Introduction
Neuroinflammation is the general term used to
describe peripheral nerve inflammation and degenerative diseases
caused by various lesions (1).
Neuroinflammation is associated with the progression of several
neurological diseases and is a sign of neurological complications.
In addition, neuroinflammation mainly occurs in the central nervous
system (brain) and is usually caused by infection, irritation or
trauma (2). Nerve tissue necrosis,
cerebral palsy, stroke, brain cancer, brain dementia, brain death
and other factors have been associated with neuroinflammation
(3).
The central nervous system mainly consists of two
types of cells, neurons and glial cells (4). Glial cells are divided into
oligodendrocytes, astrocytes and microglial cells (5). Microglial cells are immune effector
cells. Central nervous system inflammation is caused by the
activation of microglial cells and the release of cytokines and
inflammatory mediators (6).
Normally, microglial cells are in a resting state as neurons
secrete inhibitory factors (7).
During injury or disease, under the influence of pro-inflammatory
stimuli, microglial cells are activated, secrete cytokines and
chemokines, and recruit blood-derived immune cells to the central
nervous system, thus amplifying the inflammatory response of the
central nervous system (7).
However, due to the excessive release of pro-inflammatory cytokines
and neurotoxins, M1-type microglial cells may become overactivated
and produce excessive amounts of cytokines and chemokines, which in
turn, promotes neurotoxicity, damages neurons and can even lead to
neuronal death, thus aggravating inflammation of the central
nervous system (8,9).
Ginkgolide B (GB) is a terpene lactone in the
ginkgolide extracts. Currently, GB is widely used in the treatment
of neurological diseases, including cerebral ischemia (10). Emerging evidence has suggested that
GB exerts anti-inflammatory and neuroprotective effects (11,12).
GB alleviates hypoxia-induced hippocampal neuron injury in mice
through inhibiting oxidative stress and cell apoptosis (13). Furthermore, GB promotes the
differentiation of neural stem cells following cerebral
ischemia-reperfusion injury (14).
However, the effect of GB on the inflammatory response and
activation of microglial cells, to the best of our knowledge, has
not been reported.
The aim of the present study was to investigate the
effects of GB on the activation and inflammatory response of
microglial cells in vivo and in vitro, thus providing
a theoretical basis for the clinical application of GB in the
treatment of neuroinflammatory diseases.
Materials and methods
Cell culture and treatment
The murine microglial cell line BV2 was purchased
from The Cell Bank of Type Culture Collection of The Chinese
Academy of Sciences and cells were cultured in DMEM (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS (Gibco; Thermo
Fisher Scientific, Inc.) and penicillin-streptomycin (100 µg/ml;
Thermo Fisher Scientific, Inc.) at 37˚C in a humidified atmosphere
with 5% CO2. The supplemented DMEM was prepared under
aseptic conditions (DMEM:FBS, 9:1). GB (cat. no. 15291-77-7;
purity, >98%) was purchased from Shanghai Yuanye Biological
Technology Co., Ltd. A total of 100 ng/ml lipopolysaccharide (LPS;
cat. no. S1732; Beyotime Institute of Biotechnology) was used to
stimulate BV2 in six-well plates at a density of 5x105
cells/well for 6 h at 37˚C to mimic the inflammatory environment in
the brain.
Establishment of neuroinflammation
mouse model
A total of 25 male C57BL/6J mice, aged 8 weeks were
provided by the First Affiliated Hospital of Wenzhou Medical
University (Wenzhou, China). The study protocol was approved by the
Ethics Committee of the First Affiliated Hospital of Wenzhou
Medical University. All procedures were in compliance with the
National Institutes of Health Guide for the Care and Use of
Laboratory Animals. To evaluate the anti-inflammatory effects of GB
in vivo, mice were divided into the following five groups:
Control, LPS, GB 30 µmol/l + LPS, GB 60 µmol/l + LPS and GB 120
µmol/l + LPS groups (n=5/group). Mice in the GB + LPS groups were
treated daily by gavage with 30, 60 and 120 µmol/l of GB for 3
days. Mice in the LPS and control groups were treated with
intragastric administration of the same dosage of normal saline for
3 days. Following the last intragastric administration of GB or
normal saline for 1 h, the mice in the LPS groups were
intraperitoneally injected with 5 mg/kg of LPS and after 24 h, all
mice were anesthetized with intraperitoneal injection of 10%
chloral hydrate (400 mg/kg) (15,16).
The animals treated with chloral hydrate did not exhibit any
evident signs of peritonitis. Subsequently, ~0.35 ml of orbital
blood was collected from each mouse and the supernatant was
isolated by centrifugation at 300 x g at 4˚C for 10 min and stored
in a refrigerator at -80˚C. Mice were then sacrificed by cervical
dislocation and euthanasia was confirmed by the cessation of
heartbeat. Whole brains were then removed and fixed with 4%
formaldehyde for 24 h at 4˚C.
Cell Counting Kit-8 (CCK-8) assay
Cell viability was assessed using a CCK-8 assay
(Dojindo Molecular Technologies, Inc.). Cells were seeded in
96-well plates at a density of 1x103 cells/well and
cultured for 24 h. Subsequently, the cells were treated with LPS
and GB as aforementioned. Each well was then supplemented with 10
µl of CCK-8 reagent and the plates were incubated at 37˚C for 4 h.
The absorbance in each well was measured at 450 nm using a Spectra
Max 190 Enzyme standard instrument (Molecular Devices LLC).
ELISA
The secretion levels of IL-6 (cat. no. DY406), IL-1β
(cat. no. DY401) and TNF-α (cat. no. DY410) (all from R&D
Systems, Inc.) in plasma and tissues were measured using Duoset
ELISA kits according to the manufacturer's instructions.
Reverse transcription-quantitative PCR
(RT-qPCR) analysis
Total RNA was extracted from cells and tissues using
an RNA Extraction kit (cat. no. 9767) and cDNA was synthesized
using the PrimeScript® 1st Strand cDNA Synthesis kit
(cat. no. D6110A) (both from Takara Bio, Inc.). The temperature
protocol of the reverse transcription was 42˚C for 30 min, followed
by 85˚C for 5 min, according to the manufacturer's instructions.
The thermocycling conditions were as follows: 95˚C for 10 min; 30
cycles at 94˚C for 15 sec, 55˚C for 30 sec and 72˚C for 30 sec; and
72˚C for 10 min, according to the manufacturer's protocol. The PCR
reactions were carried out using a SYBR green-based system (cat.
no. RR82LR; Takara Bio, Inc.) and the gene fold changes were
calculated using the 2-ΔΔCq method (17). The primer sequences used were as
follows: Inducible nitric oxide (NO) synthase (iNOS) forward,
3'-GTCACCTACCACACCCGAGATG-5' and reverse, 3'-CGCTGGCATTCCGCACAA-5';
cyclooxygenase-2 (COX-2) forward, 3'-TGCAGTGAGCGTCAGGAG-5' and
reverse, 3'-CAAGGATTTGCTGTATGGCTGAG-5'; and GAPDH (reference gene)
forward, 3'-ATCACTGCCACCCAGAAG-5' and reverse,
3'-TCCACGACGGACACATTG-5'.
Western blot analysis
Total proteins were isolated using a RIPA lysis
buffer (Sigma-Aldrich; Merck KGaA) and their concentration was
measured using a BCA protein assay kit (Beyotime Institute of
Biotechnology). Proteins (25 µg/lane) were separated by 10%
SDS-polyacrylamide gel electrophoresis and transferred onto PVDF
membranes. Membranes were blocked with 5% skimmed milk (Beyotime
Institute of Biotechnology) for 1 h at room temperature. Following
blocking, the membranes were first incubated with primary
antibodies overnight at 4˚C and then with the corresponding goat
anti-rabbit horseradish peroxidase-conjugated secondary antibodies
(dilution, 1:5,000; cat. no. ab181658; Abcam) at room temperature
for 2 h. Finally, The ECL™ Western Blotting Analysis System
(Cytiva) and the ImageJ software (version 1.46; National Institutes
of Health) were used to detect the protein blots. In the present
study, the following primary antibodies were used: Anti-iNOS
(dilution, 1:1,000; cat. no. ab178945), anti-COX-2 (dilution,
1:1,000; cat. no. ab179800), anti-allograft inflammatory factor 1
(Iba-1; dilution, 1:1,000; cat. no. ab178846), anti-TNF-α
(dilution, 1:1,000; cat. no. ab215188), anti-IL-1β (dilution,
1:1,000; cat. no. ab2105), anti-IL-6 (dilution, 1:1,000; cat. no.
ab233706) and anti-GAPDH (dilution, 1:1,000; cat. no. ab8245) (all
from Abcam). An additional anti-GAPDH antibody (dilution, 1:1,000;
cat. no. 5174S) was obtained from Cell Signaling Technology,
Inc.
Determination of NO production
NO levels in the culture medium were directly
measured using Total Nitric Oxide Assay Kit (cat. no. S0023;
Beyotime Institute of Biotechnology). Culture supernatants (50 µl)
were mixed with 100 µl Griess reagent and incubated for 3 min at
room temperature. The absorbance of each reaction was measured at
540 nm on a microplate spectrophotometer.
Transwell assay
A Transwell chamber (24-well; 8.0-µm pore membranes;
Corning, Inc.) was used according to the manufacturer's
instructions. Briefly, the inserts had been precoated with Matrigel
(BD Biosciences) at 37˚C for 30 min. 105 cells/well were
seeded into the upper chamber in 100 µl of serum-free DMEM, while
the lower chamber was supplemented with 600 µl of DMEM supplemented
with 10% FBS as a chemoattractant for 24 h at 37˚C. Cells on the
upper surface of the membrane were removed using cotton swabs,
whereas cells on the lower chamber were fixed with 4%
paraformaldehyde for 20 min at room temperature and stained with
0.1% crystal violet solution for 30 min at room temperature.
Finally, the cells were counted under a light contrast microscope
(Olympus Corporation; magnification, x200).
Immunohistochemistry
Tissue samples were fixed in formalin for 24 h at
room temperature, embedded in paraffin, sectioned in 4-5 µm
thickness, and analyzed by immunohistochemistry. Briefly, the
samples were blocked with 5% normal goat serum (cat. no. A7007;
Beyotime Institute of Biotechnology) for 1 h at room temperature,
probed with anti-Iba-1 (dilution, 1:1,000; cat. no. ab178846;
Abcam) at 4˚C overnight and labeled with an anti-rabbit HRP
secondary antibody (cat. no. ab6721; Abcam) for 1 h at room
temperature. Antibody reactions were visualized using
3,3'-diaminobenzidine chromogen staining for 15 min at room
temperature and the slides were then counterstained with
hematoxylin for 3 min at room temperature. Stained tissue sections
were observed under a light microscope (Leica Microsystems, Inc.;
magnification, x200).
Statistical analysis
All data were analyzed with the GraphPad Prism 7.0
software (GraphPad Software, Inc.). Comparisons among multiple
groups were analyzed using one-way ANOVA followed by Tukey's post
hoc test. The data in the present study are presented as the mean ±
SD. All experiments were repeated three times independently.
P<0.05 was considered to indicate a statistically significant
difference.
Results
GB reduces the inflammatory response
and oxidative stress of LPS-induced BV2 cells
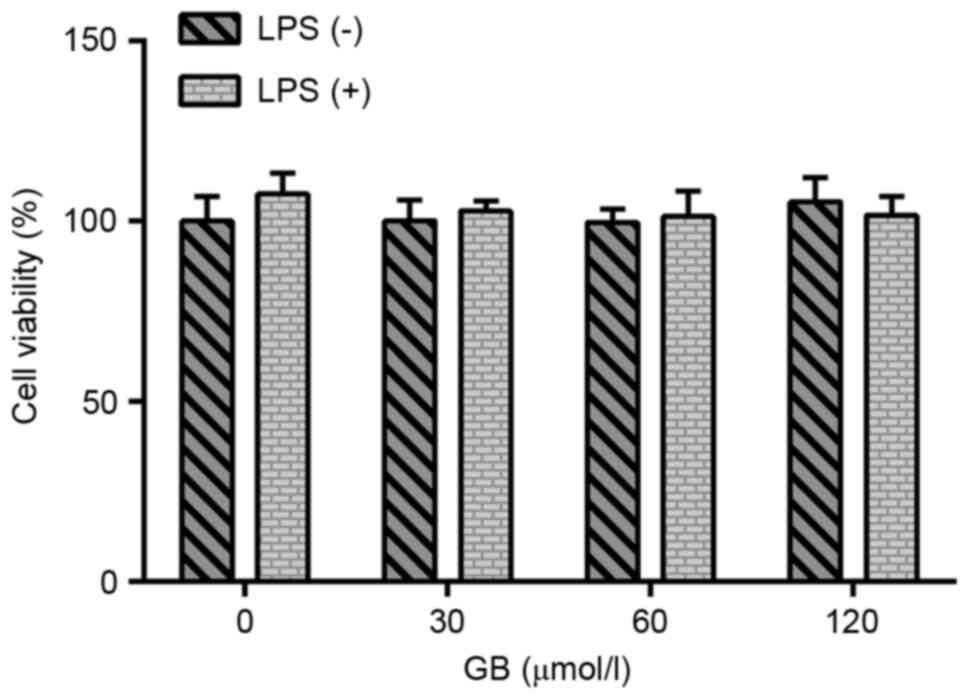
A CCK-8 assay was used to evaluate the effect of GB
at various concentrations (0, 30, 60 and 120 µmol/l) on the
activity of LPS-induced BV2 cells. The results are presented in
Fig. 1. There was no significant
difference in the viability of cells treated with or without LPS,
indicating that treatment of cells with LPS and GB at the various
concentrations had no significant effect on the activity of BV2
cells. Therefore, cells were first induced with LPS to establish
the BV2 cell model and then treated with different concentrations
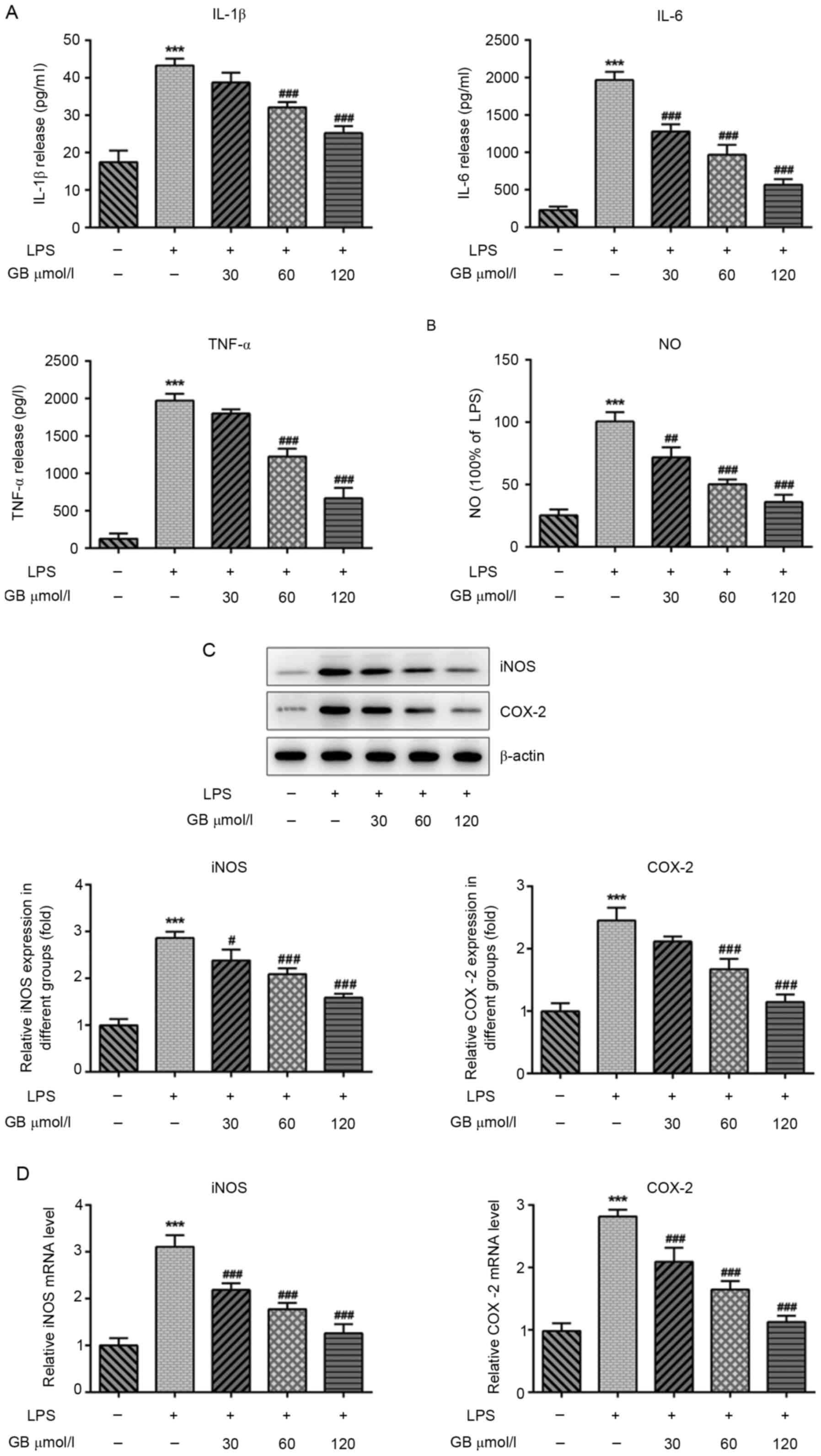
of GB. ELISAs were carried out to determine the effects of GB on
LPS-induced inflammatory factors and oxidative stress. The results
demonstrated that the LPS-induced expression levels of IL-1β, IL-6
and TNF-α were significantly increased compared with that in the
control group. The secretion levels of IL-1β, IL-6 and TNF-α in the
GB + LPS group were notably decreased in a dose-dependent manner
compared with the LPS group (Fig.
2A). Furthermore, Griess assays were used to measure the levels
of NO. Compared with the control group, the levels of NO were
significantly increased in the LPS group. Additionally, the
expression levels of NO in the GB + LPS groups were reduced in a
concentration-dependent manner compared with the LPS group
(Fig. 2B). The expression patterns
of iNOS and COX-2 were consistent with that of NO, as determined by
western blotting (Fig. 2C) and
RT-qPCR (Fig. 2D) analyses. These
findings indicated that GB attenuated the LPS-induced inflammatory
response and oxidative stress.
GB alleviates the LPS-induced
migration of BV2 cells
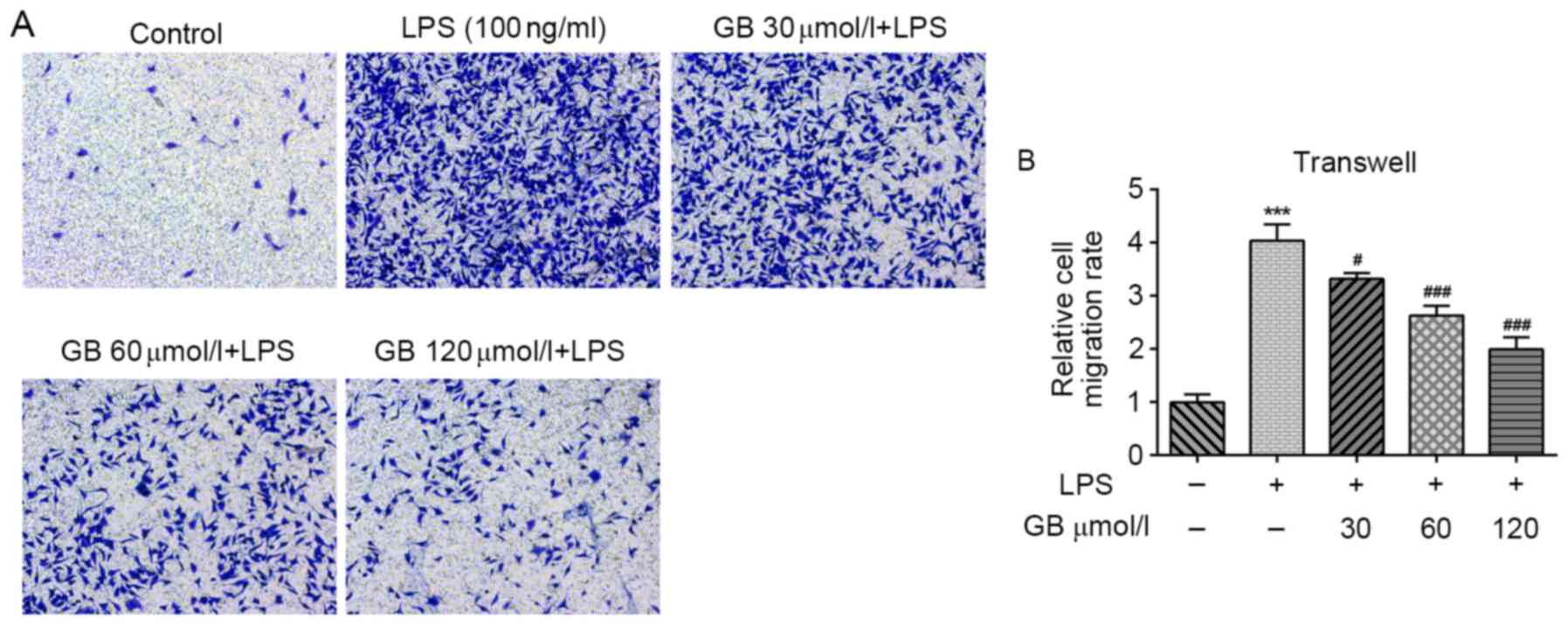
A transwell assay was carried out to evaluate the
cell migratory ability. As shown in Fig. 3A and B, compared with the untreated control
group, the cell migratory ability in the LPS group was notably
enhanced. In addition, the cell migratory ability in the GB + LPS
groups was decreased in a dose-dependent manner compared with the
LPS group. These results suggested that GB alleviated LPS-induced
BV2 cell migration.
GB reduces the activation of
microglial cells in the hippocampal dentate gyrus and striatum of
LPS-induced mice
Subsequently, the in vivo effects of GB on
the activation of BV2 cells and the inflammatory response of cells
of the hippocampal dentate gyrus and striatum microglia were
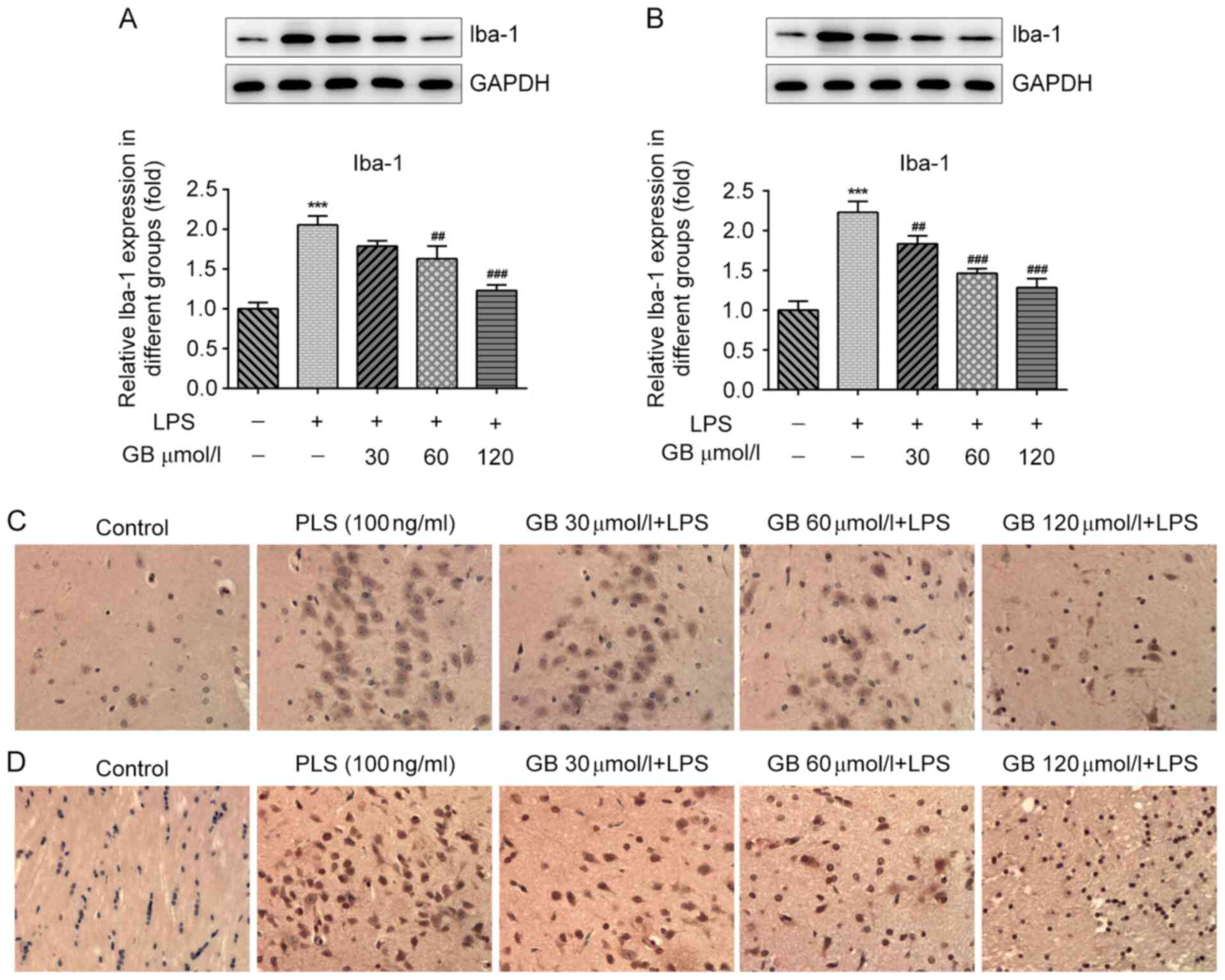
investigated in LPS-induced mice. The expression levels of Iba-1, a
microglial cell surface protein marker, were determined to evaluate
the activation status of BV2(18).
The western blotting results revealed that, compared with the
control group, the expression levels of Iba-1 in the dentate gyrus
of the hippocampus of mice were significantly increased. In
addition, compared with the LPS group, Iba-1 expression in the GB +
LPS group was notably decreased in a GB concentration-dependent
manner (Fig. 4A). The expression
status of Iba-1 in the striatum was consistent with that in the
dentate gyrus (Fig. 4B). In
addition, the expression of Iba-1 in the hippocampal dentate gyrus
(Fig. 4C) and striatum (Fig. 4D) of mice was also evaluated by
immunohistochemistry. Compared with the control group, more Iba-1
positive cells in the dentate gyrus of the hippocampus and striatum
were apparent in the LPS group. In addition, compared with the LPS
group, the number of Iba-1 positive cells in the dentate gyrus of
the hippocampus and striatum in the GB + LPS group was notably
decreased in a GB concentration-dependent manner. The results were
consistent with those observed in the western blot analysis.
Overall, the results demonstrated that GB attenuated the activation
of LPS-induced microglial cells in the hippocampal dentate gyrus
and striatum.
GB reduces the inflammatory response
in the hippocampal dentate gyrus and striatum of LPS-induced
mice
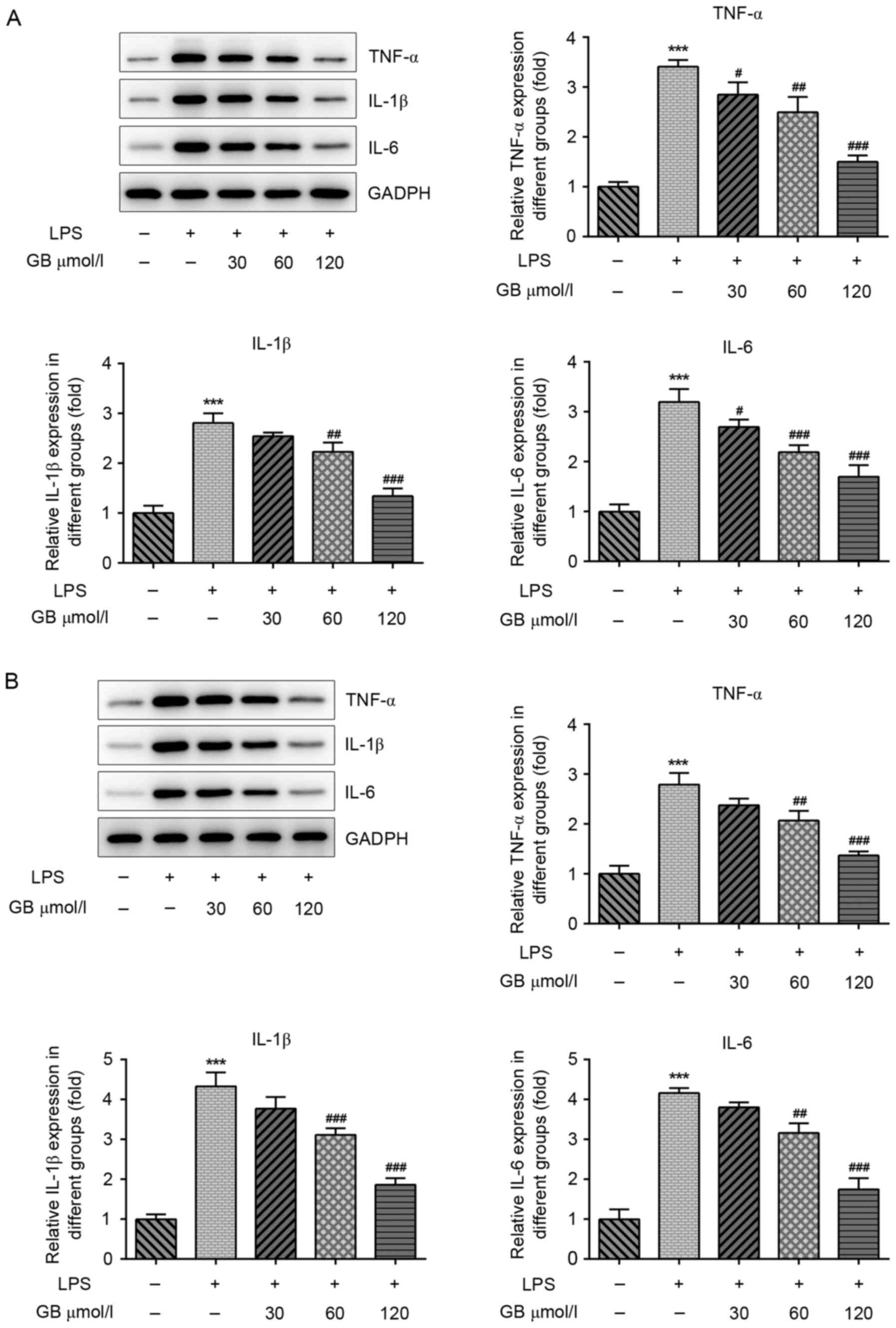
Subsequently, the expression levels of TNF-α, IL-1β
and IL-6 in the hippocampal dentate gyrus and striatum of
LPS-induced mice were also detected, (Fig. 5). The western blotting results
revealed that the protein expression levels of TNF-α, IL-1β and
IL-6 in the hippocampal dentate gyrus of LPS-induced mice were
significantly increased compared with that in the control group.
Compared with the LPS only-treated group, the expression of TNF-α,
IL-1β and IL-6 in the hippocampal dentate gyrus of mice treated
with various concentrations of GB + LPS was decreased in a
dose-dependent manner (Fig. 5A).
The expression patterns of all three cytokines in the striatum were
consistent with those observed in the hippocampal dentate gyrus
(Fig. 5B). These findings indicated
that GB reduced the expression of TNF-α, IL-1β and IL-6 in the
hippocampal dentate gyrus and striatum of LPS-induced mice.
Discussion
In the present study, BV2 microglial cells were
induced with LPS to establish a neuroinflammation cell model in
vitro. Various concentrations of GB inhibited the inflammatory
response and cell activation of LPS-induced BV2 microglial cells.
In vivo, the results demonstrated that GB attenuated the
activation and inflammatory response of microglial cells in the
hippocampal dentate gyrus and striatum in the neuroinflammation
mouse model.
Neuroinflammation is a complex and ordered process,
involving a variety of glial and peripheral immune cells of the
central nervous system (7).
Neuroinflammation is usually mediated by activated microglia and
astrocytes via the secretion of pro-inflammatory cytokines
(19). Microglia are the resident
immune cells in the brain (20). A
moderate neuroinflammatory response mediated by microglia is the
defense mechanism of the central nervous system against infection
and injury (21), while persistent
or excessive inflammatory responses can lead to progressive nerve
cell damage and dysfunction (22).
A previous study has shown that excessive neuroinflammation induced
by microglia activation is an important pathological mechanism for
the occurrence and development of neurodegenerative diseases
(23). Therefore, inhibiting the
continuous activation of microglia and reducing the excessive
release of pro-inflammatory mediators are considered to be
effective interventions for the prevention and treatment of
neurodegenerative diseases (24).
Herein, BV2 microglial cells were induced with LPS to establish a
neuroinflammation model. Subsequently, the activation of
hippocampal dentate gyrus and striatum microglia was evaluated
following LPS induction in mice to determine whether the
neuroinflammatory response was successfully induced.
NO is an important inflammatory mediator produced by
the body and its excessive production is closely associated with
various neurodegenerative diseases (25). Microglia express iNOS in the
activated state, which promotes the production of a large amount of
NO (16). Excessive NO activates
NF-κB and induces the production of inflammatory factors such as
IL-1β and TNF-α. The production of these inflammatory factors, in
turn, activates iNOS and further promotes the body to produce more
NO, thereby producing a sustained toxic effect on cells. It has
been reported that tissue trauma or infection may cause the release
of pro-inflammatory cytokines, such as peripheral and central
IL-1β, TNF-α and IL-6(26). A
previous study showed that Porphyromonas gingivalis LPS
induced neuronal inflammation in C57BL/6 mice and significantly
increased the expression levels of IL-6, TNF-α and IL-1β (27). In the present study, the expression
of TNF-α, IL-1β and IL-6 in BV2 cells, as well as the hippocampal
dentate gyrus and striatum of C57BL/6 mice, was also significantly
increased following induction with LPS.
GB exerts anti-inflammatory and antioxidant
pharmacological effects (28). In
HUVECs, GB mediates the inhibition of inflammatory cascades and
alters lipid metabolism through targeting the expression and
function of proprotein convertase subtilisin/kexin type 9(11). In addition, GB regulates myocardial
inflammation induced by ischemia-reperfusion injury via the
A20/NF-κB signaling pathway (29).
In the present study, GB inhibited the expression of
inflammation-related factors in LPS-induced microglial cells and
inhibited the activation of microglial cells, suggesting that GB
may serve a therapeutic role in LPS-induced neuroinflammation.
Attention should be paid to the following
limitations in the present study. Through in vivo and in
vitro experiments, it was demonstrated that GB inhibited
LPS-induced neuroinflammation and microglial cell activation;
however, the specific mechanism of action remains to be further
explored. In addition, the effect of GB on other cells in
neuroinflammation, such as neuronal cells, oligodendrocytes or
astrocytes, and whether GB can also inhibit the inflammation and
activation of these cells remains to be explored.
Overall, the findings of the present study revealed
that GB alleviated the inflammatory response and activation of
LPS-induced BV2 cells in vivo and in vitro, thus
providing the theoretical basis for the clinical application of GB
in the treatment of neuroinflammation.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MS wrote the manuscript and analyzed the data. YS
and YZ carried out the experiments, supervised the present study,
searched the literature and revised the manuscript. MS and YS
confirm the authenticity of all the raw data. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
The study protocol was approved by Ethics Committee
of the First Affiliated Hospital of Wenzhou Medical University
(Wenzhou, China). All the procedures were in compliance with The
National Institutes of Health Guide for the Care and Use of
Laboratory Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Klein RS, Garber C and Howard N:
Infectious immunity in the central nervous system and brain
function. Nat Immunol. 18:132–141. 2017.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Schain M and Kreisl WC: Neuroinflammation
in neurodegenerative disorders-a review. Curr Neurol Neurosci Rep.
17(25)2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Matsuda M, Huh Y and Ji RR: Roles of
inflammation, neurogenic inflammation, and neuroinflammation in
pain. J Anesth. 33:131–139. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Song I and Dityatev A: Crosstalk between
glia, extracellular matrix and neurons. Brain Res Bull.
136:101–108. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Allen NJ and Lyons DA: Glia as architects
of central nervous system formation and function. Science.
362:181–185. 2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kinney JW, Bemiller SM, Murtishaw AS,
Leisgang AM, Salazar AM and Lamb BT: Inflammation as a central
mechanism in Alzheimer's disease. Alzheimers Dement (N Y).
4:575–590. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yang QQ and Zhou JW: Neuroinflammation in
the central nervous system: Symphony of glial cells. Glia.
67:1017–1035. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Gonzalez H, Elgueta D, Montoya A and
Pacheco R: Neuroimmune regulation of microglial activity involved
in neuroinflammation and neurodegenerative diseases. J
Neuroimmunol. 274:1–13. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Burguillos MA, Deierborg T, Kavanagh E,
Persson A, Hajji N, Garcia-Quintanilla A, Cano J, Brundin P,
Englund E, Venero JL and Joseph B: Caspase signalling controls
microglia activation and neurotoxicity. Nature. 472:319–324.
2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Nabavi SM, Habtemariam S, Daglia M, Braidy
N, Loizzo MR, Tundis R and Nabavi SF: Neuroprotective effects of
ginkgolide B against ischemic stroke: A review of current
literature. Curr Top Med Chem. 15:2222–2232. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wang G, Liu Z, Li M, Li Y, Alvi SS, Ansari
IA and Khan MS: Ginkgolide B mediated alleviation of inflammatory
cascades and altered lipid metabolism in HUVECs via targeting
PCSK-9 expression and functionality. Biomed Res Int.
2019(7284767)2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Feng Z, Sun Q, Chen W, Bai Y, Hu D and Xie
X: The neuroprotective mechanisms of ginkgolides and bilobalide in
cerebral ischemic injury: A literature review. Mol Med.
25(57)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Zheng PD, Mungur R, Zhou HJ, Hassan M,
Jiang SN and Zheng JS: Ginkgolide B promotes the proliferation and
differentiation of neural stem cells following cerebral
ischemia/reperfusion injury, both in vivo and in vitro. Neural
Regen Res. 13:1204–1211. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Li MY, Chang CT, Han YT, Liao CP, Yu JY
and Wang TW: Ginkgolide B promotes neuronal differentiation through
the Wnt/β-catenin pathway in neural stem cells of the postnatal
mammalian subventricular zone. Sci Rep. 8(14947)2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Li W, Qinghai S, Kai L, Xue M, Lili N,
Jihua R, Zhengxiang L, Xiaoling L, Di G, Qi Y, et al: Oral
administration of Ginkgolide B alleviates hypoxia-induced neuronal
damage in rat hippocampus by inhibiting oxidative stress and
apoptosis. Iran J Basic Med Sci. 22:140–145. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Qu Z, Chen Y, Luo ZH, Shen XL and Hu YJ:
7-methoxyflavanone alleviates neuroinflammation in
lipopolysaccharide-stimulated microglial cells by inhibiting
TLR4/MyD88/MAPK signalling and activating the Nrf2/NQO-1 pathway. J
Pharm Pharmacol. 72:385–395. 2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Norden DM, Trojanowski PJ, Villanueva E,
Navarro E and Godbout JP: Sequential activation of microglia and
astrocyte cytokine expression precedes increased Iba-1 or GFAP
immunoreactivity following systemic immune challenge. Glia.
64:300–316. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Shabab T, Khanabdali R, Moghadamtousi SZ,
Kadir HA and Mohan G: Neuroinflammation pathways: A general review.
Int J Neurosci. 127:624–633. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ottum PA, Arellano G, Reyes LI,
Iruretagoyena M and Naves R: Opposing roles of interferon-gamma on
cells of the central nervous system in autoimmune
neuroinflammation. Front Immunol. 6(539)2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Glass CK, Saijo K, Winner B, Marchetto MC
and Gage FH: Mechanisms underlying inflammation in
neurodegeneration. Cell. 140:918–934. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ferreira ST, Clarke JR, Bomfim TR and De
Felice FG: Inflammation, defective insulin signaling, and neuronal
dysfunction in Alzheimer's disease. Alzheimers Dement. 10 (1
Suppl):S76–S83. 2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Subhramanyam CS, Wang C, Hu Q and Dheen
ST: Microglia-mediated neuroinflammation in neurodegenerative
diseases. Semin Cell Dev Biol. 94:112–120. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Tang Y and Le W: Differential roles of M1
and M2 microglia in neurodegenerative diseases. Mol Neurobiol.
53:1181–1194. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Baruch K, Kertser A, Porat Z and Schwartz
M: Cerebral nitric oxide represses choroid plexus NFκB-dependent
gateway activity for leukocyte trafficking. EMBO J. 34:1816–1828.
2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Han PF, Wei L, Duan ZQ, Zhang ZL, Chen TY,
Lu JG, Zhao RP, Cao XM, Li PC, Lv Z and Wei XC: Contribution of
IL-1β, 6 and TNF-α to the form of post-traumatic osteoarthritis
induced by ‘idealized’ anterior cruciate ligament reconstruction in
a porcine model. Int Immunopharmacol. 65:212–220. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Zhang J, Yu C, Zhang X, Chen H, Dong J, Lu
W, Song Z and Zhou W: Porphyromonas gingivalis lipopolysaccharide
induces cognitive dysfunction, mediated by neuronal inflammation
via activation of the TLR4 signaling pathway in C57BL/6 mice. J
Neuroinflammation. 15(37)2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhu PC, Tong Q, Zhuang Z, Wang ZH, Deng
LH, Zheng GQ and Wang Y: Ginkgolide B for myocardial
ischemia/reperfusion injury: A preclinical systematic review and
meta-analysis. Front Physiol. 10(1292)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhang R, Xu L, Zhang D, Hu B, Luo Q, Han
D, Li J and Shen C: Cardioprotection of ginkgolide B on myocardial
ischemia/reperfusion-induced inflammatory injury via regulation of
A20-NF-κB pathway. Front Immunol. 9(2844)2018.PubMed/NCBI View Article : Google Scholar
|