Introduction
Previous studies (1-17)
indicated the importance and various roles of ATP signaling in the
brain. Astrocytes, neurons and microglia are known to use ATP as a
transmitter molecule by releasing it into the extracellular space
to communicate with other cells (1,2). ATP
signaling has been reported to participate in synaptic strength
(3-5)
and memory formation by retaining long-term potentiation (6,7), and
can alter cerebral blood flow by increasing calcium levels in
astrocytic endfeet, which increases adjacent brain vessel diameter
(8-10).
The most investigated function of ATP signaling is the activation
of microglia to initiate inflammation (11-14).
When microglia receive an ATP signal through their surface
purinergic receptor, microglia change to amoeboid form, thus
initiating the migration to the damaged location and phagocytosis
of debris in the brain (12,15,16).
Moreover the well-known intercellular energy role of ATP in
maintaining synaptic function, which is usually derived from
glycolysis and lactate when high synaptic activity is ongoing
(17), via ATP signaling in the
extracellular space appears to be essential in both physiological
and pathological brain.
Despite numerous in vitro studies showing
various signaling functions of ATP in astrocytes, in vivo
studies demonstrating ATP release in a disease model are limited.
Melani et al (18) used
cerebral microdialysis in a rat middle cerebral artery occlusion
model and showed that the extracellular ATP value increased
1.3-fold compared with baseline levels during ischemia. Wang et
al (19) detected an increase
in ATP chemiluminescence by real-time imaging in a spinal cord
injury model. Based on these previous in vivo and in
vitro studies, combined with the well-known increase in
extracellular glutamate after traumatic brain injury (TBI)
(20), it was hypothesized that ATP
should increase in the extracellular spaces acutely after
controlled cortical impact (CCI)-induced mild injury. If ATP
release occurs after TBI, this signal might be responsible for the
inflammatory response after TBI that aggravates outcome, and could
possibly be a treatment target. The present study used a cerebral
microdialysis technique in a rat CCI model and measured the
magnitude of ATP release concomitant with changes in extracellular
glutamate. The effects of a blocker of the purinergic ATP Y1 (P2Y1)
receptor and a blocker of the store-operated calcium channel that
increases intracellular calcium levels were also investigated.
Rapid glutamate release and energy substance (e.g, glucose,
lactate) changes are well known to occur after TBI. The present
study also measured glucose and lactate levels from the perfusate
to determine their changes post-injury when the P2Y1 receptor or
store-operated calcium channels were chemically suppressed.
Materials and methods
Ethics
All experimental procedures were approved by the
UCLA Chancellor's Committee for Animal Research prior to starting
the experiments. The approval protocol number was 2008-021. All
experiments were conducted at UCLA. Efforts were made to minimize
causing pain and distress to the animals.
Animal model
Thirty eight male Sprague-Dawley rats weighing
between 313 and 413 g (mean, 355 g; 62-72 day of age) were obtained
from Charles River Laboratories, Inc., and were used in the study.
Four rats were used to determine that our selected injury
parameters did not result in cortical bleeding or tissue necrosis 1
week post-CCI and 8 rats were used in pilot work to determine the
need for an inhibitor of ATP breakdown in the microdialysis
perfusate. For animals used in the main experiment, 1 rat died
shortly after induction of anesthesia with rapid rise in body
temperature and 1 died from apparent neurogenic pulmonary
complications shortly after CCI. Rats were pair housed and
acclimated for at least 2 weeks under standard temperature and
lighting conditions (21-24˚C, 30-70% humidity, room lights on 6:00
a.m. - 6:00 p.m.), with food (Teklad; Envigo) and tap water
available ad libitum. Prior to surgery, the animals were
randomly assigned to one of four treatment groups: i) Sham injury
group with standard perfusate (sham, n=6); ii) CCI group with
standard perfusate (CCI; n=6); iii) CCI group with in situ
administration of 300 µM MRS2179 ammonium salt hydrate (M3808;
Sigma-Aldrich; Merck KGaA) (CCI-MRS2179; n=6); and iv) CCI group
with in situ administration of 1 mM 2-aminoethoxy
diphenylborinate (2-APB; D9754; Sigma-Aldrich; Merck KGaA)
(CCI-2-APB; n=6). For both drug treated groups the compounds were
administered by diffusion from the microdialysis probe membranes.
Drug concentrations are determined from the previous study and
considering the diffusion effect as explained below.
For surgery, the animal was placed in an anesthesia
box and anesthesia was induced with 3-4% isoflurane in oxygen (1.5
l/min flow rate) and the scalp was shaved. After the head was
secured in a stereotaxic frame, anesthesia was maintained with
1.5-2.0% isoflurane in oxygen delivered via nose cone on the
stereotaxic bite-bar throughout surgery. Body temperature was kept
at 37±0.5˚C throughout surgery with a rectal probe and a
thermostatically controlled heating pad (Harvard Apparatus).
Ophthalmic ointment (Bausch & Lomb) was applied to both eyes,
local anesthesia was applied to the scalp with 0.5 ml 1% lidocaine,
and the scalp was sterilized by repeated (three times) cleaning
with Betadine (Dynarex Corp.) followed by 70% ethanol. A midline
skin incision was made, and the pericranium was spread bilaterally.
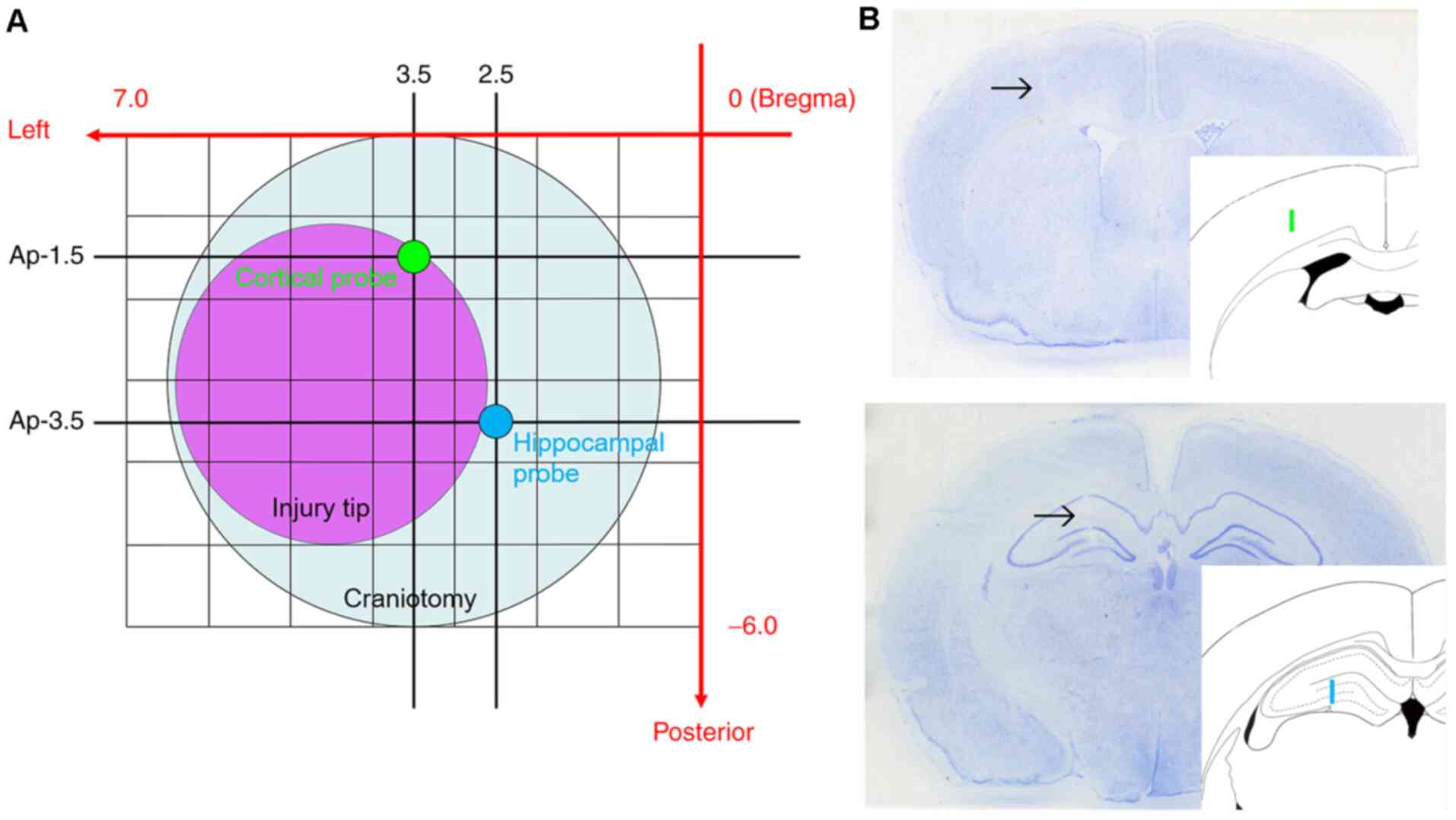
Next, a 6-mm diameter circular craniotomy was made over the left
parietal cortex in all animals, centered 3.0 mm posterior and 3.5
mm lateral from Bregma (Fig. 1).
Under microscopic guidance (M651; Leica Microsystems GmbH) the dura
mater was incised at two sites with the tip of a 27-gauge needle.
Two microdialysis probes mounted on a single micromanipulator on
the stereotaxic instrument (Kopf Instruments) were placed into the
left hemisphere. One probe was placed into the cortex adjacent to
the location of CCI. Another probe was placed into the hippocampus,
which is underneath the impact site and known to exhibit several
injury-induced changes. The final probe locations were selected
with limitation of placement through a single craniotomy and use of
a single micromanipulator. With minor deviations to avoid surface
blood vessels, the target coordinates for the cortical probe were
1.5 mm posterior, 3.5 mm lateral from Bregma and 2.2 mm depth from
cortical surface. The coordinates for the hippocampal probe were
3.5 mm posterior, 2.5 mm lateral from Bregma and 3.9 mm depth from
cortical surface (Fig. 1). After 80
min of baseline sampling (collection of four 20-min fractions) both
microdialysis probes were retracted by raising the micromanipulator
until the probes were ~10 mm above the cortical surface. Sham
injury or CCI injury was then induced, similar to what is described
in our prior studies (21-23)
but with reduced tissue compression to induce a milder injury. A
4-mm diameter flat-tipped impactor, which was angled 20˚ from
vertical to be perpendicular to the dura mater at the injury site,
was used to induce CCI centered at 3.0 mm posterior and 4.5 mm
lateral relative to Bregma (20 psi, 2.37 m/sec, 1.5 mm tissue
compression for 250 msec). Pilot work in our laboratory showed that
this injury level does not cause any gross evidence of tissue
damage at 1 week post-CCI. Immediately after injury (within ~10
sec), probes were returned to the same location by lowering the
micromanipulator, and post-injury microdialysis sampling was
performed for 3 h under isoflurane anesthesia. In total, the time
of interruption of microdialysis sampling (probe retraction, CCI
and probe re-insertion) was <30 sec. The sham injury group
underwent baseline sampling, as well as removal and replacement of
probes, but without CCI application.
Microdialysis settings
Cerebral microdialysis was performed using CMA 12
Elite probes (8010431; CMA Microdialysis AB), which have a 20,000
Da cut-off polyarylethersulfone membrane that is 1 mm in length
with a 0.5-mm membrane diameter. The perfusate used in all animals
contained 147 mM NaCl, 2.7 mM KCl, 1.2 mM CaCl2, 0.85 mM
MgCl2, 25 mM HEPES and 1 mM adenosine
5'-(α,β-methylene)diphosphate (M3763; Sigma-Aldrich; Merck KGaA) to
prevent ATP breakdown (18). Pilot
work (n=8) showed that ATP levels in the extracellular fluid
samples were not detectable if the ecto-5'-nucleotidase inhibitor
[adenosine 5'-(α,β-methylene)diphosphate] was not contained in the
perfusate. In one CCI group, 300 µM MRS2179 was added to the
perfusate to enable P2Y1 receptor blockade. For another CCI group,
1 mM 2-APB was mixed into the perfusate for blockade of
store-operated calcium channels. Drug concentrations were selected
to be 10 times higher than those used in previous in vitro
experiments where MRS2179 or 2-APB were shown to suppress calcium
responses (24-27),
assuming ~10% drug efflux from dialysate based on the probe
membrane efficiency. Before use, the pH of each perfusate was
adjusted to 7.4 and it was filtered through a sterile 0.2-µm
membrane. Prior to in vivo use, the efficiency of each probe
was measured by collecting dialysates as the probe was immersed in
a 10-µM glutamate and 10-nM ATP solution. During the experiments,
each probe was perfused with perfusate at 3 µl/min, and timing of
sample collection was performed to account for fluid volumes
contained in probe inlet and outlet tubes and the shaft dead space.
All microdialysis samples were collected in 20-min fractions, with
four samples collected in an 80-min baseline sampling period and
nine samples collected in 3 h of post-injury sampling. All
microdialysis samples were stored at -80˚C until analysis. Both ATP
and glutamate data are presented as values corrected by the
percentage of recovery of each probe. The percentage of recovery of
glutamate was substituted for correcting both glucose and lactate
values. At the end of surgery, a 0.5 ml venous blood sample was
collected from the subcutaneous vein of the neck to enable
determination of plasma glucose and lactate concentrations. The
isoflurane-anesthetized animals were sacrificed by decapitation,
and their brains were post-fixed in 4% paraformaldehyde and then
sectioned in a cryostat to enable histological staining.
Sample analyses
The concentration of ATP in each cerebral
microdialysis fraction was measured by a bioluminescent
luciferin-luciferase assay using a commercial kit (FLAAM and FLAAB;
Sigma-Aldrich; Merck KGaA) according to the manufacturer's
protocol. The signal was read with a scintillation counter (LS6500;
Beckman Coulter, Inc.). Glutamate, glucose and lactate
concentrations were measured using a CMA 600 analyzer (8001600; CMA
Microdialysis AB). Plasma glucose and lactate levels were analyzed
with YSI 2700 Select Biochemistry Analyzer (Yellow Springs
Instrument; Thermo Fisher Scientific, Inc.).
Statistical analysis
For statistical analysis, SPSS version 21 software
(IBM Corp.) was used. All data are presented as the group mean ±
standard deviation. Student's t-test was used to compare data for
body temperature and blood glucose or lactate levels between the
sham and CCI groups. For statistical analysis of the pre-injury
microdialysis values, one-way ANOVA was applied to the values of
the final samples in the baseline period (-20 min samples). When
significant differences in the baseline data were obtained across
groups (including ATP, glutamate and glucose values), the
post-injury data were analyzed using repeated measures analysis of
covariance (ANCOVA) with each pre-injury value of individual
subjects serving as covariates. For data without baseline
difference (such as lactate values), repeated measures ANOVA was
used to analyze the post-injury data. When the post-injury analyses
revealed significant differences across the groups, post hoc
Tukey's test was conducted for multiple comparisons. P<0.05 was
considered to indicate a statistically significant difference.
Results
Location of probes and physiological
parameters
As aforementioned, to avoid cortical surface
vessels, microdialysis probes could not be inserted into the
planned position for certain animals. The final cortical probe
position on average was 1.62±0.29 mm posterior and 3.53±0.33 mm
lateral from Bregma, and the final hippocampal probe position was
3.62±0.29 mm posterior and 2.53±0.33 mm lateral from Bregma. These
probe locations were very close to the initial targets, and did not
alter placement in the intended cortical or hippocampal
tissues.
The mean lowest and highest body temperature values
during surgery in the sham group were respectively 36.07±0.29˚C and
36.87±0.64˚C, and 36.07±0.27˚C and 36.97±0.61˚C in the CCI group.
The mean blood glucose levels at 3 h post-injury were 10.29±1.05 mM
in the sham group and 10.53±0.86 mM in the CCI group. The mean
blood lactate levels at the same time point were 1.27±0.31 mM in
the sham group and 1.15±0.19 mM in the CCI group. These parameters
were not significantly different between groups.
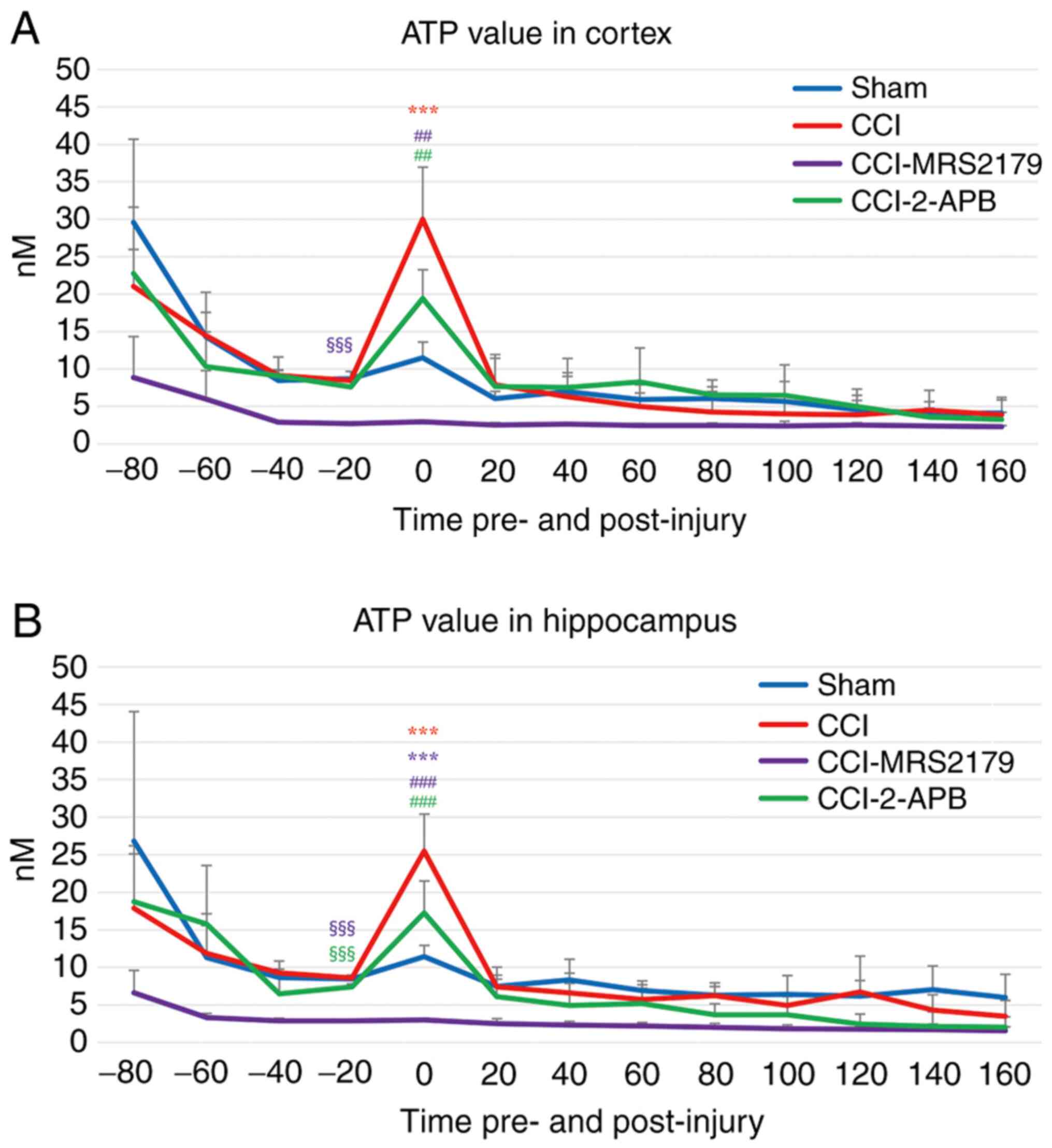
ATP changes in the extracellular
space
The ATP changes in the extracellular spaces are
represented in Fig. 2A (cortex) and
Fig. 2B (hippocampus).
The final (-20 min) pre-injury dialysate ATP values
in the cortex were 8.71±0.97 nM in the sham group, 8.50±0.20 nM in
the CCI group and 7.59±1.00 nM in the CCI-2-APB group. The
CCI-MRS2179 group had a significantly lower value (2.73±0.17 nM)
compared with that of the other three groups (P<0.001),
indicating significant effects of P2Y1 receptor blockade on
pre-injury ATP release. In the first 20 min following injury, the
ATP value of the CCI group increased to 30.01±6.95 nM which was
significantly higher (P<0.001) than that of the sham group
(11.53±2.07 nM). The ATP concentration of the CCI group increased
by 353% from the pre-injury value, while that of the sham group was
132% of pre-injury value. The ATP value of the CCI-MRS2179 group
was not significantly altered by injury, with a concentration of
2.98±0.16 nM at 20 min post-CCI (109% of pre-injury value). The ATP
value of the CCI-2-APB group increased to 19.41±3.84 nM (256% of
pre-injury value) in the first 20 min after injury, but this
increase was not significantly different from that of the sham
group. Both the CCI-MRS2179 and CCI-2-APB groups exhibited
significantly (P=0.001) lower post-injury peak ATP values compared
with those of the CCI group. The ATP values of the four groups
returned to their baseline levels within the second post-injury
fraction, with no further significant group differences.
The ATP changes in the hippocampus exhibited similar
trends to those of the cortex, with the greatest changes occurring
in the first post-injury microdialysis fraction (Fig. 2B). In the sham group, the ATP value
changed from 8.47±0.22 to 11.43±1.50 nM (135% of pre-injury value).
In the CCI group, the pre-injury value of 8.59±0.40 nM increased to
25.49±4.92 nM (297% of pre-injury value), and this peak value was
significantly higher (P<0.001) than that of the sham group. The
-20 min pre-injury ATP value of the CCI-MRS2179 group was
significantly reduced (P<0.001) compared with that of the other
three groups. In this group, the ATP value changed from 2.88±0.07
nM pre-injury to 3.05±0.21 nM (106% of pre-injury value) in 20 min
after CCI, with a significantly lower peak value compared with that
of the sham (P<0.001) and CCI (P<0.001) groups. The
pre-injury ATP value in hippocampus of the CCI-2-APB group was
significantly lower (P<0.001) than that of the sham and CCI
groups. The ATP values of the CCI-2-APB group changed from
7.40±0.41 nM pre-injury to 17.25±4.26 nM (233% of pre-injury
value), and the 20 min post-CCI peak value was significantly lower
than that of the CCI group (P<0.001). From the second
post-injury fraction onward, the ATP value of all groups returned
to the baseline level without any significant difference between
the groups.
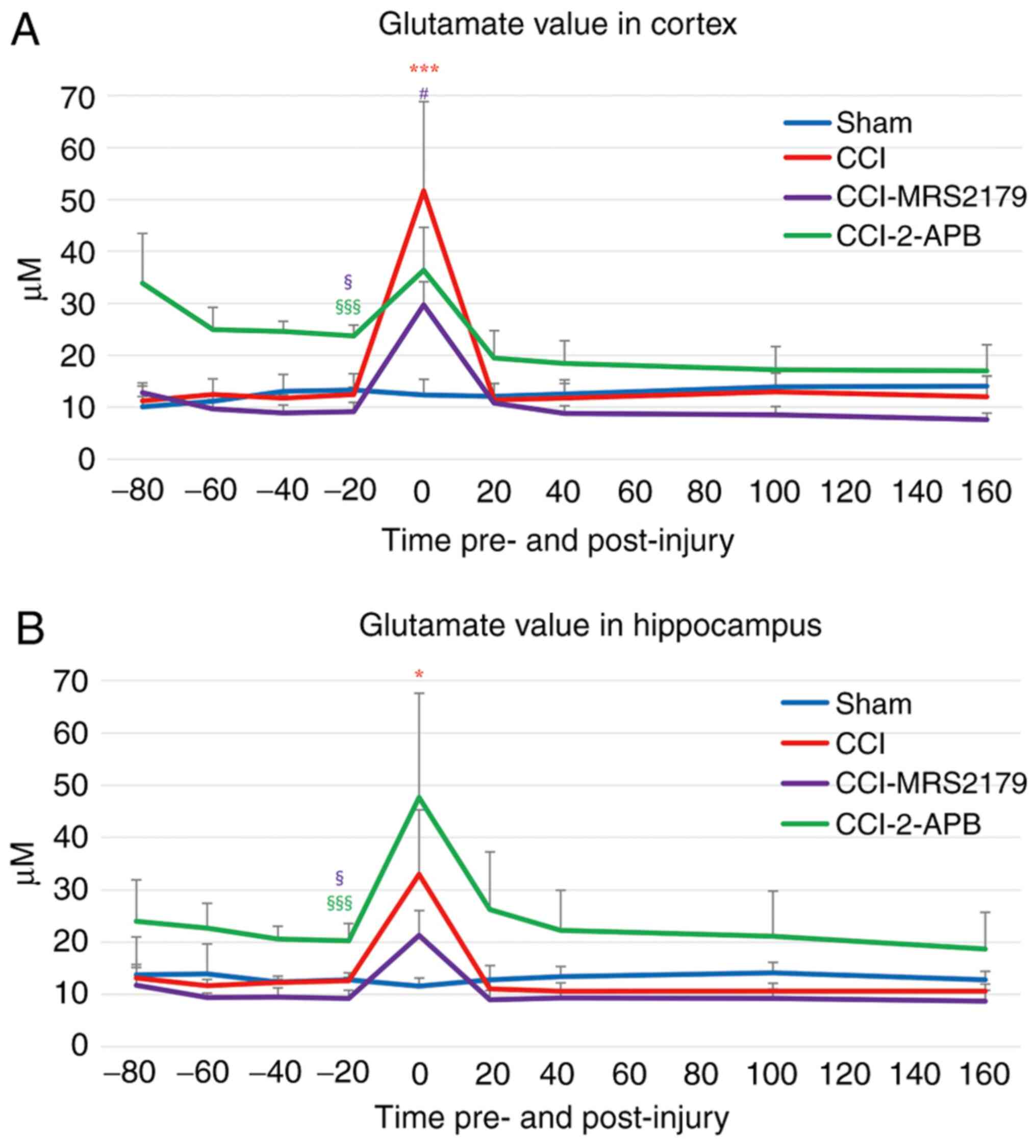
Glutamate changes in the extracellular
space
The glutamate changes in the extracellular spaces
are represented in Fig. 3A (cortex)
and Fig. 3B (hippocampus).
In the cortex, the -20 min pre-injury glutamate
levels of the sham and CCI groups were 13.29±3.18 and 12.49±1.38
µM, respectively. The pre-injury glutamate value of the CCI-MRS2179
group (9.13±1.82 µM) was significantly reduced (P=0.030), while
that of the CCI-2-APB group (23.70±2.12 µM) was significantly
increased (P<0.001), compared with that of the sham group,
demonstrating the pre-injury effects of P2Y1 receptor or store
operated calcium channel blockade. In the first fraction after
injury, the cortical glutamate concentration of the CCI group
reached 51.63±17.18 µM (413% of pre-injury value), which was
significantly higher (P<0.001) than the concentration observed
in the sham group (93% of pre-injury value; 12.38±3.01 µM). The
glutamate values for the CCI-MRS2179 and CCI-2-APB groups increased
to 29.71±4.46 µM (325% of pre-injury value) and 36.39±8.23 µM (154%
of pre-injury value), respectively, in the first 20 min
post-injury, but these values were not significantly different from
those of the sham group. The post-injury peak glutamate values of
the CCI-MRS2179 group were significantly lower (P=0.037) than those
of the CCI group. In the second fraction after injury, the cortical
glutamate values of the four groups returned to the baseline level,
with no further significant group differences.
In the hippocampus (Fig.
3B), the -20 min pre-injury glutamate levels were 12.78±1.37
and 12.67±0.61 µM in the sham and CCI groups, respectively. In the
cortex, the pre-injury value of the CCI-MRS2179 group (9.22±1.61
µM) was significantly lower (P=0.027), and the pre-injury value of
the CCI-2-APB group (20.27±3.31 µM) was significantly higher
(P<0.001) in the hippocampus than the pre-injury glutamate
levels in the sham group. In the first fraction following injury,
the peak value of the CCI group increased significantly (260% of
pre-injury value; 32.98±12.32 µM; P=0.040) compared with that of
the sham group (91% of pre-injury value; 11.61±1.53 µM). The
post-injury glutamate peak for the CCI-MRS2179 group was 21.30±4.74
µM (231% of pre-injury value), and that of the CCI-2-APB group was
47.64±19.94 µM (235% of pre-injury value), with neither of these
increases being significant compared with the values exhibited by
the sham group. In the second fraction post-injury and thereafter,
the values for glutamate in the hippocampus of the four groups
returned to the baseline level without any significant group
differences.
The post-injury glutamate peaks of the MRS2179 and
2-APB groups increased to 154-325% of pre-injury value in the
cortex and hippocampus, but ANCOVA showed that these changes were
not significantly different compared with the values exhibited by
the sham group (P=0.148 in cortex and P=0.990 in hippocampus). The
significance of these post-CCI peak values was eliminated due to
the significant -20 min pre-injury levels being set as a covariate
in these groups.
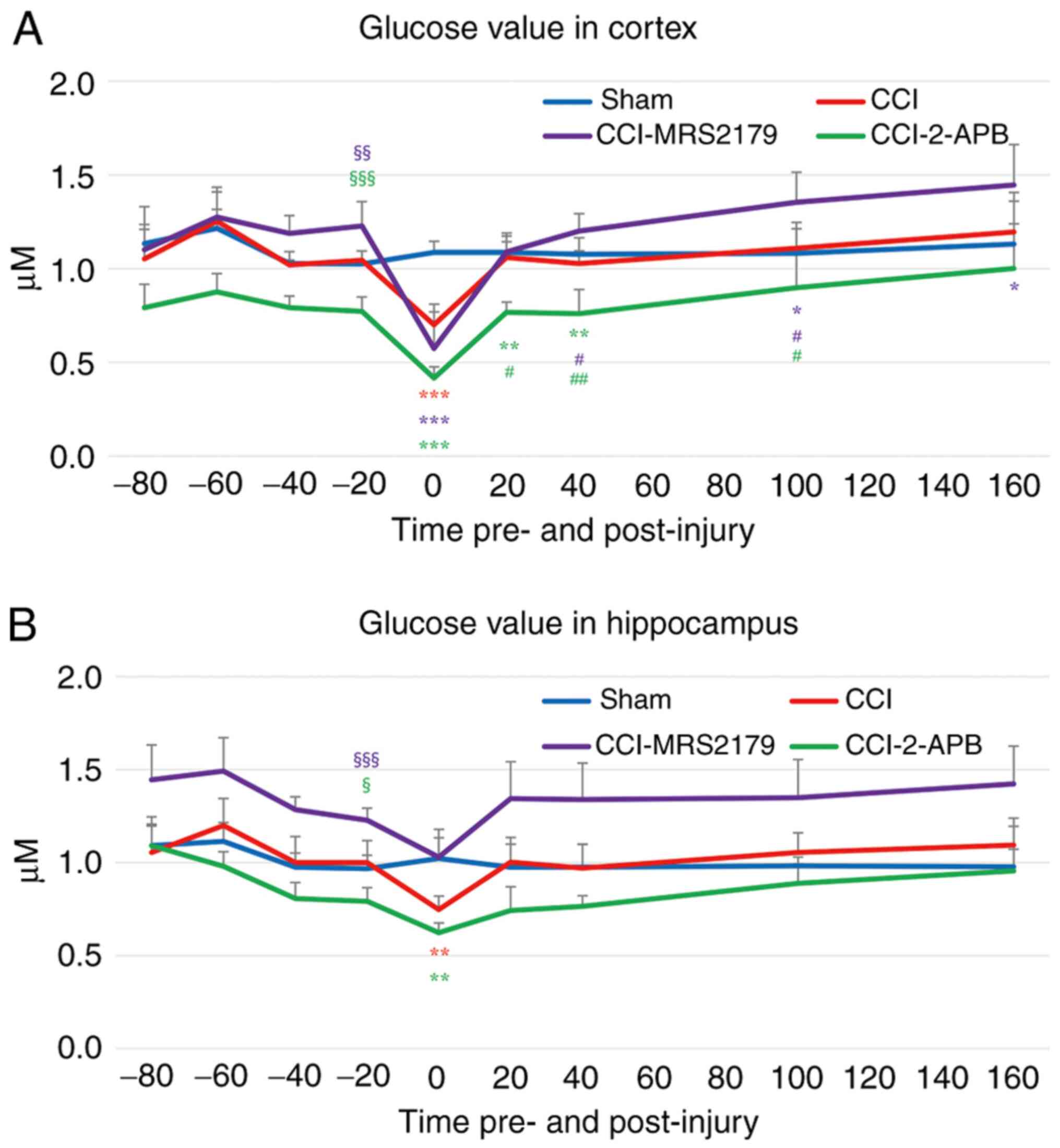
Glucose changes in the extracellular
space
The glucose concentrations in the extracellular
space are represented in Fig. 4A
(cortex) and Fig. 4B
(hippocampus).
The -20 min pre-injury glucose level in the cortex
of the sham group was 1.02±0.07 µM, and in the CCI group it was
1.04±0.05 µM. The CCI-MRS2179 group had significantly higher
(P=0.004) pre-injury glucose value (1.23±0.13 µM) and the CCI-2-APB
group had a significantly lower (P<0.001) pre-injury glucose
value (0.77±0.08 µM) compared with those of the sham controls.
After injury, in the first fraction, the glucoses value decreased
to 0.70±0.11 (67% of pre-injury value), 0.57±0.19 (47% of
pre-injury value) and 0.42±0.06 µM (54% of pre-injury value) in the
CCI, CCI-MRS2179 and CCI-2-APB groups, respectively. These post-CCI
values were significantly reduced (P<0.001) compared with those
of the sham group (106% of pre-injury value; 1.09±0.06 µM). The
cortical glucose values of the CCI group returned to near baseline
from the second post-injury fraction onward, without any
significant differences versus the sham group. The cortical glucose
values of the CCI-MRS2179 group started to recover after the second
fraction post-injury and became significantly higher compared with
those of the sham group at 100-120 min (P=0.016) and at 160-180 min
after injury (P=0.041). The glucose level in cortical dialysates of
the CCI-2-APB group started to recover from the second fraction
post-injury but remained significantly lower than the levels in the
sham controls at 20-40 min (P=0.004) and 40-60 min (P=0.001) after
CCI.
The hippocampal glucose levels of the four groups
showed similar trends to those observed in cortex (Fig. 4B). The -20 min pre-injury glucose
concentrations in the sham and CCI groups were 0.97±0.07 and
1.00±0.12 µM, respectively. The CCI-MRS2179 group had significantly
higher (P<0.001) glucose values (1.23±0.07 µM), and the
CCI-2-APB group had significantly lower (P=0.010) glucose values
(0.79±0.07 µM) compared with those of the sham group. A post-injury
decrease was observed in extracellular glucose in the hippocampus
of the CCI group (75% of pre-injury value; 0.75±0.07 µM), which was
significant (P=0.001), compared with that of the sham group (106%
of pre-injury value; 1.02±0.16 µM). In the CCI-MRS2179 group,
glucose levels decreased to 1.03±0.10 µM (84% of pre-injury value)
in the first post-injury fraction, which did not differ from those
of the sham group. The first 20 min post-injury glucose levels of
the CCI-2-APB group were 0.62±0.05 µM (79% of pre-injury value),
which were significantly reduced (P=0.002) compared with those in
the sham group. In the second fraction post-injury, the glucose
levels in hippocampal dialysates returned to baseline level ranges,
with no significant difference between the four groups
thereafter.
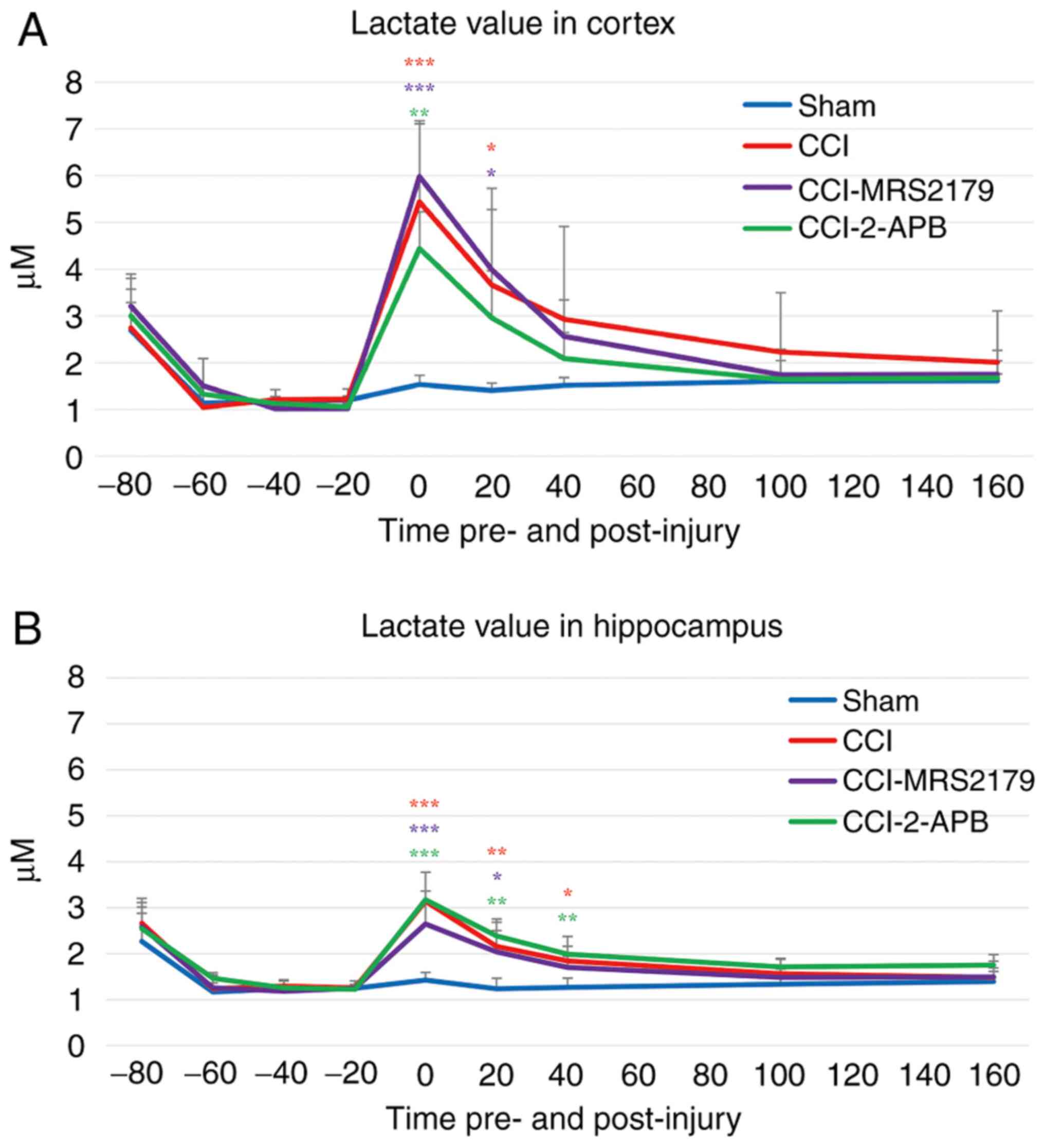
Lactate changes in the extracellular
space
The lactate data from the cerebral microdialysis
samples are represented in Fig. 5A
(cortex) and Fig. 5B
(hippocampus).
The -20 min pre-injury lactate levels in the cortex
of the sham, CCI, CCI-MRS2179 and CCI-2-APB groups were 1.20±0.04,
1.21±0.07, 1.02±0.42 and 1.05±0.07 µM, respectively. There was no
significant difference between the four groups for pre-injury
lactate levels. In the CCI group, the post-injury cortical lactate
value reached 5.44±1.73 µM (449% of pre-injury value) in the first
fraction and 3.67±2.06 µM in the second fraction, with these values
being significantly different (P<0.001 and P=0.037,
respectively) compared with the sham group values of 1.54±0.19 µM
(129% of pre-injury value) and 1.41±0.15 µM. In the CCI-MRS2179
group, cortical lactate in the first post-injury fraction was
5.98±1.13 µM (588% of pre-injury value), and it was 3.99±1.29 µM in
the second fraction. Both values were significantly elevated
(P<0.001 and P=0.021, respectively) compared with the findings
in the sham group. The lactate value of the CCI-2-APB group
increased significantly (P=0.001) above sham levels to 4.44±0.78 µM
(442% of pre-injury value) in the first fraction, but it was not
significantly different from that of the sham group in the second
fraction post-injury.
In the hippocampus, the -20 min pre-injury lactate
values of the sham, CCI, CCI-MRS2179 and CCI-2-APB groups were
1.25±0.04, 1.26±0.15, 1.25±0.04 and 1.22±0.11 µM, respectively
(Fig. 5B). There was no significant
difference between the four groups for pre-injury lactate. In the
CCI group, the lactate concentrations in the hippocampus increased
to 3.14±0.63 (249% of pre-injury value), 2.16±0.60 and 1.84±0.54 µM
in the first, second and third post-injury fractions, respectively,
with these levels being significantly increased (P<0.001,
P=0.006 and P=0.028, respectively) compared with those of the sham
group (114% of pre-injury value; 1.42±0.17, 1.24±0.22 and 1.26±0.21
µM, respectively). In the CCI-MRS2179 group, the lactate values
(212% of pre-injury value; 2.65±0.48 and 2.03±0.47 µM) were
significantly higher (P<0.001 and P=0.019) than those of the
sham group for the first two post-injury fractions. In the
CCI-2-APB group, the post-injury lactate levels in the hippocampus
increased to 3.17±0.19 µM (259% of pre-injury value) in the first
fraction, 2.39±0.30 µM in the second fraction and 1.99±0.17 µM in
the third fraction. These values were all significantly higher than
the lactate levels in the sham group (P<0.001, P=0.001 and
P=0.005, respectively).
Discussion
A major finding of the present study is that a large
quantity of ATP was released into the extracellular spaces in the
cortex and hippocampus immediately after mild experimental TBI. In
addition, it was demonstrated that this ATP release was restricted
by in situ pre-injury administration of the P2Y1 receptor
blocker MRS2179 or the store-operated calcium channel blocker
2-APB. Treatment with 2-APB increased the pre-injury extracellular
glutamate level, but both MRS2179 and 2-APB reduced the extent of
glutamate release induced by trauma. The pre-injury extracellular
glucose level was increased by MRS2179 and decreased by 2-APB, but
the post-CCI decrease in glucose levels was not altered by either
P2Y1 or store-operated calcium channel blockade. Anaerobic
metabolism, which is reflected by an increase in extracellular
lactate after CCI, was not substantially affected by either MRS2179
or 2-APB treatment.
Although it is now recognized that a variety of
brain cells release ATP (28-30),
it seems likely that part of the increase in ATP observed in the
present study derives from astrocytes. It is known that astrocytes
increase their calcium levels and become activated astrocytes in a
wide range including areas that are not directly damaged in
response to environmental changes. In this process,
gliotransmitters ATP, D-serine and glutamate are used as a
paracrine signal to communicate with surrounding astrocytes,
synapses and microglia. The P2 receptor that receives ATP signals
is known to exist in the majority of brain cells. The major P2
receptor subtype to receive ATP signals during gliotransmission is
known to be the P2Y1 receptor (24,31).
Molnar et al (32) reported
that the P2Y1 receptor blocker MRS2179 successfully restricted
gliotransmission in a rat brain slice model. In other studies,
administration of MRS2179 restricted calcium increase and ATP
release of astrocytes, while stimulation of P2Y1 receptors
potentiated ATP release (33,34).
To increase the intracellular calcium level, astrocytes release
calcium from the endoplasmic reticulum or import calcium from the
extracellular space through calcium channels on the cell membrane.
In addition to the voltage dependent calcium channel,
store-operated calcium channels that are activated by calcium
depletion in the endoplasmic reticulum play an important role in
ATP release (35). The blocker
2-APB has been shown to decrease calcium entry through the
store-operated calcium channel, which results in the attenuation of
calcium signaling (36).
Astrocytes are activated by several stimuli such as
glutamate, ischemia and mechanical stress. Koizumi et al
(37) applied mechanical stress to
cultured astrocytes and demonstrated an increase in ATP
bioluminescence signaling. Those results showed that mechanically
stimulated astrocytes create a calcium wave that spreads
concentrically at 21±1.7 µm/sec. It is important to clarify the
role of astrocytes and their effects after TBI, since astrocytes
play a complex role in several functions of the brain such as
inflammation, blood flow regulation, metabolism, plasticity,
epilepsy and mental disorders (38,39).
Of note, the source of ATP detected in the present study may derive
from cells other than astrocytes, such as microglia,
oligodendrocytes, Muller cells, Bergmann glia and neurons, which
have all been reported to release ATP (28-30).
In addition, P2Y1 receptors and store-operated calcium channels are
not astrocyte specific.
The present results revealed that a large quantity
of ATP was released into the extracellular space immediately after
mild experimental TBI, which is consistent with previous studies on
increased ATP signaling in other in vivo injury models
(18,19). However, the extracellular levels
reported in the present study were markedly low compared with the
reported ATP values within the cell (40). After injury, astrocytes probably
release gliotransmitters to increase the activity of neighboring
astrocytes and/or to initiate the microglial response to maintain
electrolyte levels in the extracellular space and to remove debris
from the brain (12,15). Besides this beneficial effect of
microglia, in TBI, it is known that the insult caused by trauma
will cause excessive inflammation that can lead to a harmful
secondary injury (41).
An unexpected finding of the present study is that
MRS2179 reduced extracellular ATP within the baseline sampling
period to an extremely low value, although this result is
consistent with a previous study reporting that administration of
MRS2179 decreased the frequency of astroglial calcium oscillation
in a rat epilepsy model (42). The
low levels of ATP in the CCI-MRS2179 group both pre- and
post-injury suggest that P2Y1 receptor blockade may induce certain
adverse effects if used long term, and future studies should
examine short-term, post-injury treatment. Preventing the
activation of astrocytes for therapeutic purpose needs to be
carefully evaluated, since astrocytes have both beneficial and
harmful aspects in TBI.
The present study also showed that the
store-operated calcium channel was required to release ATP after
relatively mild CCI injury. The store-operated calcium channel,
together with the endoplasmic reticulum, is known to be essential
for astrocytes to control the intracellular calcium level (43). The pre-injury ATP value of the
CCI-2-APB group was significantly higher than that of the
CCI-MRS2179 group, and it was similar to that of the sham and CCI
groups. The 2-APB compound is known to block the activation of
astrocytes in lipopolysaccharide-stimulated cultured astrocytes
(44). The present results suggest
that, in physiological conditions, P2Y1 signaling is required, but
not the store-operated calcium channel, to maintain homeostasis for
glutamate and electrolyte preservation. However, when greater
signal transduction is required in response to an insult such as
trauma, not only P2Y1 signaling but also the store-operated calcium
channel are essential to release sufficient ATP signals into the
extracellular space.
One limitation of the present study is the use of
microdialysis for the study of ATP levels, since the mechanical
insertion of the 0.5-mm diameter probe into the brain parenchyma
constitutes a stab wound injury. Astrocytes are known to release
ATP and create a calcium wave even after mild mechanical stimuli to
a single cell (45). In the current
study the ATP values of the first fraction (-80 min) of baseline
sampling are considered to represent the effects of this stab
wound. Notably, the ATP values were higher in all groups at that
time compared with those at the final baseline (-20 min), and P2Y1
receptor blockade reduced ATP levels within the first 20 min of
sampling period. The smaller post-injury ATP peak in the sham group
likely represents ATP release due to further mechanical stimulation
caused by retraction and re-insertion of probes. As this same
stimulus was provided along with mild TBI to the CCI, CCI-MRS2179
and CCI-2-APB groups, post-injury ATP values must be interpreted
with consideration of this ‘probe injury’ effect, with a portion of
the total ATP peak observed post-injury being attributed to
stimulation from probe re-insertion and a portion attributed to the
mild TBI. The observed increased in ATP after CCI does not appear
to simply reflect ATP leakage from mechanically injured cells,
since the microdialysis probes were inserted into the cortex at the
anterior edge of the contusion site as well as in the more remote,
underlying hippocampus, and in both sampling sites the post-CCI ATP
release was restricted by the P2Y1 receptor blocker and the
store-operated calcium channel blocker. In the present study, a
mild injury level that does not cause visible or histological
contusion was used to minimize any ATP leakage from mechanically
disrupted cells. The use of this mild TBI may explain why the peak
in post-injury ATP levels was similar to the peak observed after
the initial probe insertions for baseline sampling. Future studies
should determine if the post-injury ATP peak is higher or more
enduring to last longer if a moderate-to-severe TBI is administered
at levels sufficient to cause cerebral contusion.
Consistent with previous studies on TBI (20,46),
extracellular glutamate was transiently increased in the current
study. The results of the cortical microdialysis probe showed that
inhibition of the P2Y1 receptor decreased the extracellular
glutamate release after injury. Astrocytes are known to control the
neuronal glutamate release in physiological brain (47,48),
and astrocytes themselves release glutamate as a transmitter into
the extracellular space (49).
MRS2179 probably inactivated astrocytes and neurons, which
restricted glutamate release, thus resulting in lowered glutamate
concentrations in the extracellular space. Glutamate toxicity is a
well-known aggravating factor in cerebral infarct or TBI (50). Thus, reduced cortical glutamate with
MRS2179 treatment suggests that modification of gliotransmission
after injury may have a therapeutic potential to decrease secondary
injury. It is also noteworthy that the pre-injury glutamate levels
increased in the 2-APB-treated group. The increase in extracellular
glutamate in the 2-APB-treated group may reflect that
store-operated calcium channels are essential to re-pump the
glutamate from the extracellular space, which may be associated
with the function of metabotropic glutamate receptors (51).
The current results also confirm previous studies on
early decrease in extracellular glucose with concomitant increase
in extracellular lactate after brain trauma, which is likely to be
due to increased glutamatergic activity with anaerobic glycolysis
(52,53). The baseline glucose and lactate
levels in the present study were consistent with previously
reported values, but were slightly lower compared with those of a
freely moving rat microdialysis model (54,55).
The extracellular glucose levels were increased by MRS2179
treatment, although the relative reduction in glucose post-CCI was
not altered by this P2Y1 receptor blockade. The lactate levels were
not altered by MRS2179 treatment. The higher glucose concentrations
of the CCI-MRS2179 group may represent the P2Y1 receptor blockade
effects on lowering the metabolic activity of cells, although the
post-injury changes suggesting neuronal hyperactivity (decreased
glucose with increased lactate) were not different among the CCI
groups. A previous in vitro study showed that ATP
stimulation of the P2 receptor on astrocytes activates the
phosphoinositide 3 kinase/Akt pathway, which in turn depletes
intracellular glucose, thus leading to glucose import into the cell
via the glucose transporter (56).
Blockade of the P2 receptor with MRS2179 may represent the
inactivation of this pathway, which resulted in the elevated
extracellular glucose level observed in the current study. In
contrast to treatment with MRS2179, 2-APB treatment led to a
decrease in pre-injury extracellular glucose levels. However,
store-operated calcium channel blockade did not exacerbate nor
attenuate the post-CCI induced reduction in glucose or increase in
lactate compared with those of CCI alone. Blocking the
store-operated calcium channel in several injury models has been
reported to be beneficial. For example, 2-APB increased ischemic
tolerance in a neuron astrocyte co-culture model (57), and inhibition of store-operated
calcium channels restored mitochondrial membrane potential, reduced
cytochrome c release and prevented cell death in a
hyperglycemia-induced in vitro neuronal injury model
(58). In contrast to the current
results, previous studies indicated that store-operated calcium
channels function to increase glucose uptake from the extracellular
space, and blockade of these channels with 2-APB will increase the
extracellular glucose concentration (57,59).
Further studies will be required to explain these contradictory
findings and to clarify the roles of store-operated calcium
channels on cerebral metabolism or energy status following
injury.
In conclusion, the present results demonstrated that
post-traumatic ATP signaling can be altered to certain degree by
pharmacological interventions. Further studies on pharmacological
ways of altering the activation states of astrocytes for treating
TBI are warranted.
Acknowledgements
Not applicable.
Funding
Funding: The current study was supported by the UCLA Brain
Injury Research Center.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
NM designed the study and RLS obtained funding. NM
and SSG performed the experiments and NM, SSG and RLS analyzed the
samples. NM and RLS confirm the authenticity of all raw data. NM
and RLS wrote the manuscript. All authors read, edited and approved
the final manuscript.
Ethics approval and consent to
participate
All experimental procedures were approved by the
UCLA Chancellor's Committee for Animal Research. This research was
conducted in accordance with the National Institutes of Health
guide for the care and use of laboratory animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bazargani N and Attwell D: Amines,
astrocytes, and arousal. Neuron. 94:228–231. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Harada K, Kamiya T and Tsuboi T:
Gliotransmitter release from astrocytes: Functional, developmental,
and pathological implications in the brain. Front Neurosci.
9(499)2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
De Pittà M, Brunel N and Volterra A:
Astrocytes: Orchestrating synaptic plasticity? Neuroscience.
323:43–61. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Lalo U, Bogdanov A and Pankratov Y: Age-
and experience-related plasticity of ATP-mediated signaling in the
neocortex. Front Cell Neurosci. 13(242)2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Schwarz Y, Zhao N, Kirchhoff F and Bruns
D: Astrocytes control synaptic strength by two distinct
v-SNARE-dependent release pathways. Nat Neurosci. 20:1529–1539.
2017.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Alberini CM, Cruz E, Descalzi G, Bessieres
B and Gao V: Astrocyte glycogen and lactate: New insights into
learning and memory mechanisms. Glia. 66:1244–1262. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Henneberger C, Papouin T, Oliet SH and
Rusakov DA: Long-term potentiation depends on release of D-serine
from astrocytes. Nature. 463:232–236. 2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Cai C, Zambach SA, Fordsmann JC, Lonstrup
M, Thomsen KJ, Jensen AGK and Lauritzen M: In vivo
three-dimensional two-photon microscopy to study conducted vascular
responses by local ATP ejection using a glass micro-pipette. J Vis
Exp. 148:2019.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Haydon PG and Carmignoto G: Astrocyte
control of synaptic transmission and neurovascular coupling.
Physiol Rev. 86:1009–1031. 2006.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Takano T, Tian GF, Peng W, Lou N, Libionka
W, Han X and Nedergaard M: Astrocyte-mediated control of cerebral
blood flow. Nat Neurosci. 9:260–267. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
11
|
Corps KN, Roth TL and McGavern DB:
Inflammation and neuroprotection in traumatic brain injury. JAMA
Neurol. 72:355–362. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Davalos D, Grutzendler J, Yang G, Kim JV,
Zuo Y, Jung S, Littman DR, Dustin ML and Gan WB: ATP mediates rapid
microglial response to local brain injury in vivo. Nat Neurosci.
8:752–758. 2005.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Jassam YN, Izzy S, Whalen M, McGavern DB
and El Khoury J: Neuroimmunology of traumatic brain injury: Time
for a paradigm shift. Neuron. 95:1246–1265. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Karve IP, Taylor JM and Crack PJ: The
contribution of astrocytes and microglia to traumatic brain injury.
Br J Pharmacol. 173:692–702. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Koizumi S, Shigemoto-Mogami Y, Nasu-Tada
K, Shinozaki Y, Ohsawa K, Tsuda M, Joshi BV, Jacobson KA, Kohsaka S
and Inoue K: UDP acting at P2Y6 receptors is a mediator of
microglial phagocytosis. Nature. 446:1091–1095. 2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Haynes SE, Hollopeter G, Yang G, Kurpius
D, Dailey ME, Gan WB and Julius D: The P2Y12 receptor regulates
microglial activation by extracellular nucleotides. Nat Neurosci.
9:1512–1519. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Winkler U, Seim P, Enzbrenner Y, Köhler S,
Sicker M and Hirrlinger J: Activity-dependent modulation of
intracellular ATP in cultured cortical astrocytes. J Neurosci Res.
95:2172–2181. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Melani A, Turchi D, Vannucchi MG, Cipriani
S, Gianfriddo M and Pedata F: ATP extracellular concentrations are
increased in the rat striatum during in vivo ischemia. Neurochem
Int. 47:442–448. 2005.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wang X, Arcuino G, Takano T, Lin J, Peng
WG, Wan P, Li P, Xu Q, Liu QS, Goldman SA and Nedergaard M: P2X7
receptor inhibition improves recovery after spinal cord injury. Nat
Med. 10:821–827. 2004.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Hinzman JM, Wilson JA, Mazzeo AT, Bullock
MR and Hartings JA: Excitotoxicity and metabolic crisis are
associated with spreading depolarizations in severe traumatic brain
injury patients. J Neurotrauma. 33:1775–1783. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Moro N, Ghavim SS, Harris NG, Hovda DA and
Sutton RL: Pyruvate treatment attenuates cerebral metabolic
depression and neuronal loss after experimental traumatic brain
injury. Brain Res. 1642:270–277. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Shijo K, Sutton RL, Ghavim SS, Harris NG
and Bartnik-Olson BL: Metabolic fate of glucose in rats with
traumatic brain injury and pyruvate or glucose treatments: A NMR
spectroscopy study. Neurochem Int. 102:66–78. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Taylor AN, Tio DL, Paydar A and Sutton RL:
Sex differences in thermal, stress, and inflammatory responses to
minocycline administration in rats with traumatic brain injury. J
Neurotrauma. 35:630–638. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kawamura M, Gachet C, Inoue K and Kato F:
Direct excitation of inhibitory interneurons by extracellular ATP
mediated by P2Y1 receptors in the hippocampal slice. J Neurosci.
24:10835–10845. 2004.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Bowser DN and Khakh BS: Vesicular ATP is
the predominant cause of intercellular calcium waves in astrocytes.
J Gen Physiol. 129:485–491. 2007.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Heinke B and Sandkuhler J: Group I
metabotropic glutamate receptor-induced Ca(2+)-gradients in rat
superficial spinal dorsal horn neurons. Neuropharmacology.
52:1015–1023. 2007.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Singaravelu K, Lohr C and Deitmer JW:
Regulation of store-operated calcium entry by calcium-independent
phospholipase A2 in rat cerebellar astrocytes. J Neurosci.
26:9579–9592. 2006.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Fields RD: Nonsynaptic and nonvesicular
ATP release from neurons and relevance to neuron-glia signaling.
Semin Cell Dev Biol. 22:214–219. 2011.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Illes P, Burnstock G and Tang Y:
Astroglia-derived ATP modulates CNS neuronal circuits. Trends
Neurosci. 42:885–898. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Loiola EC and Ventura AL: Release of ATP
from avian Müller glia cells in culture. Neurochem Int. 58:414–422.
2011.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Muller MS and Taylor CW: ATP evokes
Ca2+ signals in cultured foetal human cortical
astrocytes entirely through G protein-coupled P2Y receptors. J
Neurochem. 142:876–885. 2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Molnar T, Dobolyi A, Nyitrai G, Barabas P,
Heja L, Emri Z, Palkovits M and Kardos J: Calcium signals in the
nucleus accumbens: Activation of astrocytes by ATP and succinate.
BMC Neurosci. 12(96)2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Svobodova I, Bhattaracharya A, Ivetic M,
Bendova Z and Zemkova H: Circadian ATP release in organotypic
cultures of the rat suprachiasmatic nucleus is dependent on P2X7
and P2Y receptors. Front Pharmacol. 9(192)2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Guo H, Liu ZQ, Zhou H, Wang ZL, Tao YH and
Tong Y: P2Y1 receptor antagonists mitigate oxygen and glucose
deprivationinduced astrocyte injury. Mol Med Rep. 17:1819–1824.
2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Koss DJ, Riedel G and Platt B:
Intracellular Ca2+ stores modulate SOCCs and NMDA
receptors via tyrosine kinases in rat hippocampal neurons. Cell
Calcium. 46:39–48. 2009.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Papanikolaou M, Lewis A and Butt AM:
Store-operated calcium entry is essential for glial calcium
signalling in CNS white matter. Brain Struct Funct. 222:2993–3005.
2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Koizumi S, Fujishita K, Tsuda M,
Shigemoto-Mogami Y and Inoue K: Dynamic inhibition of excitatory
synaptic transmission by astrocyte-derived ATP in hippocampal
cultures. Proc Natl Acad Sci USA. 100:11023–11028. 2003.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Diniz C, Rodrigues M, Casarotto PC,
Pereira VS, Crestani CC and Joca SRL: Monoamine involvement in the
antidepressant-like effect induced by P2 blockade. Brain Res.
1676:19–27. 2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Asadollahi M and Simani L: The diagnostic
value of serum UCHL-1 and S100-B levels in differentiate epileptic
seizures from psychogenic attacks. Brain Res. 1704:11–15.
2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Lerchundi R, Huang N and Rose CR:
Quantitative imaging of changes in astrocytic and neuronal
adenosine triphosphate using two different variants of ATeam. Front
Cell Neurosci. 14(80)2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Burda JE, Bernstein AM and Sofroniew MV:
Astrocyte roles in traumatic brain injury. Exp Neurol. 275:305–315.
2016.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Wellmann M, Alvarez-Ferradas C, Maturana
CJ, Saez JC and Bonansco C: Astroglial Ca2+-dependent
hyperexcitability requires P2Y1 purinergic receptors and
pannexin-1 channel activation in a chronic model of epilepsy. Front
Cell Neurosci. 12(446)2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Sakuragi S, Niwa F, Oda Y, Mikoshiba K and
Bannai H: Astroglial Ca2+ signaling is generated by the
coordination of IP3R and store-operated Ca2+
channels. Biochem Biophys Res Commun. 486:879–885. 2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Li JH, Zhao ST, Wu CY, Cao X, Peng MR, Li
SJ, Liu XA and Gao TM: Store-operated Ca2+ channels
blockers inhibit lipopolysaccharide induced astrocyte activation.
Neurochem Res. 38:2216–2226. 2013.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Charles AC, Dirksen ER, Merrill JE and
Sanderson MJ: Mechanisms of intercellular calcium signaling in
glial cells studied with dantrolene and thapsigargin. Glia.
7:134–145. 1993.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Obrenovitch TP and Urenjak J: Is high
extracellular glutamate the key to excitotoxicity in traumatic
brain injury? J Neurotrauma. 14:677–698. 1997.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Jourdain P, Bergersen LH, Bhaukaurally K,
Bezzi P, Santello M, Domercq M, Matute C, Tonello F, Gundersen V
and Volterra A: Glutamate exocytosis from astrocytes controls
synaptic strength. Nat Neurosci. 10:331–339. 2007.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Parri R and Crunelli V: Astrocytes target
presynaptic NMDA receptors to give synapses a boost. Nat Neurosci.
10:271–273. 2007.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Rose CR, Felix L, Zeug A, Dietrich D,
Reiner A and Henneberger C: Astroglial glutamate signaling and
uptake in the hippocampus. Front Mol Neurosci.
10(451)2017.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Dorsett CR, McGuire JL, DePasquale EA,
Gardner AE, Floyd CL and McCullumsmith RE: Glutamate
neurotransmission in rodent models of traumatic brain injury. J
Neurotrauma. 34:263–272. 2017.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Gonzalez-Sanchez P, Del Arco A, Esteban JA
and Satrustegui J: Store-operated calcium entry is required for
mGluR-dependent long term depression in cortical neurons. Front
Cell Neurosci. 11(363)2017.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Carpenter KL, Jalloh I and Hutchinson PJ:
Glycolysis and the significance of lactate in traumatic brain
injury. Front Neurosci. 8(112)2015.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Zeiler FA, Thelin EP, Helmy A, Czosnyka M,
Hutchinson PJA and Menon DK: A systematic review of cerebral
microdialysis and outcomes in TBI: Relationships to patient
functional outcome, neurophysiologic measures, and tissue outcome.
Acta Neurochir (Wien). 159:2245–2273. 2017.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Jones DA, Ros J, Landolt H, Fillenz M and
Boutelle MG: Dynamic changes in glucose and lactate in the cortex
of the freely moving rat monitored using microdialysis. J
Neurochem. 75:1703–1708. 2000.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Clausen F, Hillered L and Gustafsson J:
Cerebral glucose metabolism after traumatic brain injury in the rat
studied by 13C-glucose and microdialysis. Acta Neurochir (Wien).
153:653–658. 2011.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Prebil M, Chowdhury HH, Zorec R and Kreft
M: Changes in cytosolic glucose level in ATP stimulated live
astrocytes. Biochem Biophys Res Commun. 405:308–313.
2011.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Tanaka M, Kawahara K, Kosugi T, Yamada T
and Mioka T: Changes in the spontaneous calcium oscillations for
the development of the preconditioning-induced ischemic tolerance
in neuron/astrocyte co-culture. Neurochem Res. 32:988–1001.
2007.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Xu Z, Xu W, Song Y, Zhang B, Li F and Liu
Y: Blockade of store-operated calcium entry alleviates high
glucose-induced neurotoxicity via inhibiting apoptosis in rat
neurons. Chem Biol Interact. 254:63–72. 2016.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Olianas MC, Dedoni S and Onali P:
Involvement of store-operated Ca(2+) entry in activation of
AMP-activated protein kinase and stimulation of glucose uptake by
M3 muscarinic acetylcholine receptors in human neuroblastoma cells.
Biochim Biophys Acta. 1843:3004–3017. 2014.PubMed/NCBI View Article : Google Scholar
|