Introduction
Parkinson's disease (PD), the second-most common
neurodegenerative disorder that affects 2-3% of the population ≥65
years of age in the world, is characterized by quiescent tremor,
motor retardation, myotonia and postural balance disorder (1). However, this disease has two main
pathological characteristics: Massive degeneration and loss of
dopaminergic neurons in substantia nigra and formation of Lewis
bodies (1,2). Currently, the pathogenesis of PD is
still unclear but mitochondrial damage leading to overproduction of
reactive oxygen species (ROS) is an important cause of the
activation of microglia and the loss of dopaminergic neurons
(3,4). In addition, overwhelming evidence
indicates that mitochondrial disorder and oxidative stress serve
important roles in the development of PD (5,6).
Furthermore, activated microglia, which can lead to oxidative
stress, increase the level of ROS and directly or indirectly
leading to the death of dopaminergic neurons in the substantia
nigra, which is one of the pathological characteristics of PD
(5). Activated microglia may thus
be a key target for the treatment of PD.
Rotenone, an inhibitor of mitochondrial complex I,
was first used in PD research in the 1980s (7). Research on the application of rotenone
to treat Parkinson's disease has continued to increase (8). In terms of molecular mechanism,
rotenone induces mitochondrial dysfunction and increases ROS
production, which are implicated in the degeneration of
dopaminergic neurons (9). Previous
studies have reported that rotenone activates microglia cells by
causing mitochondrial dysfunction and oxidative stress via
inhibiting oxidative respiratory chain complex and increasing the
production of ROS. Therefore rotenone treatment of cells can also
be used as an in vitro model of PD (10,11).
Dl-butylphthalide (NBP), a compound isolated from
Chinese celery, was approved by the China Food and Drug
Administration for the treatment of acute ischemic stroke (12). Therapy using NBP has been
recommended by Chinese guidelines for cerebral collateral
circulation in ischemic stroke (13). The mechanisms of NBP in ischemic
stroke treatment may be mediated through different processes
including anti-oxidant activity, protection of mitochondria,
anti-inflammation, anti-thrombosis and anti-apoptosis (14). A recent review indicated that the
therapeutic effect of NBP is not limited to cerebrovascular
diseases but also treats neurodegeneration diseases (15). Our previous study reviewed the
neuroprotective mechanism of NBP and found that reducing oxidative
stress is one of the most important mechanisms (16). However, the mechanism through which
NBP inhibits the microglial oxidative stress is not completely
understood. Much less is known about its neuroprotection and
regulation of the underlying signaling pathways.
Therefore, the present study aimed to investigate
whether NBP exerts neuroprotective effects on rotenone-induced
mitochondrial dysfunction and oxidative stress in BV2 cells. To
elucidate the effects of the drug in vitro more fully,
rotenone was used to induce damage to mitochondria in BV2 cells.
Then, the effects of NBP on rotenone-induced morphological changes
in microglia, mitochondrial dysfunction and ROS production and the
underlying signaling pathways were examined. The results may aid in
the development of novel treatment strategies for Parkinson's
disease.
Materials and methods
Reagents
Rotenone was purchased from Sigma-Aldrich (Merck
KGaA) and NBP was provided by China Shijiazhuang Pharmaceutical
company. The Mitochondrial Membrane Potential Assay kit (with JC-1)
was purchased from Elabscience Biotechnology.
Dichlorodihydrofluorescein diacetate (DCFH-DA) and Cell Counting
Kit-8 (CCK-8) were obtained from Beyotime Institute of
Biotechnology. The antibodies against Kelch-like ECH-associated
protein 1 (Keap1), nuclear respiratory factor-2 (Nrf2) and heme
oxygenase-1 (HO-1) were obtained from Wanleibio and antibodies
against β-tubulin and lamin B were purchased from Cell Signaling
Technology, Inc. The NE-PER nuclear and cytoplasmic extraction
reagent kit was obtained from Thermo Fisher Scientific, Inc.
Cell culture
BV2 microglial cells were provided by the Cell
Culture Center of the Chinese Academy of Medical Sciences. Cells
were maintained in Dulbecco's modified Eagle's medium (DMEM;
HyClone; Cytiva) high glucose, supplemented with 10%
heat-inactivated fetal bovine serum (FBS; Biological Industries) in
a 5% CO2 incubator set at 37˚C. On reaching
approximately 80% confluency, the cells were passaged at 1:3 ratio
using trypsin (HyClone; Cytiva) solution (0.25% trypsin without
EDTA).
Determination of cell viability
BV2 cell viability under different concentrations of
rotenone and NBP was assessed using the CCK-8 assay. Briefly, BV2
cells were seeded in 96-well plates (Mettler-Toledo Rainin, LLC) at
a density of 3,000 cells/well for 24 h. The medium was then
replaced with DMEM with 10% FBS containing different doses of
rotenone or NBP (3 replicate wells for each treatment). After 24 h,
10 µl CCK-8 reagent was added to each well. The absorbance was then
recorded within 4 h at 450 nm using the Model 550 microplate reader
(Bio-Rad Laboratories, Inc.). Data were collected from three
independent experiments.
Detection of mitochondrial membrane
potential
BV2 cells were seeded in 6-well plates at a density
of 4x105 cells/well for 24 h, followed by treatment with
rotenone (0.05 µM) with or without NBP (200 µM) for 24 h. Next, the
cells were harvested by using trypsin (0.25% trypsin without EDTA),
resuspended in phosphate buffered saline (PBS), immediately stained
with JC-1 (1 mg/ml in DMSO) and incubated at 37˚C for 30 min in
darkness. After washing twice in ice-cold PBS, the cells were
analyzed using the CytoFLEX flow Cytometer (Beckman Coulter, Inc.).
Data were collected from three independent experiments.
Measurement of intracellular ROS
level
Intracellular ROS generation was examined using the
DCFH-DA method. BV2 cells were seeded at a density of
4x105 cells/well in 6-wells plates (Mettler-Toledo
Rainin, LLC) for 24 h, followed by treatment with rotenone (0.05
µM) with or without NBP (200 µM)for 24 h. The cells were then
incubated with 10 µM DCFH-DA for 30 min at 37˚C in 5%
CO2. Next, the cells were washed thrice with PBS and
visualized under a fluorescence microscope (Leica, Germany). Images
were captured through band pass filters of 505-530 nm (10X).
Finally, the fluorescence intensity of each group was analyzed by
ImageJ 1.8 software (National Institutes of Health).
Preparation of nuclear extract
BV2 cells were seeded at a density of
4x105 cells/well in 6-well plates for 24 h, followed by
treatment with rotenone (0.05 µM) with or without NBP (200 µM) for
another 24 h. The cells were then used for nuclear extract
preparation using the NE-PER nuclear and cytoplasmic extraction
reagent kit (Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions. The concentration of the extracted
protein was evaluated using a bicinchoninic acid (BCA) protein
assay kit (Beyotime Institute of Biotechnology).
Western blotting
In each group, proteins (20 µg for cytoplasmic
protein and 5 µg for nuclear protein) were separated on 10%
SDS-polyacrylamide gels (Beyotime Institute of Biotechnology) and
then transferred onto polyvinylidene difluoride membranes (EMD
Millipore). Subsequently, the membranes were blocked with Tris
buffered saline with 5% Tween-20 containing 5% non-fat milk for 1 h
at room temperature. The levels of Keap1 (cat. no. WL03285;
Wanleibio), Nrf2 (cat. no. WL02135; Wanleibio) and HO-1 (cat. no.
WL02400; Wanleibio) were detected by immunoblotting using specific
primary and secondary antibodies. β-tubulin (cat. no. 2128S; Cell
Signaling Technology, Inc.) and Lamin B (cat. no. 13435S; Cell
Signaling Technology, Inc.) were used as controls to ensure equal
loading of cell lysates. All primary antibodies were rabbit
antibodies and were used at a concentration of 1:1,000. The
secondary antibody (cat. no. IH-0011; Beijing Dingguo Changsheng
Biotechnology Co., Ltd.) used was an anti-rabbit antibody and was
used at a concentration of 1:5,000. The primary antibody was
incubated at 4˚C for 20 h and the secondary antibody was incubated
at room temperature for 1 h. ECL (cat. no. 32106; Thermo Fisher
Scientific) was used to develop the bands. The band intensity was
analyzed using ImageJ 1.8 software. Data were analyzed by one-way
analysis of variance (ANOVA).
Statistical analysis
All images were analyzed with ImageJ 1.8 software.
Statistical analysis was performed by Dunnett test following
one-way ANOVA using GraphPad Prism 8 software (GraphPad Software
Inc.). All data were expressed as mean ± standard deviation from
three independent experiments. P<0.05 was considered to indicate
a statistically significant difference.
Results
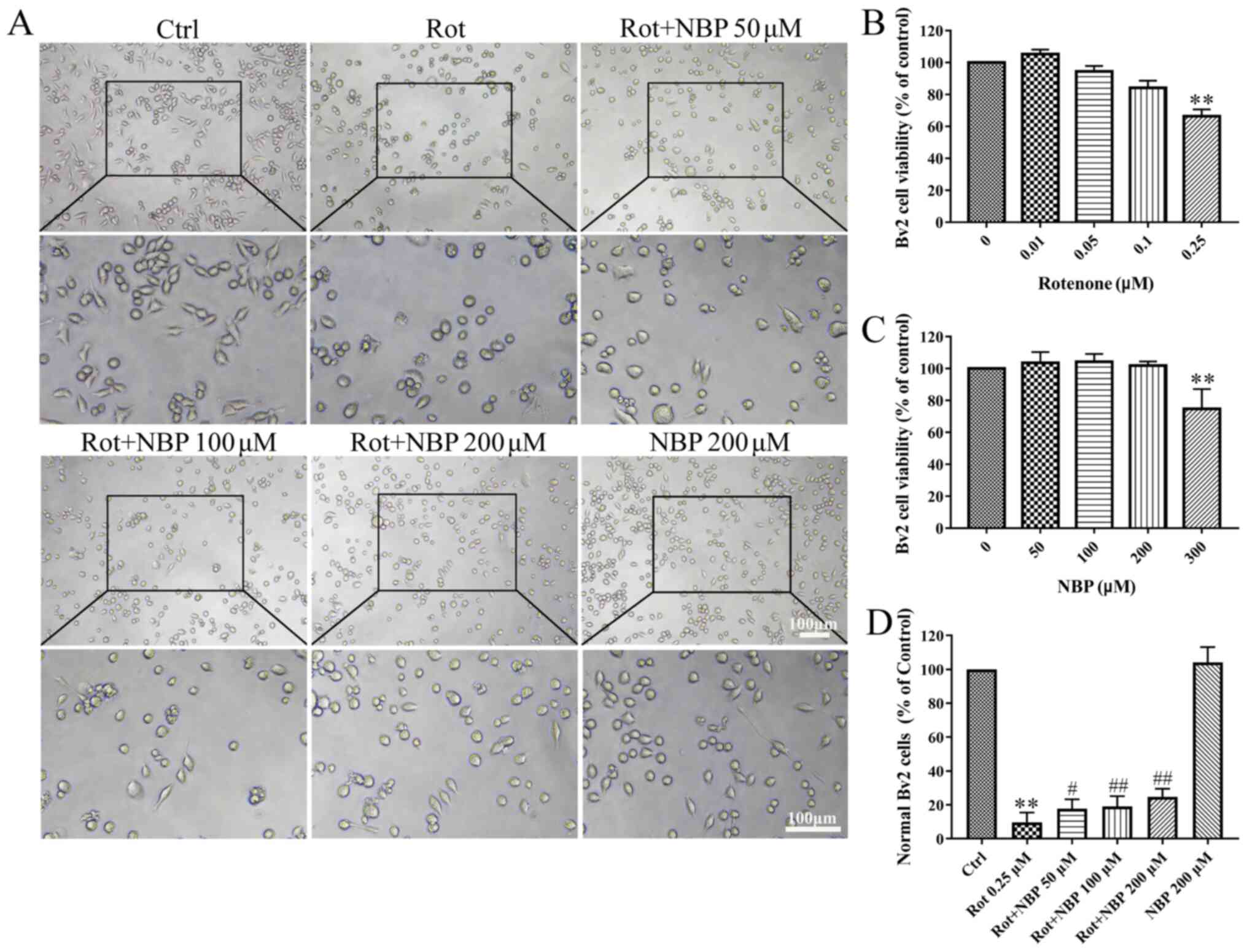
Effect of different concentrations of
NBP and rotenone in BV2 cells
First, the cytotoxicity of NBP and rotenone in BV2
cells was determined. The concentration of rotenone chosen was 0.25
µM with or without different dose of NBP for determination of the
neuroprotective effect of NBP on BV2 cells. The results showed that
NBP significantly protected microglia cells from rotenone-induced
morphological changes. The degree of protection was dependent on
the concentration of NBP as shown in Fig. 1A and D. Cell viability of the control was set at
100% as shown in Fig. 1B and
C. Therefore, Rotenone
significantly diminished survival rates of BV2 cells while NBP
rescued that in a dose-dependent manner.
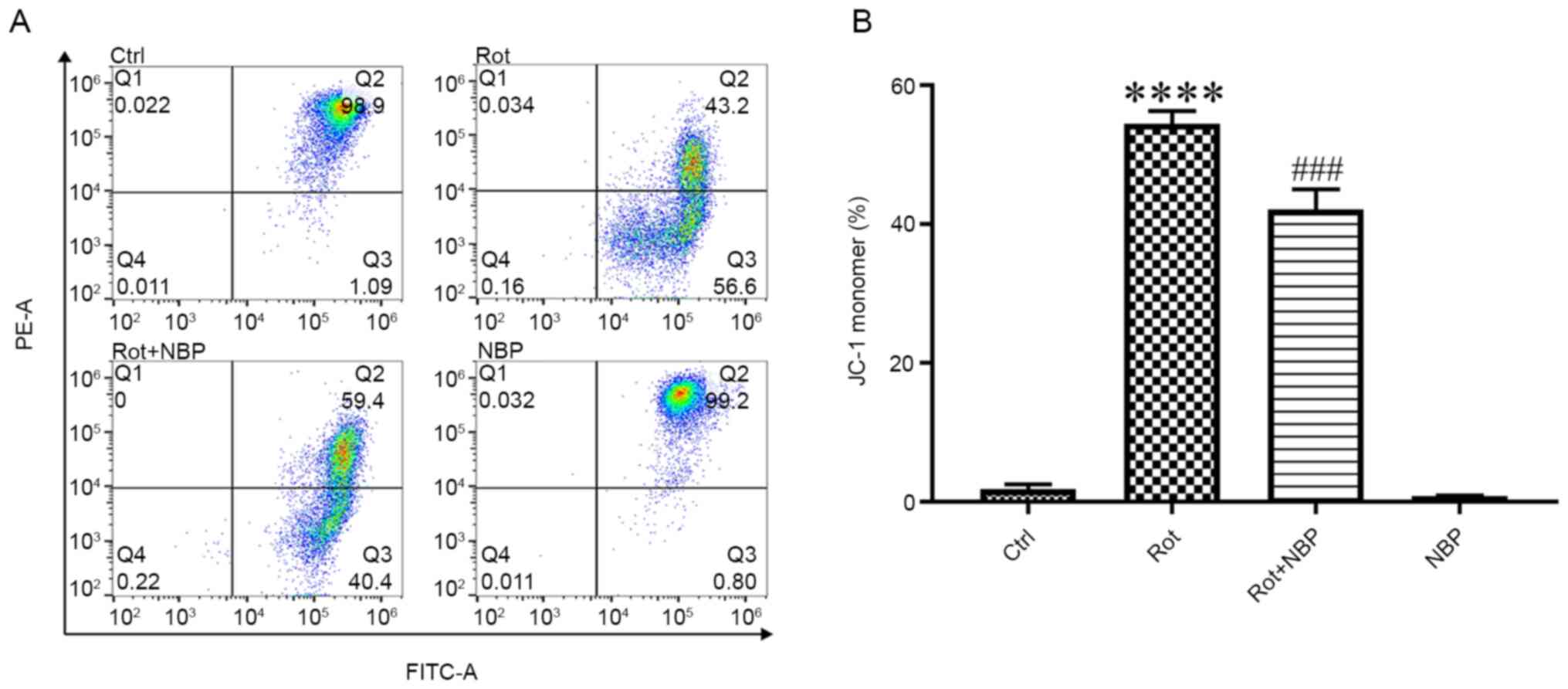
NBP protects mitochondrial
function
The mitochondrial membrane potential (MMP) controls
ATP synthesis and the generation of ROS. A decrease in MMP
represents a decrease in mitochondrial function (2). When stained with JC-1 dye,
phycoerythrin A fluorescence of mitochondria indicates the
formation of J-aggregates at high negative MMP and fluorescein
isothiocyanate A fluorescence of mitochondria indicates the
formation of JC-1 monomers at low MMP. The results of the present
study showed that treatment of cells with rotenone (0.05 µM) for 24
h caused the MMP reduction and NBP significantly inhibited the
formation of JC-1 monomers in rotenone-treated BV2 cells (Fig. 2).
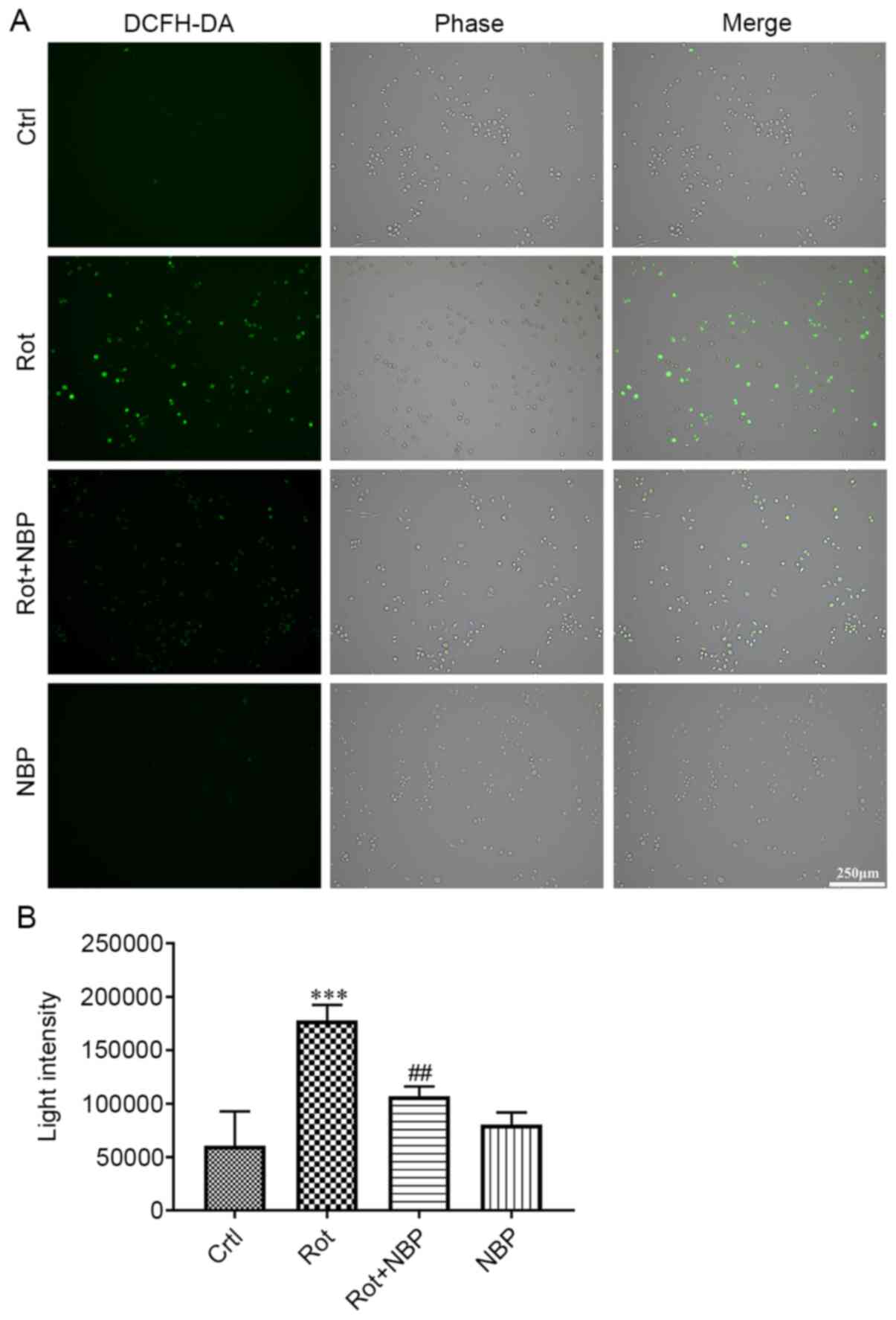
NBP reduces intracellular ROS in
rotenone-treated BV2 cells
The present study attempted to verify whether NBP
reduced intracellular ROS generation in rotenone-treated BV2 cells.
As shown in Fig. 3, the
intracellular ROS of BV2 cells increased following treatment with
rotenone for 24 h. However, BV2 cells treated with rotenone and NBP
for 24 h showed a clear drop in ROS levels when compared with cells
treated with rotenone alone. Treatment of cells with NBP alone had
no effect on ROS levels when compared with the control group.
NBP activates Keap1/Nrf2/HO-1
signaling pathway
The Keap1/Nrf2/HO-1 signaling pathway was examined
by western blotting. The expression of this pathway in the
cytoplasm s shown in Fig. 4A.
Following treatment with rotenone, the level of Keap1 in BV2 cells
decreased significantly compared with the control group as shown in
Fig. 4B. However, on inclusion of
NBP in rotenone treated cells, the level of Keap1 decreased
significantly (Fig. 4B). By
contrast, following rotenone treatment, the level of HO-1 in BV2
cells decreased significantly compared with control group, whereas
on addition of NBP, the level of HO-1 increased significantly
compared with cells treated with rotenone alone (Fig. 4C). Treatment of cells with rotenone
had no significant effect on the level of Nrf2 in cytoplasm
compared with control group. However, addition of NBP to rotenone
treated cells significantly decreased the level of Nrf2 in
cytoplasm compared with cells treated with rotenone alone (Fig. 4D). Additionally, treatment of cells
with rotenone, significantly increased the level of Nrf2 in nucleus
compared with control group, but addition of NBP to rotenone
treated cells significantly increased the level of Nrf2 in nucleus
compared with cells treated with rotenone alone (Fig. 4E and F).
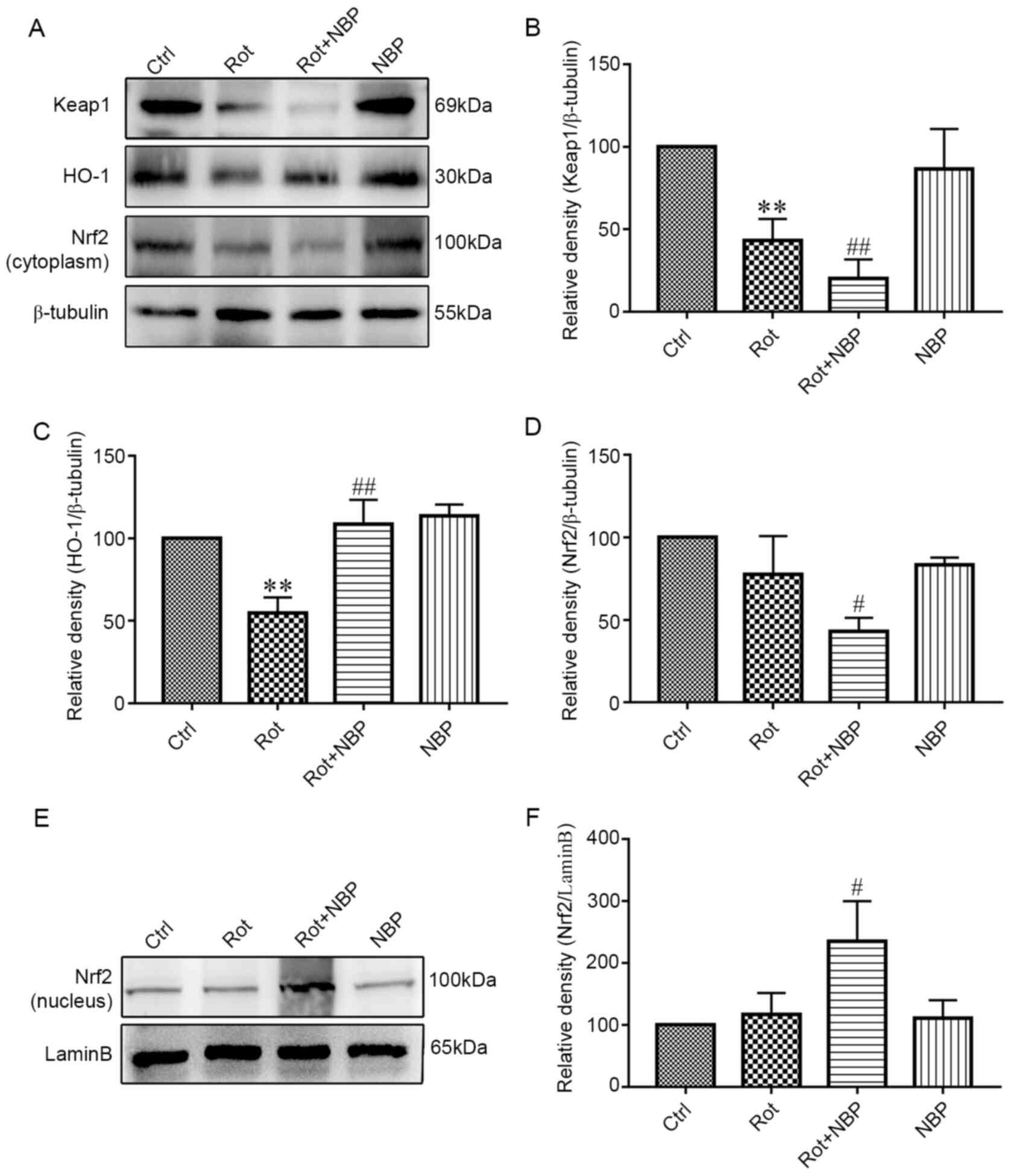 | Figure 4Effect of NBP on Keap1/Nrf2/HO-1
signaling pathway in rotenone-induced BV2 microglia. (A) The
results of cytoplasmic protein expression of Keap1/Nrf2/HO-1
signaling pathway. (B) Inhibition of the levels of Keap1 in the
cytoplasm by rotenone. Adding NBP significantly enhanced this
inhibition but NBP alone had no effect on Keap1 expression. (C)
Rotenone inhibited the levels of HO-1 but adding NBP eliminated the
inhibition. When used alone, NBP had no effect on HO-1 expression.
(D) Rotenone inhibited the levels of Nrf2 and adding NBP
significantly enhanced this effect. However, NBP alone had no
effect on Nrf2 expression. (E) The results of nuclear protein
expression indicated that Nrf2 was promoted into the nucleus. (F)
Rotenone increased the levels of Nrf2 in the nuclear and adding NBP
significantly enhanced this effect. However, NBP alone had no
effect on Nrf2 expression. The density of bands was analyzed by
ImageJ and the data was analyzed by one-way analysis of variance.
All data were expressed as mean ± standard deviation for three
independent experiments **P<0.01 vs. the control
group; #P<0.05, ##P<0.01 vs. the
Rotenone group. NBP, dl-butylphthalide; Keap1, Kelch-like
ECH-associated protein 1; Nrf2, nuclear respiratory factor-2; HO-1,
heme oxygenase-1; Ctrl, control; rot, rotenone. |
Discussion
In recent years, NBP has been shown to have powerful
effects against oxidative stress in different models. For instance,
NBP inhibits oxidative stress in K141N-induced SH-SY5Y cells and in
LPS-induced rats through activation of the Keap1/Nrf2/antioxidant
response element (ARE) signaling pathway (17,18).
Similarly, NBP reduces oxidative damage to provide neuroprotection
in mice following tumor brain injury and in rats following carbon
monoxide poisoning (19,20). In addition, NBP protects brain
tissue against cerebral ischemia-reperfusion injury by its
antioxidant activity via ERK signaling (21). NBP also works against
H2O2-induced injury in neural stem cells by
activation of the PI3K/Akt pathway (22). NBP increases the superoxide
dismutase and catalase activity and reduces malondialdehyde
activity in experimental autoimmune myositis model (23). Despite the above findings, it is
unclear how NBP regulates microglia in vitro and the
mechanisms involved remain to be elucidated. Additionally, little
is known about its effect on the oxidative stress model of
Parkinson's disease.
The pathogenesis of PD is closely associated with
mitochondrial dysfunction and oxidative stress (24). Mitochondria are the production
factory for ATP in cells. When the mitochondrion is damaged, there
is a decrease of ATP, which causes dysfunction of the sodium
potassium pump. This results in electrolyte and water balance
disorder in the cell, causing difficulties in maintaining normal
cell morphology (25). Thus,
microglia are unable maintain the original spindle shape and become
rounded following rotenone treatment (Fig. 1A). Following NBP treatment, the
number of normal BV2 cells increased significantly, which suggested
that NBP might protect mitochondrial function.
Mitochondria are surrounded by two layers of
membrane, the inner and the outer membrane. The inner mitochondrial
membrane (IMM) is implicated in mitochondrial energy conversion
whereas the outer mitochondrial membrane is the principal platform
for mitochondrial signaling (26).
In the process of respiratory oxidation, the energy produced by
mitochondria is stored in the IMM as electrochemical potential
energy. This results in asymmetric distribution of protons and
other ions on both sides of the inner membrane, which forms MMP,
maintaining MMP is essential for ATP production (27). Therefore, mitochondrial function can
be assessed indirectly by detecting MMP (28).
Mitochondrial dysfunction leads to excessive
increase in intracellular ROS and activates microglia (10). ROS generated by mitochondria can
induce rapid depolarization of MMP which further stimulates ROS
generation resulting in an amplified ROS signal leading to further
mitochondrial dysfunction (3).
Thus, the level of MMP and ROS indicate the mitochondrial function
to an extent. Therefore, in the present study, the analyses of MMP
and ROS indicated that the mitochondrial function of BV2 cells was
preserved by treatment with NBP.
Nrf2 is a transcription factor responsible for
reducing oxidative stress (29). In
normal conditions, Nrf2 is sequestered in cytoplasm by Keap1, which
keeps it in a resting state (30).
This explains why the level of Nrf2 in the nucleus did not change
when NBP was used alone. Under stressful condition, especially
under oxidative stress, Nrf2 separates from Keap1 and transfers to
the nucleus where it activates ARE and increases transcription of
Nrf2-regulated genes (31). This
increases the expression of HO-1 which, in turn, downregulates the
level of intracellular ROS (32).
Thus, Keap1/Nrf2/ARE signaling pathway is an important target for
reducing oxidative stress (33).
The present study investigated whether NBP reduced
intracellular ROS levels in BV2 cells via the Keap1/Nrf2/HO-1
signaling pathway. When rotenone was used alone, the level of HO-1
decreased, which may give a false impression. This is because the
early rise of ROS consumed the original HO-1 in the cells and the
decrease in intracellular expression level of Keap1 and Nrf2 was
not statistically significant. This suggests that the pathway may
be at the beginning of activation and so the new HO-1 had yet to be
produced. The cells treated with rotenone and NBP (Rot+NBP) showed
that NBP treatment promoted Nrf2 entry into the nucleus and
increased HO-1 expression in rotenone treated BV2 cells. In
addition, the data demonstrated that NBP treatment reduced the
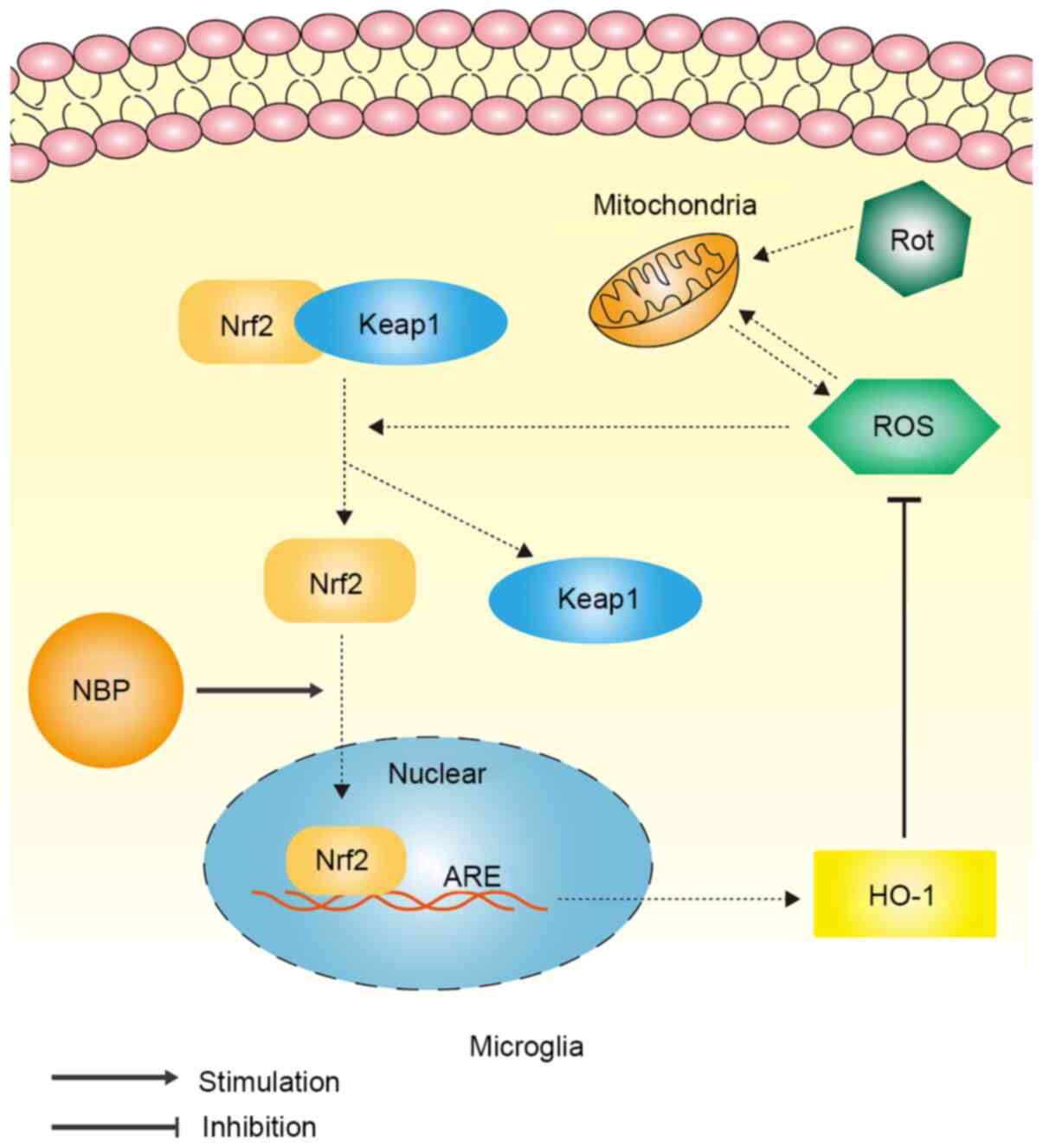
level of the Nrf2 inhibitory protein, Keap1. The overall mechanism
by which NBP inhibited rotenone-induced oxidative stress in
microglia is illustrated in Fig.
5.
In conclusion, the present study demonstrated that
NBP inhibits rotenone-induced oxidative stress in microglia via
Keap1/Nrf2/HO-1 signaling pathway. Therefore, the results supported
the notion that NBP treatment could decrease oxidative stress and
might have considerable value as a therapeutic agent against
PD.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by grants from the Natural
Science Foundation of China (grant nos. 81200930 and 82071568), the
Training program for outstanding young teachers in higher education
institutions of Guangdong Province (grant nos. YQ2015024) and the
Fundamental Research Funds for the Central Universities (grant nos.
21617482).
Availability of data and materials
The datasets analyzed during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
RL and LZ were the major contributors in writing the
manuscript and designing of the experiments. ZZ and RZ were
responsible for designing and conducting experiments. JZ and SX
were responsible for cell culturing and sample extractions. LZ and
WB confirmed the authenticity of all the raw data. WB played a
major role in the data analysis and experimental co-ordination. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Poewe W, Seppi K, Tanner CM, Halliday GM,
Brundin P, Volkmann J, Schrag AE and Lang AE: Parkinson disease.
Nat Rev Dis Primers. 3(17013)2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Nicholls DG: Mitochondrial membrane
potential and aging. Aging Cell. 3:35–40. 2004.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ganguly G, Chakrabarti S, Chatterjee U and
Saso L: Proteinopathy, oxidative stress and mitochondrial
dysfunction: Cross talk in Alzheimer's disease and Parkinson's
disease. Drug Des Devel Ther. 11:797–810. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Dias V, Junn E and Mouradian MM: The role
of oxidative stress in Parkinson's disease. J Parkinsons Dis.
3:461–491. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Subramaniam SR and Chesselet MF:
Mitochondrial dysfunction and oxidative stress in Parkinson's
disease. Prog Neurobiol. 106-107:17–32. 2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Heikkila RE, Nicklas WJ, Vyas I and
Duvoisin RC: Dopaminergic toxicity of rotenone and the
1-methyl-4-phenylpyridinium ion after their stereotaxic
administration to rats: Implication for the mechanism of
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity. Neurosci
Lett. 62:389–394. 1985.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Johnson ME and Bobrovskaya L: An update on
the rotenone models of Parkinson's disease: Their ability to
reproduce the features of clinical disease and model
gene-environment interactions. Neurotoxicology. 46:101–116.
2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Maturana MG, Pinheiro AS, de Souza TL and
Follmer C: Unveiling the role of the pesticides paraquat and
rotenone on α-synuclein fibrillation in vitro. Neurotoxicology.
46:35–43. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ye J, Jiang Z, Chen X, Liu M, Li J and Liu
N: Electron transport chain inhibitors induce microglia activation
through enhancing mitochondrial reactive oxygen species production.
Exp Cell Res. 340:315–326. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Yuan YH, Sun JD, Wu MM, Hu JF, Peng SY and
Chen NH: Rotenone could activate microglia through NFkB associated
pathway. Neurochem Res. 38:1553–1560. 2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Xu ZQ, Zhou Y, Shao BZ, Zhang JJ and Liu
C: A systematic review of neuroprotective efficacy and safety of
DL-3-N-butylphthalide in ischemic stroke. Am J Chin Med.
47:507–525. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Chinese Society of Cerebral Blood Flow and
Metabolism. The Chinese guidelines for the evaluation and
management of cerebral collateral circulation in ischemic stroke
(2017). Zhonghua Nei Ke Za Zhi. 56:460–471. 2017.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
14
|
Wang S, Ma F, Huang L, Zhang Y and Peng Y,
Xing C, Feng Y, Wang X and Peng Y: Dl-3-n-butylphthalide (NBP): A
promising therapeutic agent for ischemic stroke. CNS Neurol Disord
Drug Targets. 17:338–347. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Huang L, Wang S, Ma F, Zhang Y and Peng Y,
Xing C, Feng Y, Wang X and Peng Y: From stroke to neurodegenerative
diseases: The multi-target neuroprotective effects of
3-n-butylphthalide and its derivatives. Pharmacol Res. 135:201–211.
2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Luo R, Wangqin R, Zhu L and Bi W:
Neuroprotective mechanisms of 3-n-butylphthalide in
neurodegenerative diseases. Biomed Rep. 11:235–240. 2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhao CY, Lei H, Zhang Y, Li L, Xu SF, Cai
J, Li PP, Wang L, Wang XL and Peng Y: L-3-n-Butylphthalide
attenuates neuroinflammatory responses by downregulating JNK
activation and upregulating Heme oxygenase-1 in
lipopolysaccharide-treated mice. J Asian Nat Prod Res. 18:289–302.
2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yang XD, Cen ZD, Cheng HP, Shi K, Bai J,
Xie F, Wu HW, Li BB and Luo W: L-3-n-butylphthalide protects HSPB8
K141N mutation-induced oxidative stress by modulating the
mitochondrial apoptotic and Nrf2 pathways. Front Neurosci.
11(402)2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Liu Z, Wang H, Shi X, Li L, Zhou M, Ding
H, Yang Y, Li X and Ding K: DL-3-n-butylphthalide (NBP) provides
neuroprotection in the mice models after traumatic brain injury via
Nrf2-ARE signaling pathway. Neurochem Res. 42:1375–1386.
2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Li Q, Cheng Y, Bi M, Lin H, Chen Y, Zou Y,
Liu Y, Kang H and Guo Y: Effects of N-butylphthalide on the
activation of Keap1/Nrf-2 signal pathway in rats after carbon
monoxide poisoning. Environ Toxicol Pharmacol. 40:22–29.
2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhu BL, Xie CL, Hu NN, Zhu XB and Liu CF:
Inhibiting of GRASP65 phosphorylation by DL-3-N-butylphthalide
protects against cerebral ischemia-reperfusion injury via ERK
signaling. Behav Neurol. 2018(5701719)2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Wang S, Huang L, Zhang Y and Peng Y, Wang
X and Peng Y: Protective effects of L-3-n-butylphthalide against
H2O2-induced injury in neural stem cells by activation of PI3K/Akt
and mash1 pathway. Neuroscience. 393:164–174. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chen J, Wang J, Zhang J and Pu C:
3-n-Butylphthalide reduces the oxidative damage of muscles in an
experimental autoimmune myositis animal model. Exp Ther Med.
14:2085–2093. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Bose A and Beal MF: Mitochondrial
dysfunction in Parkinson's disease. J Neurochem. 139 (Suppl
1):216–231. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Pivovarov AS, Calahorro F and Walker RJ:
Na(+)/K(+)-pump and neurotransmitter membrane receptors. Invert
Neurosci. 19(1)2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Giacomello M, Pyakurel A, Glytsou C and
Scorrano L: The cell biology of mitochondrial membrane dynamics.
Nat Rev Mol Cell Biol. 21:204–224. 2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Zorova LD, Popkov VA, Plotnikov EY,
Silachev DN, Pevzner IB, Jankauskas SS, Babenko VA, Zorov SD,
Balakireva AV, Juhaszova M, et al: Mitochondrial membrane
potential. Anal Biochem. 552:50–59. 2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Sakamuru S, Attene-Ramos MS and Xia M:
Mitochondrial membrane potential assay. Methods Mol Biol.
1473:17–22. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Mitsuishi Y, Motohashi H and Yamamoto M:
The Keap1-Nrf2 system in cancers: Stress response and anabolic
metabolism. Front Oncol. 2(200)2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lu MC, Ji JA, Jiang ZY and You QD: The
Keap1-Nrf2-ARE pathway as a potential preventive and therapeutic
target: An update. Med Res Rev. 36:924–963. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Loboda A, Damulewicz M, Pyza E, Jozkowicz
A and Dulak J: Role of Nrf2/HO-1 system in development, oxidative
stress response and diseases: An evolutionarily conserved
mechanism. Cell Mol Life Sci. 73:3221–3247. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Chau LY: Heme oxygenase-1: Emerging target
of cancer therapy. J Biomed Sci. 22(22)2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Chen QM and Maltagliati AJ: Nrf2 at the
heart of oxidative stress and cardiac protection. Physiol Genomics.
50:77–97. 2018.PubMed/NCBI View Article : Google Scholar
|