MA and Ware LB: The alveolar epithelium can
initiate the extrinsic coagulation cascade through expression of
tissue factor. Thorax. 62:608–616. 2007.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Liu B, Wu Y, Wang Y, Cheng Y, Yao L, Liu
Y, Qian H, Yang H and Shen F: NF-κB p65 Knock-down inhibits TF,
PAI-1 and promotes activated protein C production in
lipopolysaccharide-stimulated alveolar epithelial cells type II.
Exp Lung Res. 44:241–251. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Liu B, Wang Y, Wu Y, Cheng Y, Qian H, Yang
H and Shen F: IKKβ regulates the expression of coagulation and
fibrinolysis factors through the NF-κB canonical pathway in
LPS-stimulated alveolar epithelial cells type II. Exp Ther Med.
18:2859–2866. 2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Mori N, Yamada Y, Ikeda S, Yamasaki Y,
Tsukasaki K, Tanaka Y, Tomonaga M, Yamamoto N and Fujii M: Bay
11-7082 inhibits transcription factor NF-kappaB and induces
apoptosis of HTLV-I-infected T-cell lines andprimary adult T-cell
leukemia cells. Blood. 100:1828–1834. 2002.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Gonzales JN, Lucas R and Verin AD: The
acute respiratory distress syndrome: Mechanisms and perspective
therapeutic approaches. Austin J Vasc Med. 2(1009)2015.PubMed/NCBI
|
|
20
|
Ware LB and Matthay MA: The acute
respiratory distress syndrome. N Engl J Med. 342:1334–1349.
2000.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Camprubí-Rimblas M, Tantinyà N, Bringué J,
Guillamat-Prats R and Artigas A: Anticoagulant therapy in acute
respiratory distress syndrome. Ann Transl Med. 6(36)2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Glas GJ, Van Der Sluijs KF, Schultz MJ,
Hofstra JJ, Van Der Poll T and Levi M: Bronchoalveolar hemostasis
in lung injury and acute respiratory distress syndrome. J Thromb
Haemost. 11:17–25. 2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kasthuri RS, Glover SL, Boles J and
Mackman N: Tissue factor and tissue factor pathway inhibitor as key
regulators of global hemostasis: Measurement of their levels in
coagulation assays. Semin Thromb Hemost. 36:764–771.
2010.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Liu RM: Oxidative stress, plasminogen
activator inhibitor 1, and lung fibrosis. Antioxid Redox Signal.
10:303–319. 2008.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Song M, Fang F, Dai X, Yu L, Fang M and Xu
Y: MKL1 is an epigenetic mediator of TNF-α-induced proinflammatory
transcription in macrophages by interacting with ASH2. FEBS Lett.
591:934–945. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
He Z, Du L, Ke Y, Wen C and Zhang Y:
PP2ACα of alveolar macrophages is a novel protective factor for
LPS-induced acute respiratory distress syndrome. Inflammation.
42:1004–1014. 2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Qi D, Wand D, Zhang C, Tang X, He J, Zhao
Y, Deng W and Deng X: Vaspin protects against LPS-induced ARDS by
inhibiting inflammation, apoptosis and reactive oxygen species
generation in pulmonary endothelial cells via the Akt/GSK-3β
pathway. Int J Mol Med. 40:1803–1817. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kim YS, Kim JS, Kwon JS, Jeong MH, Cho JG,
Park JC, Kang JC and Ahn Y: BAY 11-7082, a nuclear factor-κB
inhibitor, reduces inflammation and apoptosis in a rat cardiac
ischemia-reperfusion injury model. Int Heart J. 51:348–353.
2010.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Lampiasi N, Azzolina A, D'Alessandro N,
Umezawa K, McCubrey JA, Montalto G and Cervello M: Antitumor
effects of dehydroxymethylepoxyquinomicin, a novel nuclear
factor-kappaB inhibitor, in human liver cancer cells are mediated
through a reactive oxygen species-dependent mechanism. Mol
Pharmacol. 76:290–300. 2009.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhang A, Wang K, Ding L, Bao X, Wang X,
Qiu X and Liu J: Bay11-7082 attenuates neuropathic pain via
inhibition of nuclear factor-kappa B and nucleotide-binding
domain-like receptor protein 3 inflammasome activation in dorsal
root ganglions in a rat model of lumbar disc herniation. J Pain
Res. 10:375–382. 2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Gamble C, McIntosh K, Scott R, Ho KH,
Plevin R and Paul A: Inhibitory kappa B Kinases as targets for
pharmacological regulation. Br J Pharmacol. 165:802–819.
2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zheng C, Yin Q and Wu H: Structural
studies of NF-κB signaling. Cell Res. 21:183–195. 2011.PubMed/NCBI View Article : Google Scholar
|